

Журнал неорганической химии, 2019, T. 64, № 12, стр. 1294-1303
Новые молекулярные хемосенсоры на основе ниобий(V) 5,10,15,20-(тетра-4-трет-бутилфенил)порфина для обнаружения VOCs
Е. В. Моторина 1, Т. Н. Ломова 1, *, Е. Г. Можжухина 1, М. С. Груздев 1
1 Институт химии растворов им. Г.А. Крестова РАН
153045 Иваново, ул. Академическая, 1, Россия
* E-mail: tnl@isc-ras.ru
Поступила в редакцию 06.05.2019
После доработки 29.05.2019
Принята к публикации 17.06.2019
Аннотация
Изучены реакции пиридина (Py) с (5,10,15,20-(тетра-4-трет-бутилфенил)порфинато)трихлорониобием(V) (Nb(Cl)3TtBuPP) и H+-связанным Nb(Cl)3TtBuPP (Nb(Cl)3TtBuPP…H+…Cl−) в среде толуола методами спектроскопии (УФ, видимая, ИК-, ЯМР 1H, масс-спектрометрия, флуоресценция), химической термодинамики и кинетики. Взаимодействие представляет собой последовательность двух- и односторонних реакций связывания двух молекул пиридина, природа которого определяется химическим строением исходного ниобий(V)порфирина. Получено полное количественное описание реакций и определены параметры спектров промежуточных и конечных продуктов, на основании чего установлена химическая структура последних. Обоснована перспектива использования Nb(Cl)3TtBuPP и Nb(Cl)3TtBuPP…H+…Cl− в качестве оптических и флуоресцентных хемосенсоров VOCs (высоколетучие органические соединения) и азотистых оснований – “строительных блоков” фармацевтических препаратов, компонентов пищи и загрязнителей окружающей среды − с параметрами: константа устойчивости комплекса с Py K = (1.99 ± 0.3) × 104 л2/моль2 и (2.8 ± 0.5) × 102 л/моль, относительный оптический отклик A = 0.91 и 0.35, предел обнаружения Py 1.74 × 10–3 и 4.05 × 10–4 моль/л соответственно. Результаты полезны для использования при дизайне DSSCs (сенсибилизированные красителем солнечные элементы), так как изученная реакция является модельной в отношении самосборки донорно-акцепторных систем на основе металлопорфиринов и пиридильных производных наноформ углерода.
ВВЕДЕНИЕ
В настоящее время координационная химия ниобия активно изучается; в научной литературе представлено огромное количество работ по синтезу, идентификации и реакциям лигандного обмена комплексов с координационной сферой переменного состава, в структуру которых не входит порфириновый лиганд [1–7]. Описаны комплексы с сорбционной активностью по отношению к красителям [8] и способностью к встраиванию кислорода в ниобий-фосфорные связи с высвобождением синтетически значимых фосфорсодержащих молекул [9]. Особый интерес представляют смешанные порфиринсодержащие комплексы ниобия, в которых центральный атом чаще всего находится в степени окисления +5. Благодаря своим уникальным свойствам смешанные комплексы порфиринов являются мощным инструментом молекулярного распознавания. Связывание иона металла с плоским порфириновым макроциклом обеспечивает дополнительные места для аксиальной координации, что можно использовать для детектирования малых органических молекул, электро- и химического катализа. Для комплексов O=NbVLP и O=NbIVP (Р – дианионы тетрафенилпорфина (ТРР), октаэтилпорфина (OEP), тетра-4-толилпорфирина (TTP), L – AcO− или Acac−) описаны механизмы окисления-восстановления в неводных растворах [10]. В обзоре [11] изучена пространственная и электронная структура Cl3NbPc (Рс – фталоцианин-дианион). Описан пример использования NbIVPc в качестве активного слоя при разработке новых катализаторов. В [12] представлены разработки химических зондов на основе порфиринов для катионов, анионов, ионных пар, летучих органических химикатов, нитроароматических соединений, газов, активных форм кислорода.
В настоящее время в хемосенсорике, биологической химии, физиологии и медицине термин “сенсор” ассоциируется с молекулой или молекулярным устройством. В том случае, если молекула дает отчетливый отклик (измеримый сигнал) при связывании определяемого объекта, мы имеем дело с хемосенсором (ХС) [13]. Комплексы порфиринов, в которых не достигнуто максимально возможное координационное число центрального иона, могут быть использованы в качестве оптических ХС [14–17].
Ранее в наших работах толуольные растворы комплексов ниобия(V), молибдена(V), вольфрама(V), циркония(IV), гафния(IV), марганца(III) с порфиринами были использованы для обнаружения летучих органических соединений (VOCs) и гетероциклических компонентов пищевых продуктов [18, 19], а также при изучении самособирающихся систем с фотоиндуцированным электронным транспортом (РЕТ) [20]. Цель настоящей работы – поиск и исследование новых химических VOCs-сенсоров на основе комплексов порфиринов. Для этого синтезированы комплексы 5,10,15,20-(тетра-4-трет-бутилфенил)21H,23H-порфина (Н2TtBuPP) c Nb(V) (формулы I, II), исследовано их строение, спектральные свойства и реакции с органическим основанием – пиридином (Py). Оптические характеристики в контролируемых условиях (CI,II, CPy, λраб, T (K), среда − толуол) были получены путем измерения электронных спектров поглощения (ЭСП) комплексов Nb в толуоле и в смесях толуол−Py с нарастающей концентрацией последнего.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
(5,10,15,20-(Тетра-4-трет-бутилфенил)порфинато)трихлорониобий(V) (Cl)3NbTtBuPP (I) получали по известной методике: 0.03 г (0.049 ммоль) H2TtВuPP, 0.068 г (0.25 ммоль) NbCl5 и 8 мл бензонитрила кипятили с обратным холодильником (464 K) в течение 1 ч. Бензонитрил отгоняли из реакционной смеси под вакуумом. Далее смесь растворяли в хлороформе и проводили двукратную хроматографию с использованием CHCl3. Вначале раствор в CHCl3 помещали на колонку с Al2O3. Получили три последовательно идущие зоны: коричневую, серо-коричневую (небольшие количества примесей неустановленного состава) и ярко-оранжевую (основной конечный продукт). Ярко-оранжевая зона адсорбируется в верхней части колонки и смывается смесью хлороформ–этанол (20 : 1). Вторую хроматографию вещества этой зоны проводили на колонке с силикагелем, в качестве элюента вначале использовали CHCl3, а затем смесь хлороформ–этанол (20 : 1). Выход (Cl)3NbTtBuPP – 60%. ЭСП в толуоле (λmax, нм (lg ε)): 334.0 (4.26), 419.0 (5.14), 551.0 (4.01). ИК‑спектр (KBr, ν, см–1): колебания бензольных колец 722, 741 (γC–H); 1070, 1109 (δC–H); 1500, 1520, 1607 (νC=C), 2961 (νC–H); колебания пиррольных колец 801, 812 (γC–H); 995 (C3–C4, νC–N, δC–H); 1342 (νC–N); 1461 (νC=N); 1395, 1363, 1267, 1203 (–С(СН3)3); Nb–Cl 438; Nb–N 528. ЯМР 1H (400 MГц; δ, м. д.; J, Гц; CDCl3): 8.64 (с, 8Hβ); 7.73 (д, J = 7.9 Гц, 8Hо, 8Hм); 1.66 (д, J = 11.74 Гц, ${\text{36}}{{{\text{H}}}_{{^{t}{\text{Bu}}}}}$). MS (MALDI-TOF) (m/z): 945.81 [М−3Cl + O]+.
H+-Связанный (5,10,15,20-(тетра-4-трет-бутилфенил)порфинато)-трихлорониобий(V) Nb(Cl)3TtBuPP…H+…Cl− синтезировали по методике [21]. Пары HCl пропускали через толуольный раствор Nb(Cl)3TtBuPP в течение 30 мин. Толуол отгоняли из реакционной смеси под вакуумом. Выход Nb(Cl)3TtBuPP…H+…Cl− составил ∼100%. ЭСП в толуоле (λmax, нм): 313.0 (4.69), 426.0 (5.70), 542.0 (3.31). ИK-спектр (KBr, ν, см–1): колебания бензольных колец 722, 743 (γC–H); 1109, 1072 (δC–H); 1501, 1609 (νC=C); 2962 (νC-H); колебания пиррольных колец 801, 813 (γC–H); 996 (C3–C4, νC–N, δC–H); 1342 (νC–N); 1462 (νC=N); 1394, 1363, 1268, 1204 (–С(СН3)3); Nb–N 527; Nb–Cl 440 [25]. ЯМР 1H (400 МГц; δ, м. д.; CDCl3): 8.67 (с, 8Hβ); 7.72 (уш. с, 8Hо, 8Hм); 4.07 (с, 1${\text{H}}_{{\text{H}}}^{{\text{ + }}}$); 1.64 (с, ${\text{36}}{{{\text{H}}}_{{^{t}{\text{Bu}}}}}$). MS (MALDI-TOF), m/z: 945.92 [М−3Cl + O]+.
Хлорид дихлоро(5,10,15,20-(тетра-4-трет-бутилфенил)порфинато) (дипиридин)ниобия(V) [Cl2(Py)2NbTtBuPP]+Cl− синтезировали по реакции (Cl)3NbTtBuPP с Py в толуоле при 298 K в течение 3 ч с выходом, близким к 100%. ЭСП в толуоле (λmax, нм): 429, 523, 584. ИK-спектр (KBr, ν, см–1): колебания бензольных колец 723, 743 (γC–H); 1071, 1109 (δC–H); 1499, 1606 (νC=C); 2961 (νC–H); колебания пиррольных колец 801, 812 (γC–H); 995 (C3–C4, νC–N, δC–H); 1341 (νC–N); 1462 (νC=N); 1394, 1363, 1267, 1207 (‒С(СН3)3); Nb–N 525; Nb–Cl 440. ЯМР 1H (400 МГц; δ, м. д.; J, Гц; CDCl3): 8.67 (с, 8Hо); 8.64 (с, 8Hβ, 4Hо (Py)); 7.72 (д, J = 8.4 Гц, 8Hм, 4Hм (Py)); 7.43 (уш. с, 2Hп (Py)), 1.64 (с, ${\text{36}}{{{\text{H}}}_{{^{t}{\text{Bu}}}}}$).
H+-Связанный трихлоро(5,10,15,20-(тетра-4-трет-бутилфенил)порфинато)(пиридин)ниобий(V) Cl3(Py)NbTtBuPP…H+…Py синтезировали из Nb(Cl)3TtBuPP…H+…Cl− по реакции с Py в толуоле при 298 K в течение 3 ч с выходом, близким к 100%. ЭСП в толуоле (λmax, нм): 420, 554, 592. ИК-спектр (KBr, ν, см–1): колебания бензольных колец 721, 742 (γC–H); 1108, 1070, (δC–H); 1499, 1608 (νC=C); 2962 (νC–H); колебания пиррольных колец 801, 813 (γC–H); 995 (C3–C4, νC–N, δC–H); 1341 (νC–N); 1461 (νC=N); 1394, 1363, 1267, 1203 (–С(СН3)3); Nb–N 526; Nb–Cl 438. ЯМР 1H (400 МГц; δ, м. д.; J, Гц; CDCl3): 8.65 (с, 8Hβ); 8.40 (с, 8Hо, 4Hо (Py)); 7.72 (с, 8Hм), 7.62 (с, 4Hм, 2Hп (Py)); 4.06 (с, ${\text{1}}{{{\text{Н}}}_{{{{{\text{Н}}}^{ + }}}}}$); 1.66 (д, J = 12.20 Гц, ${\text{36}}{{{\text{H}}}_{{^{t}{\text{Bu}}}}}$).
Равновесия и скорости реакций I и II с Py исследовали спектрофотометрически соответственно методами молярных отношений и избыточных концентраций. Растворы I, II и Py в свежеперегнанном толуоле готовили непосредственно перед использованием во избежание образования перекисей в среде растворителя. Оптическую плотность раствора I (II) с концентрацией 2.45 × × 10−6 моль/л (2.84 × 10−6 моль/л) и концентрацией основания 1.74 × 10−3–6.2 × 10−1 моль/л (4.05 × × 10−4−2.7 × 10−1 моль/л) измеряли на рабочей длине волны 417 нм (424 нм) в начальный момент времени (τ = 0) и во времени. Растворы термостатировали с погрешностью ±0.1 K при 298 K в закрытых кварцевых кюветах в специальной ячейке спектрофотометра. Данные спектрофотометрического титрования в концентрационном и временнóм полях (метод зависимого от времени спектрофотометрического титрования [22]) использовали для определения в одной экспериментальной серии констант равновесия, скоростей односторонних реакций и времени установления равновесия (по минимальной ошибке в численном значении константы равновесия, рассчитанной при разных значениях τ).
Константы равновесий двухсторонних реакций определяли по формуле (1), выведенной с использованием законов действующих масс и Бугера–Ламберта–Бера, математическое выражение которого записывали для смеси двух окрашенных соединений – I (II) и продукта его реакции с Py.
(1)
$K = \frac{{\frac{{{{A}_{p}} - {{A}_{{\text{0}}}}}}{{{{A}_{\infty }} - {{A}_{{\text{0}}}}}}}}{{1 - \frac{{{{A}_{p}} - {{A}_{{\text{0}}}}}}{{{{A}_{\infty }} - {{A}_{{\text{0}}}}}}}}\frac{1}{{{{{\left( {{{C}_{{\text{L}}}} - C_{{{\text{MP}}}}^{0}\frac{{{{A}_{p}} - {{A}_{{\text{0}}}}}}{{{{A}_{\infty }} - {{A}_{{\text{0}}}}}}} \right)}}^{n}}}},$Константы скорости односторонних реакций при различных концентрациях Py рассчитывали по уравнению формального первого порядка при условии избытка Py по отношению к ниобий(V)порфирину:
(2)
${{k}_{{{\text{эф}}}}} = \frac{1}{\tau }\ln \frac{{{{A}_{{\text{0}}}} - {{A}_{\infty }}}}{{{{A}_{\tau }} - {{A}_{\infty }}}},$Оптимизацию величин K и kэф с определением их средних квадратичных отклонений проводили методом наименьших квадратов (МНК) в программе Microsoft Excel. Относительная ошибка в определении K и kэф не превышала соответственно 27 и 10%. Число присоединяемых молекул Py (n в уравнении (1)) определяли по тангенсу угла наклона прямой в координатах lgI−lg${{C}_{{{\text{Py}}}}}$, где I = = Aр − A0/A∞ − Aр, порядок реакции по Py – из зависимости lg kэф−lg ${{C}_{{{\text{Py}}}}}$.
Py “ч. д. а.” высушивали в течение 2 сут над гранулами KOH, затем перегоняли (tкип = = 115.3°С). Толуол осушали гидроксидом калия и перед использованием перегоняли (tкип = = 110.6°С). Содержание воды определяли титрованием по Фишеру, оно не превышало 0.01%.
ЭСП, ИК-, ЯМР 1H и масс-спектры записывали соответственно на спектрофотометре UV-Vis Agilent 8453, спектрометрах VERTEX 80v, AVANCE-500 и масс-спектрометре Shimadzu AXIMA Confidence. Спектры флуоресценции регистрировали в термостатируемой ячейке на основе элемента Пелтье при 298 K. В качестве источника света использовали облучатель Иволга ОМС-1 (Люмикс, Россия) с монохроматором ЛМ-4 (Люмикс, Россия).
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
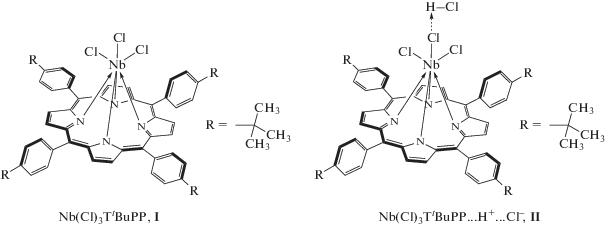
Химическое строение полученных комплексов (формулы I, II) установлено по параметрам ЭСП, ИК-, ЯМР 1H и масс-спектров. ЭСП комплексов I и II (рис. 1) относятся к спектрам “гисо-типа” [23], что подтверждает формальный заряд +5 и электронную конфигурацию 4d0 центрального атома в обоих комплексах. Различие состоит в батохромном и гипсохромном смещении полос B(0, 0) и Q(0, 1) соответственно от 419 до 426 и от 551 до 542 нм в спектре комплекса II по сравнению со спектром I. В ИК-спектрах обоих комплексов, демонстрирующих высокое сходство, проявляются сигналы характеристических колебаний пиррольных и бензольных колец, трет-бутильных групп, а также связей Nb–N и Nb–Cl. Особенностью спектра комплекса II является небольшое уменьшение относительной (по сравнению с самым интенсивным сигналом в спектре с частотой 995 см−1) интенсивности поглощения, отвечающего колебаниям Nb–Cl, и появление малоинтенсивного низкочастотного сигнала при 408 см−1 (рис. 2б, кривая 1), которые указывают на различия в координационной сфере комплексов по аксиальной оси. Характерно, что в спектрах как I, так и II не обнаруживаются сигналы оксолиганда. Последний, однако, появляется в образцах вместо трех хлорид-ионов при снятии (независимо от присутствия или отсутствия матрицы) масс-спектров (рис. 3), демонстрирующих единственный сигнал молекулярного иона [O=NbTtBuPP]+. Это не удивительно с учетом известного факта, что лиганды слабого поля, каковым является Cl−, всегда отсутствуют в молекулярном ионе аксиально координированных порфириновых комплексов [23]. Наконец, очень небольшое (0.02–0.03 м. д.) слабопольное смещение сигналов β-протонов в спектре ЯМР 1H комплекса II по сравнению с I и обратное − для сигналов протонов трет-бутильных групп, указывающее, соответственно, на усиление в магнитном поле кольцевого тока в макроцикле и уменьшение эффективного отрицательного заряда на заместителях в мезо-фенилах, является индикатором ослабления аксиальных взаимодействий в комплексе II. Об этом свидетельствует также расщепление сигнала протонов трет-бутильных групп в спектре I и соответствующий одиночный синглет в спектре соединения II, что является результатом существенного нарушения копланарности макроцикла из-за смещения Nb в сторону аксиальных лигандов, уменьшающегося при ослаблении аксиальных взаимодействий. С учетом получения соединения II обработкой комплекса I соляной кислотой и процедуры его выделения в твердом виде комплексу II следует приписать химическую структуру с водородно-связанным координированным хлорид-ионом (Nb(Cl)3TtBuPP…H+…Cl−, формула II).
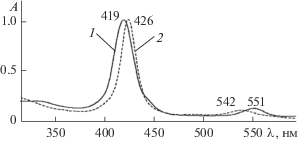
Рис. 2.
ИК-спектры в таблетках с KBr до (1) и после обработки Nb(Cl)3TtBuPP (а) и Nb(Cl)3TtBuPP…H+…Cl− (б) пиридином (2).
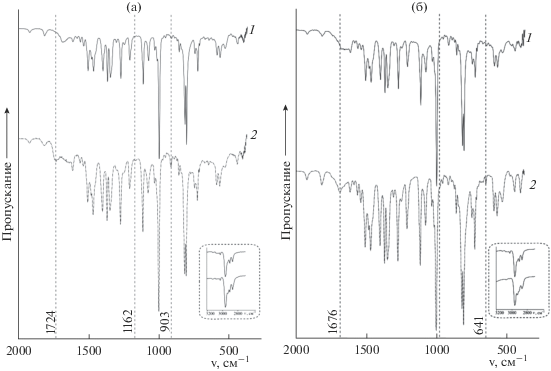

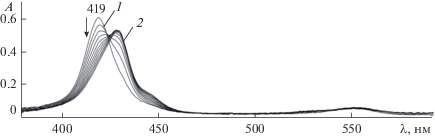
Хемосенсорную активность соединений I и II в отношении Py исследовали в свежеперегнанном толуоле путем сравнения их ЭСП до и после добавления основания в различной концентрации. Отчетливый оптический отклик, который дают молекулы комплексов при связывании Py, демонстрирует рис. 4. Видно, что оптический ответ ниобий(V)порфиринов зависит от их химического строения. В области поглощения B-полосы проявляются более выраженные изменения по мере увеличения концентрации Py. Для комплекса I наблюдается батохромное смещение полосы от 419 до 429 нм и гипсохромное от 551 до 523 нм в длинноволновой области спектра. В случае соединения II добавление Py индуцирует гипсохромное смещение В-полосы от 426 до 420 нм и появление нового малоинтенсивного поглощения при 554 нм. В случае ранее изученной [19] системы с ХС (Cl)О=NbТPP (III) в диапазоне низких концентраций Py (4.96 × 10–4−2.98 × 10–2 моль/л) происходит лишь закономерное уменьшение интенсивности максимума при 417 нм, однако затем (концентрация Py от 2.98 10–2 до 3.1 моль/л) картина спектрального отклика изменяется. Таким образом, важно отметить, что три комплекса ниобия(V) представляют разные оптические отклики на одно и то же основание. Относительный оптический отклик Аопт для соединений I, II и III составил 0.91 (диапазон концентраций Py 1.74 × × 10−3–6.2 × 10−1 моль/л), 0.35 (СPy = 4.05 × 10−4–2.7 × 10−1 моль/л) и 0.42 (СPy = 4.96 × 10−4–2.98 × × 10−2 моль/л), т.е. наиболее чувствительным ХС оказался Nb(Cl)3TtBuPP.
Рис. 4.
Электронные спектры поглощения при τ = 0 Nb(Cl)3TtBuPP (С = 2.45 × 10−6 моль/л) (1) и Nb(Cl)3TtBuPP…H+…Cl− (С = 2.84 × 10−6 моль/л) (3) в толуоле и с добавками пиридина 6.2 × 10–1 (2) и 2.7 × 10–1 (4) моль/л.
На рис. 5, 6 и в табл. 1 приведены результаты спектрофотометрического титрования толуольных растворов I и II пиридином для соответствующих диапазонов концентраций последнего. Из этих данных следует, что реакция с Py проходит в одну обратимую стадию с участием двух (соединение I) или одной (соединение II) молекул Py. В равновесных смесях во времени фиксируются медленные процессы, для которых с использованием оригинальной методики сбора данных по зависимому от времени титрованию определены константы скорости k и порядки по Py (табл. 1).
Рис. 5.
Кривые спектрофотометрического титрования Nb(Cl)3TtBuPP (а) и Nb(Cl)3TtBuPP…H+…Cl– (б) с Py в толуоле при 298 K.
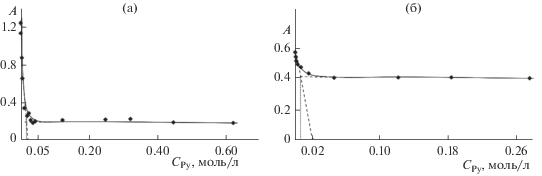
Рис. 6.
Зависимость lg((Ap − A0)/(A∞ − Aр)) – lgCL для реакций Nb(Cl)3TtBuPP (а) и Nb(Cl)3TtBuPP…H+…Cl– (б) с Py в толуоле при 298 K. (R2 = 0.965 (а), R2 = 0.995 (б)).
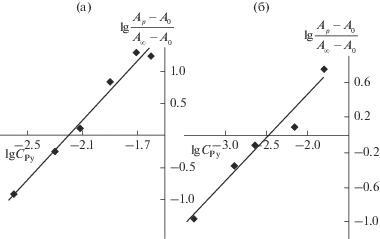
Таблица 1.
Константы равновесия K, число реагирующих молекул Py (tg α), скорости k и порядки реакций по Py (m) для Nb(Cl)3TtBuPP, Nb(Cl)3TtBuPP…H+…Cl–, (Cl)О=NbTPP. Толуол, 298 K
| MP | K | tg α | k, с–1 | m |
|---|---|---|---|---|
| Nb(Cl)3TtBuPP | (1.99 ± 0.3) × 104 л2/моль2 | 2.25 | 1.94 × 10–4 | 0 |
| (Nb(Cl)3TtBuPP…H+…Cl– | (2.8 ± 0.5) × 102 л/моль | 0.99 | 2.18 × 10–4 | ≈1.0 |
| (Cl)О=NbTPP | K1 = (2.2 ± 0.6) × 105 л2/моль2 | 2.15 | 6.92 × 10–5 | 0 |
| K2 = 5.19 ± 1.44 л/моль | 0.90 | 4.81 × 10–5 | 0 |
Спектрофотометрический мониторинг изменения оптических полос поглощения от взаимодействия с Py вместе с количественными параметрами равновесий и односторонних реакций позволяет записать следующую схему рассматриваемых превращений:
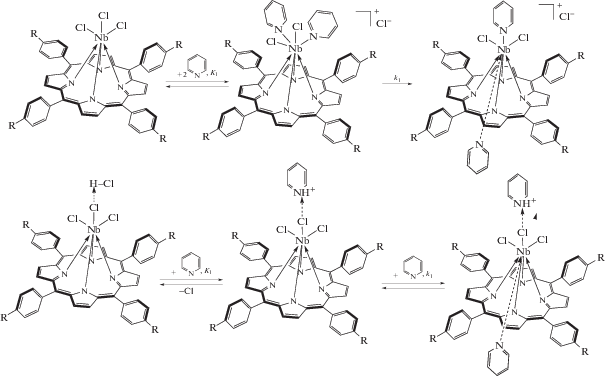
Схема 1. Простые реакции в ходе взаимодействия Nb(Cl)3TtBuPP (верх) и Nb(Cl)3TtBuPP…H+…Cl− (низ) с пиридином.
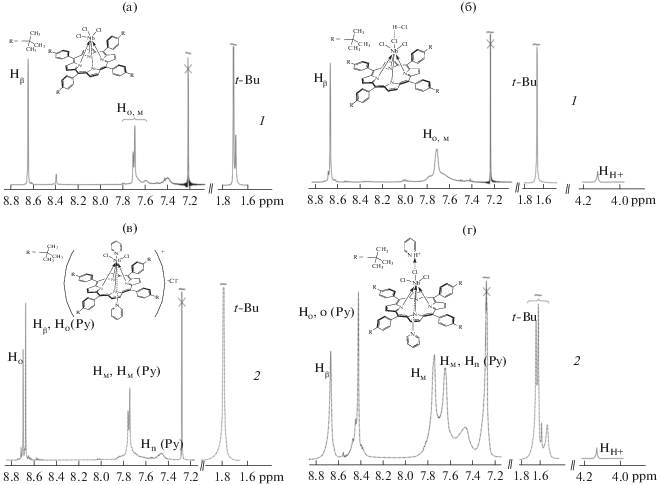
Видно, что присоединение двух молекул Py комплексами I и II различается по природе взаимодействий и строению продукта. В комплексе I имеет место вытеснение одного из хлорид-ионов во вторую координационную сферу, в то время как в продукте реакции соединения II хлорид-ион остается в первой координационной сфере, будучи связанным водородной связью с протоном сопряженной кислоты пиридина. Оба продукта, образующихся при избытке Py – более слабого по сравнению с хлорид-ионом основания, устойчивы и могут быть выделены в твердом виде и исследованы спектрально. Образцы продуктов реакций для снятия ИК-спектров [Cl2(Py)2NbtBuPP]+Cl− (IV) и Cl3(Py)NbtBuP…H+…Py (V) были получены вакуумной отгонкой пиридина из смеси состава, соответствующего точке эквивалентности при титровании (когда весь исходный комплекс оттитрован). Новые сигналы с частотами колебаний координированного Py представлены в табл. 2 (рис. 2, кривые 2) [24]. Изменение структуры координационного узла за счет связывания Py подтверждают и спектры ЯМР 1Н. На рис. 7 (кривые 2) представлены спектры ЯМР 1Н соединений IV и V, полученные добавлением пиридина (в концентрации, соответствующей точке эквивалентности при титровании) к растворам I и II в CDCl3. Образование донорно-акцепторных комплексов I и II с Py сопровождается появлением в спектрах новых характерных сигналов протонов координированного Py. Так, наблюдается сильный слабопольный сдвиг орто-протонов фенильных заместителей от 7.73 (I) до 8.67 м. д. (IV) и от 7.72 (II) до 8.4 м. д. (V), причем новый синглет при 8.4. м. д. − общий для орто-протонов мезо-заместителей макроциклического лиганда и координированного основания. Сигналы трет-бутильной группы регистрируются при 1.66 м. д. в случае I, однако под воздействием основания происходит небольшой сильнопольный сдвиг до 1.64 м. д. (рис. 7а, 2). В спектре соединения V наблюдается сильнопольный сдвиг Hβ сигналов относительно соединения II от 8.67 до 8.65 м. д. Протоны трет-бутильной группы соединения V проявляются в спектре в виде уширенных сигналов в области 1.66–1.64 м. д. Таким образом, картина трансформации спектров ЯМР 1Н комплексов I и II после взаимодействия с Py не противоречит идее более копланарного расположения Nb в плоскости экваториального лиганда за счет появления транс-лиганда Py и ослабления (в комплексе I) аксиального связывания и связанного с этим усиления кольцевого тока в макроцикле при регистрации спектра.
Таблица 2.
Сигналы координированного Py в ИК-спектрах соединений [Cl2(Py)2NbtBuPP]+Cl− и Cl3(Py)NbtBuP…H+…Py и соответствующие им частоты чистого Py
| [Cl2(Py)2NbtBuPP]+Cl− | Cl3(Py)NbtBuP…H+…Py | ||
|---|---|---|---|
| ν, см–1 (Py) | ν, см–1 (Pyкоорд) | ν, см–1 (Py) | ν, см–1 (Pyкоорд) |
| 1718 | 1724 | 1681 | 1676 |
| 1148 | 1162 | 974 | 968 |
| 940 | 903 | 701 | 641 |
Рис. 7.
Спектры ЯМР 1Н в СDCl3 до (1) и после обработки пиридином (2) для соединений Nb(Cl)3TtBuPP (а) и Nb(Cl)3TtBuPP…H+…Cl− (б).
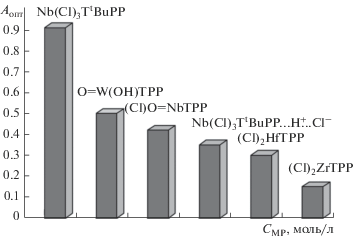
На рис. 8 представлена диаграмма чувствительности обнаружения Py с использованием ХС I, II и III и изученных ранее О=W(OH)ТPP, (Cl)2HfTPP и (Cl)2ZrТPP. Видно, что повышение формального заряда от М4+ к М5+ приводит к значительному увеличению чувствительности металлорганических сенсоров и эффективности детектирования гетероциклического основания. Так, из диаграммы следует, что наиболее высокую сенсорную активность проявляют пятизарядные оксокомплексы, в то же время соединение I проявляет свойства самого эффективного ХС в отношении Py. Минимальный предел определения концентрации Py (Сlim), найденный в соответствии с рекомендациями ИЮПАК [25–27] как абсцисса в точке пересечения линейных участков калибровочной кривой A−f(CPy), для ХС I, II и III составляет 1.24 × 10−2, 5.0 × 10−3 и 4.9 × × 10−3 моль/л.
Рис. 8.
Диаграмма чувствительности сенсоров Nb(Cl)3TtBuPP, О=W(ОН)TPP, (Cl)O=NbTPP, Nb(Cl)3TtBuPP…H+…Cl–, (Cl)2HfTPP, (Cl)2ZrTPP по отношению к Py.
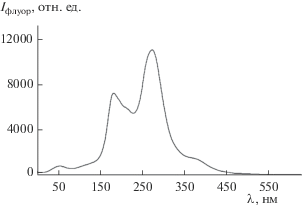
Предварительные исследования, в которых Py был заменен на замещенный аналог 1-метил-2-(пиридин-4'-ил)-3,4-фуллеро[60]пирролидин (PyF) (рис. 9), показали прохождение в смеси комплекс I–PyF–толуол самосборки, что может быть использовано для получения донорно-акцепторной системы, пригодной для создания систем с PET в оптоэлектронике. Установлено также, что соединение I обладает флуоресцентными свойствами и является флуорофором (рис. 10). Известно, что для флуоресцентного ХС очень важно правильно подобрать составляющие части, самый удачный вариант – флуорофор–мостик–рецептор. В нашем случае флуорофор – макроцикл, рецептор – комплексообразователь, отвечающий за селективное распознавание Py благодаря наличию свободных координационных мест; мостик заменяет связь М–N. Надо заметить, что его наличие не обязательно [28], так как связывание детектируемого основания может иметь другую природу.
ЗАКЛЮЧЕНИЕ
Результаты спектрального, термодинамического и кинетического исследования взаимодействия Py с Nb(Cl)3TtBuPP и его H+-связанным аналогом Nb(Cl)3TtBuPP…H+…Cl− показали, что оно представляет собой сложную последовательность двух- и односторонних реакций связывания двух молекул пиридина, природа которого определяется химическим строением исходного ниобий(V)порфирина. Nb(Cl)3TtBuPP по величине относительного оптического отклика на присутствие Py Аопт = 0.91 является наиболее чувствительным ХС среди изученных ранее металлопорфиринов. Для характеристики возможностей методики с точки зрения количественного анализа нами определен минимальный концентрационный предел определения VOCs-основания − для сенсора Nb(Cl)3TtBuPP Сlim ≈ 1.24 × 10−2 моль/л, для Nb(Cl)3TtBuPP…H+…Cl−Сlim ≈ 5.0 × 10−3 моль/л. Высокие показатели хемосенсорной активности и чувствительности позволяют рекомендовать изученные металлопорфирины для использования в качестве новых эффективных оптических и, вероятно, флуоресцентных ХС VOCs-основания Py. Экспериментально показана также реакционная способность Nb(Cl)3TtBuPP по отношению к пиридину, модифицированному фуллеренсодержащим заместителем, обещающая перспективу в создании на основе порфиринов ниобия(V) донорно-акцепторных систем со свойством PET.
Список литературы
Ziolek M., Sobczak I. // Catalysis Today. 2017. V. 285. P. 211. https://doi.org/10.1016/j.cattod.2016.12.013
Carmalt C.J., Dinnage C.W., Parkin I.P. et al. // Inorg. Chem. 2002. V. 41. № 14. P. 3668. https://doi.org/10.1021/ic020097l
Kilgore U.J., Tomaszewski J., Fan H. et al. // Organometallics. 2007. V. 26. № 25. P. 6132. https://doi.org/10.1021/om7008233
Matsuo T., Kawaguchi H. // Inorg. Chem. 2002. V. 41. № 23. P. 6090. https://doi.org/10.1021/ic025882c
Bayot D., Devillers M. // Coord. Chem. Rev. 2006. V. 250. № 19-20. P. 2610. https://doi.org/10.1016/j.ccr.2006.04.011
Rolf W. Berg. // Coord. Chem. Rev. 1992. V. 113. P. 1.
Poursaberi T., Ganjali M.R., Hassanisadi M. // Talanta. 2012. V. 101. P. 128.
Yuan Lv-Bing, He Yan-Ping, Zhang Lei et al. // Inorg. Chem. 2018. V. 57. № 8. P. 4226. https://doi.org/10.1021/acs.inorgchem.7b03265
Morello G.R., Cundari T.R. // Organometallics. 2016. V. 35. № 20. P. 3624. https://doi.org/10.1021/acs.organomet.6b00679
Anderson J.E., Liu Y.H., Guilard R. et al. // Inorg. Chem. 1986. V. 25. № 21. P. 3786. https://doi.org/10.1021/ic00241a017
Wong E.W.Y., Walsby C.J., Storr T. et al. // Inorg. Chem. 2010. V. 49. P. 3343. https://doi.org/10.1021/ic902409n
Hosoowi Lee, Kyeong-Im Hong, Woo-Dong Jang. // Coord. Chem. Rev. 2018. V. 354. P. 46. https://doi.org/10.1016/j.ccr.2017.06.008
Callan J.F., de Silva A.P., Magri D.C. // Tetrahedron. 2005. V. 61. № 38. P. 8551.
Tiening Jin, Junchao Zhou, Hao-Yu Greg Lin et al. // Anal. Chem. 2019. V. 91. № 1. P. 817. https://doi.org/10.1021/acs.analchem.8b03004
Colombelli A., Manera M.G., Borovkov V. // Sensors and Actuators B: Chem. 2017. V. 246. P.1039. https://doi.org/10.1016/j.snb.2017.01.192
Lomova T.N., Klyueva M.E., Berezin B.D. // Russ. J. Chem. Chem. Technol. 1988. V. 31. № 12. P. 75. [Ломова Т.Н., Клюева М.Е., Березин Б.Д. // Изв. вузов. Серия: Химия и хим. технология. 1988. Т. 31. № 12. С. 75.]
Tipugina M.Y., Lomova T.N. // Russ. J. Phys. Chem. 2002. V. 76. № 4. P. 567. [Типугина М.Ю., Ломова Т.Н. // Журн. физ. химии. 2002. Т. 76. С. 653.]
Lomova T.N., Motorina E.V., Ovchenkova E.N. et al. // Russ. Chem. Bull. 2007. V. 56. № 4. P. 660. [Ломова Т.Н., Моторина Е.В., Овченкова Е.Н. и др. // Изв. АН. Сер. хим. 2007. Т. 56. № 4. С. 636.] https://doi.org/10.1007/s11172-007-0105-1
Motorina E.V., Mozhzhukhina E.G., Lomova T.N. // J. Struct. Chem. 2018. V. 59. № 8. Р. 1880. [Моторина Е.В., Можжухина Е.Г., Ломова Т.Н. // Журн. структур. химии. 2018. Т. 59. Вып. 8. С. 1942.] https://doi.org/10.1134/S0022476618080164
Motorina E.V., Lomova T.N., Klyuev M.V. // Mendeleev Commun. 2018. V. 28. P. 426. https://doi.org/10.1016/j.mencom.2018.07.029
Matsuda Y., Murakami Y. // Coord. Chem. Rev. 1988. V. 92. P. 157.
Ломова Т.Н. Теоретические и экспериментальные методы химии растворов (Проблемы химии растворов). М.: Проспект, 2011.
Ломова Т.Н. Аксиально координированные металлопорфирины в науке и практике. М.: Красанд, 2018.
Большаков Г.Ф., Глебовская Е.А. Таблицы частот инфракрасных спектров гетероорганических соединений. Л.: Химия, 1968.
Гармаш А.В., Сорокина Н.М. Метрологические основы аналитической химии. М., 2017.
Дворкин В.И. Метрология и обеспечение качества количественного химического анализа. М.: Химия, 2001.
Электронный ресурс // Предел обнаружения, предел определения и границы определяемых содержаний. https://studref.com/504076/matematika_himiya_fizik/predel_obnaruzheniya_predel_opre-deleniya_graniny_opredelyaemyh_soderzhaniy
Dubonosov A.D., Tsukanov A.V., Tolpygin I.E. et al. // Bulletin of the Southern Scientific Center. 2013. V. 9. P. 70. [Дубоносов А.Д., Цуканов А.В., Толпыгин И.Е. и др. // Вестник Южного научного центра. 2013. Т. 9. С. 70.]
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии