

Журнал неорганической химии, 2021, T. 66, № 3, стр. 396-401
Влияние щелочной среды на гидротермальный синтез бемита
И. В. Козерожец a, *, Г. П. Панасюк a, Е. А. Семенов a, М. Г. Васильев a, Г. Е. Никифорова a, И. Л. Ворошилов a
a Институт общей и неорганической химии им. Н.С. Курнакова РАН
119991 Москва, Ленинский пр-т, 31, Россия
* E-mail: irina135714@yandex.ru
Поступила в редакцию 03.08.2020
После доработки 05.10.2020
Принята к публикации 09.10.2020
Аннотация
Методами рентгенофазового анализа, ИК-спектроскопии, Брунауэра–Эммета–Теллера, дифференциальной сканирующей калориметрии и сканирующей электронной микроскопии исследовано превращение гиббсита в бемит при гидротермальной обработке при 200°С в 1.5 мас. %-ном растворе NaOH. Определены стадии процесса. Установлено, что превращение гиббсита в бемит в щелочной среде осуществляется за 1 ч и сопровождается расщеплением гиббсита на пластины бемита со средним размером частиц ~1 мкм. Показано, что при гидротермальной обработке гиббсита в 1.5 мас. %-ном растворе NaOH в течение 24 ч образуются частицы бемита пластинчатого габитуса, что позволяет рекомендовать синтезированный порошок бемита в качестве присадки к маслам, в производстве бетона, антипиренов, а также в качестве промежуточной фазы при синтезе порошков α-Al2O3.
ВВЕДЕНИЕ
Современное производство заинтересовано в получении высококачественного сырья с заданными свойствами (форма, размер частиц, насыпная плотность порошков, фазовый и примесный состав и т.д.). Метод гидротермальной обработки является широко распространенным промышленным методом синтеза, позволяющим получать порошки оксидов металлов с заданными характеристиками путем варьирования параметров обработки в автоклаве [1–12]. Так, при гидротермальной обработке смеси Zn(NO3)2 совместно с (CH2)6N4 в нейтральной среде образуются игольчатые наноструктуры ZnO длиной до 3 мкм; обработка этой же смеси в щелочной среде приводит к формированию тонких гексагональных пластин ZnO размером ~5 мкм с толщиной ~300 нм [13]. Варьирование прекурсора и условий синтеза при гидротермальной обработке способствует получению частиц MoO3 различных морфологий: ленты [14], стержни [15], листы [16] и т.д. На форму и размер синтезируемых частиц может оказывать влияние внесение различных добавок в реакционную среду, например, внесение хлорида хрома (>1 мас. %) приводит к изменению среды, что находит отражение в форме и размерах синтезированных частиц MoO3 [17].
Особенно стоит отметить влияние рН среды на гидротермальный синтез бемита из различных прекурсоров. Так, при гидротермальной обработке гиббсита или γ-Al2O3 в 1.5 мас. %-ном растворе HCl в течение 6 ч формируются игольчатые частицы бемита, размер которых зависит от выбранного прекурсора [18–20]. Гидротермальная обработка в 1.5 мас. %-ном растворе HCl также позволяет осуществлять очистку исходного прекурсора, что находит отражение в работе [21]. Согласно работам [18, 20, 22], частицы бемита игольчатой морфологии могут использоваться в качестве сорбентов для извлечения металлов из сточных вод промышленных предприятий, а также в качестве носителя для катализаторов за счет более высокой удельной площади поверхности (60 м2/г) по сравнению с бемитом, синтезированным в нейтральной и щелочной среде (не более 5 м2/г) [23]. Гидротермальная обработка гиббсита в воде позволяет получать частицы бемита изометрического габитуса со средним размером частиц ~1 мкм.
Однако стоит отметить, что несмотря на то, что гидротермальная обработка прекурсоров для синтеза бемита в настоящее время имеет довольно широкое распространение в производстве, до сих пор нет единого понимания сущности процесса, приводящего к синтезу бемита с теми или иными свойствами в зависимости от условий обработки.
Цель работы – исследование процесса синтеза бемита при температуре гидротермальной обработки гиббсита 200°С в щелочной среде 1.5 мас. %-ного водного раствора NaOH.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В работе в качестве прекурсора использовали гиббсит марки МДГА производства ООО “Пикалево” и NaOH марки “ос. ч.”. Обработку гиббсита массой 0.35 г осуществляли в автоклавах объемом 20 см3 в тефлоновом вкладыше в среде 1.5 мас. %-ного раствора NaOH объемом 7 мл при температуре 200°C в течение разного промежутка времени. Коэффициент заполнения автоклава составлял 35%. После завершения нагрева автоклавы охлаждали проточной водой, а полученные продукты извлекали из вкладышей, промывали дистиллированной водой и высушивали при 100°С. Использование тефлонового вкладыша позволяет избежать загрязнения продукта примесями в процессе синтеза, которые возникают за счет коррозии стенок стального контейнера [21]. Синтезированные продукты исследовали методами рентгенофазового анализа на дифрактометре Bruker D8 Advance, сканирующей электронной микроскопии (СЭМ) на приборе Сам Scan-S2; ИК-спектроскопии на ИК-фурье-спектрометре Nexus Nicolett, дифференциальной сканирующей калориметрии на приборе SDT Q600 (скорость нагрева 10 град/мин, температурный интервал 20–1000°С). Определение удельной поверхности осуществляли методом низкотемпературной адсорбции азота с использованием анализатора АТХ-06.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Порошок гиббсита представляет собой монофазный образец (рис. 1, кривая 1) со средним размером частиц ~3 мкм (рис. 2а). Удельная площадь поверхности менее 5 м2/г. В ИК-спектре гиббсита (рис. 3, кривая 1) присутствуют полосы, соответствующие межслоевым (3617, 3523 см–1) и внутрислоевым колебаниям ОН-групп (3455, 3370 см–1), и полосы, отвечающие колебаниям δ(OH) (1020, 965 см–1) и Al–O (780, 739, 665 см–1) [19, 20, 23].
Рис. 1.
Дифрактограммы образцов, полученных после обработки гиббсита в 1.5 мас. %-ном водном растворе NaOH при 200°С: 1 – исходный гиббсит; 2 – обработка в течение 0.5 ч (гиббсит и бемит); 3 – обработка в течение 1 ч (бемит); 4 – обработка в течение 3 ч (бемит); 5 – обработка в течение 6 ч (бемит); 6 – обработка в течение 24 ч (бемит).
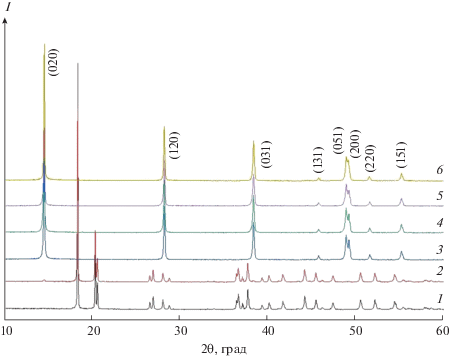
Рис. 2.
СЭМ-изображения гиббсита (а) и бемита, полученного после гидротермальной обработки гиббсита в 1.5 мас. %-ном растворе NaOH при 200°С, τ = 0.5 ч (б, в), 1 ч (г), 24 ч (д).

Рис. 3.
ИК-спектры гиббсита исходного (1) и после обработки при 200°С в 1.5 мас. %-ном растворе NaOH в течение 0.5 ч (2) (гиббсит и бемит); 1 ч (3) (бемит).
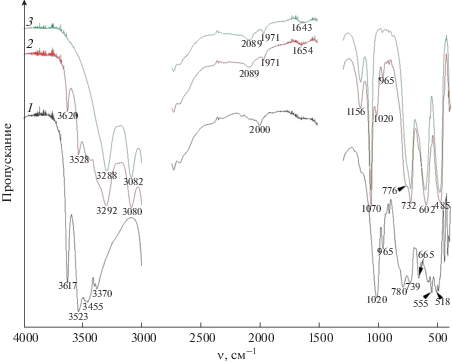
Гидротермальная обработка гиббсита при 200°С в 1.5 мас. %-ном водном растворе NaOH в течение 0.5 ч приводит к формированию смеси фаз бемита и гиббсита (рис. 1, кривая 2). При этом на дифрактограмме появляется рефлекс бемита, соответствующий межплоскостному расстоянию d020 = 6.13 Å, а в ИК-спектре (рис. 3, кривая 2) этого образца присутствуют полосы, относящиеся к межслоевым колебаниям ОН-групп гиббсита (3620, 3528 см–1), и пологое плечо, переходящее в полосы при 3292 и 3080 см–1, соответствующие валентным колебаниям ОН-групп бемита. Полоса поглощения при 3080 см–1 соответствует колебаниям ОН-групп бемита, которые связаны с катионами Al3+, входящими в состав краевых фрагментов плоскости (201). Полоса поглощения при 3292 см–1 относится к мостиковым ОН-группам бемита, которые принадлежат плоскости (001) бемита. Полосы, отвечающие колебаниям δ(OH) гиббсита, присутствуют и в низкочастотной области при 1020 и 965 см–1. Полосы при 1156 и 1070 см–1 соответствуют асимметричным и симметричным деформационным колебаниям связи δas(Al–OH) и δs(Al–OH) бемита соответственно. Полосы поглощения при 732, 602 и 485 см–1 относятся к деформационным колебаниям связи Al–O–Al в бемите. Особое внимание стоит уделить появлению хорошо выраженной полосы деформационных колебаний молекулярной воды при 1654 см–1, что указывает на активное взаимодействие гидротермального раствора со структурой гиббсита. Удельная площадь поверхности для этого образца также менее 5 м2/г. СЭМ-изображения образца, полученного после гидротермальной обработки гиббсита в щелочной среде в течение 0.5 ч позволяют обнаружить расслоение исходной частицы гиббсита на тонкие пластины (рис. 2б, 2в) за счет интеркаляции молекул воды в межслоевые промежутки структуры гиббсита. Об этом свидетельствует появление полосы деформационных колебаний молекулярной воды при 1654 см–1.
Гидротермальная обработка гиббсита при 200°С в 1.5 мас. %-ном растворе NaOH в течение 1 ч приводит к формированию монофазного образца бемита (рис. 1, кривая 3) пластинчатого габитуса со средним размером частиц 2 мкм (рис. 2г). Удельная площадь поверхности синтезированного бемита менее 5 м2/г. В ИК-спектре присутствуют только полосы, характерные для структуры бемита (рис. 3, кривая 3).
Более продолжительная гидротермальная обработка гиббсита при 200°С в щелочной среде 1.5 мас. %-ного водного раствора NaOH в течение 3, 6 и 24 ч не приводит к изменению фазового состава образца (рис. 1), пластинчатый габитус частиц бемита сохраняется (рис. 2д).
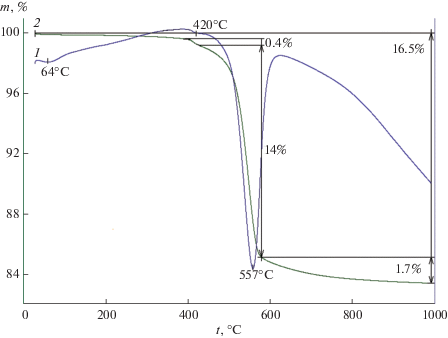
Результаты термогравиметрического исследования бемита, полученного при гидротермальной обработке гиббсита при 200°С в 1.5 мас. %-ном водном растворе NaOH в течение 24 ч представлены на рис. 4. На кривых ДТА и ТГ фиксируется эндотермический предэффект при 420°С, соответствующий удалению слабосвязанной воды, и эндотермический эффект с максимумом при 557°С, отвечающий дегидратации бемита с образованием γ-Al2O3. Потеря массы, соответствующая тепловому эффекту удаления слабосвязанной воды, равна 0.4 мас. %, а тепловому эффекту дегидратации бемита при удалении структурной воды – 14 мас. %. Общая потеря массы при выделении воды из образца составляет 16.5 мас. %, что превышает стехиометрическое значение для бемита, равное 15.016 мас. %. В составе синтезированного бемита отмечается повышенное содержание слабосвязанной воды, свидетельствующее о разупорядоченном состоянии его структуры.
Рис. 4.
Кривые ДТА (1) и ТГ (2) бемита, полученного после гидротермальной обработки гиббсита при 200°С в щелочной среде 1.5 мас. %-ного водного раствора NaOH, τ = 24 ч.
Таким образом, обработка гиббсита в растворе NaOH обусловливает форму и размер частиц образующегося бемита, а также высокую скорость превращения гиббсит → бемит при гидротермальной обработке. Так, процесс превращения гиббсита в бемит при 200°С в 1.5 мас. %-ном водном растворе NaOH завершается за 1 ч, в то время как превращение в тех же условиях в кислой (1.5 мас. %-ный водный раствор HСl) и нейтральной (вода) среде требует большего времени обработки – 12 и 2 ч соответственно. Согласно дифрактограммам (рис. 1), не происходит образования алюминатов натрия или любых других соединений, что косвенно указывает на твердофазный механизм процесса превращения гиббсит → → бемит в данных условиях. Результаты термогравиметрического исследования синтезированного бемита зафиксировали повышенное содержание поверхностно-связанной воды в образце. В соответствии с работами [24–30], слабосвязанная поверхностная вода стабилизирует метастабильное промежуточное состояние в ходе твердофазной перестройки структуры прекурсора [24].
Полученные ранее [18–20, 22–24, 31, 32] и в настоящей работе экспериментальные результаты позволяют заключить, что форма и размер образующихся частиц бемита зависят от комплекса параметров, к которым относится концентрация щелочи, влияние поверхностно-активной воды на структуру прекурсора, время и температура гидротермальной обработки. Результаты работы представляют интерес не только со стороны физико-химического описания сложного многостадийного процесса, но также могут иметь широкое применение в промышленности при подборе условий для синтеза частиц бемита заданной формы и размера.
ЗАКЛЮЧЕНИЕ
При исследовании синтеза бемита в условиях гидротермальной обработки гиббсита в 1.5 мас. %-ном растворе NaOH при 200°С установлены стадии процесса. Показано, что в щелочной среде расщепление гиббсита на пластины бемита со средним размером частиц ~1 мкм завершается за 1 ч без изменения удельной площади поверхности. Пластинчатые частицы бемита образуются при расслоении исходной частицы гиббсита на отдельные кристаллиты с последующим их сшиванием при формировании структуры бемита.
Список литературы
Cai W., Li H., Zhang G. // J. Phys. Chem. Solids. 2010. V. 71. № 4. P. 515. https://doi.org/10.1016/j.jpcs.2009.12.025
Carneiro J., Tobaldi D.M., Hajjaji W. et al. // Waste Managem. 2018. V. 80. P. 371. https://doi.org/10.1016/j.wasman.2018.09.032
Cui W., Zhang X., Pearce C.I. et al. // Environ. Sci. Technol. 2019. V. 53. № 18. P. 11043. https://doi.org/10.1021/acs.est.9b02693
Filho R.W., Rocha G.D., Montes C.R. et al. // Mater. Res. 2016. V. 19. № 3. P. 659. https://doi.org/10.1590/1980-5373-MR-2016-0019
Stolin A.M., Bazhin P.M., Konstantinov A.S. et al. // Ceram. Int. 2018. V. 44. № 12. P. 13815. https://doi.org/10.1016/j.ceramint.2018.04.225
Simonenko T.L., Ivanova V.M., Simonenko N.P. et al. // Russ. J. Inorg. Chem. 2019. V. 64. P. 1753. https://doi.org/10.1134/S0036023619140080
Egorova S.R., Mukhamed’yarova A.N., Nesterova O.V. et al. // Coatings. 2018. V. 8. № 1. P. 30. https://doi.org/10.3390/coatings8010030
He T., Xiang L., Zhu S. et al. // CrystEngComm. 2009. № 11. P. 1338. https://doi.org/10.1039/B900447P
He S., Lin H. // Nanoscale. 2019. № 11. P. 10348. https://doi.org/10.1039/C9NR02148E
Huang L., Yang Z., He Y. et al. // J. Hazardous Mater. 2020. V. 394. № 122555. https://doi.org/10.1016/j.jhazmat.2020.122555
Li Huo, Fu-Hui Liao, Jun-Ran Li et al. // Chem. Eng. Commun. 2010. V. 197. № 5. P. 684. https://doi.org/10.1080/00986440903287809
Jiao W., Wu X., Xue T. et al. // Cryst. Growth Des. 2016. V. 16. № 9. P. 5166. https://doi.org/10.1021/acs.cgd.6b00723
Kloprogge J.T., Hickey L., Frost R.L. // J. Solid State Chem. 2004. V. 177. P. 4047. https://doi.org/10.1016/j.jssc.2004.07.010
Zhou L., Yang L., Yuan P. et al. // J. Phys. Chem. C. 2010. V. 114. № 49. P. 21868. https://doi.org/10.1021/jp108778v
Wang Q., Sun J., Wang Q. et al. // J. Mater. Chem. A. 2015. V. 3. № 9. P. 5083. https://doi.org/10.1039/c5ta00127g
Li G., Jiang L., Pang S. et al. // J. Phys. Chem. B. 2006. V. 110. № 48. P. 24472. https://doi.org/10.1021/jp064855v
Li Y. // Physica E. 2017. V. 94. P. 22. https://doi.org/10.1016/j.physe.2017.07.010
Panasyuk G.P., Kozerozhets I.V., Semenov E.A. et al. // Inorg. Mater. 2019. V. 55. № 9. P. 929. https://doi.org/10.1134/S0020168519090139
Panasyuk G.P., Kozerozhets I.V., Voroshilov I.L. et al. // Russ. J. Phys. Chem. A. 2015. V. 89. № 4. P. 592. https://doi.org/10.1134/S0036024415040196
Panasyuk G.P., Belan V.N., Voroshilov I.L. et al. // Theor. Found. Chem. Eng. 2013. V. 47. № 4. P. 415. https://doi.org/10.1134/S0040579513040143
Panasyuk G.P., Semenov E.A., Kozerozhets I.V. et al. // Dokl. Chem. 2018. V. 483. P. 272. https://doi.org/10.1134/S0012500818110022
Kozerozhets I.V., Panasyuk G.P., Semenov E.A. et al. // Theor. Found. Chem. Eng. 2020. V. 45. № 3. P. 345. https://doi.org/10.1134/S0040579520030082
Panasyuk G.P., Belan V.N., Voroshilov I.L. et al. // Inorg. Mater. 2010. V. 46. № 7. P. 747. https://doi.org/10.1134/S0020168510070113
Panasyuk G.P., Kozerozhets I.V., Semenov E.A. et al. // Inorg. Mater. 2019. V. 55. № 9. P. 920. https://doi.org/10.1134/S0020168519090127
Tavakoli A.H., Maram P.S., Widgeon S.J. et al. // J. Phys. Chem. C. 2013. V. 117. P. 17123. https://doi.org/10.1021/jp405820g
Navrotsky A. // Geochem. Trans. 2003. № 4. P. 34. https://doi.org/10.1039/b308711e
McHale J.M., Auroux A., Perrotta A.J. et al. // Science. 1997. V. 277. P. 788. https://doi.org/10.1126/science.277.5327.788
Calvin J.J., Rosen P.F., Ross N.L. et al. // J. Mater. Res. 2019. P. 416. https://doi.org/10.1557/jmr.2019.33
Castro R.H.R., Ushakov S.V., Gengembre L. et al. // Chem. Mater. 2006. V. 18. P. 1867. https://doi.org/10.1021/cm052599d
Mu R., Zhao Z.J., Dohnálek Z. et al. // Chem. Soc. Rev. 2017. V. 46. P. 1785. https://doi.org/10.1039/c6cs00864j
Li Z., Liu G., Li X. et al. // Cryst. Growth Des. 2019. V. 19. P. 1778.
Santos P.D., Coelho A.C., Santos H. et al. // Mater. Res. Ibero Am. J. Mater. 2009. V. 12. P. 437.
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии