

Журнал неорганической химии, 2021, T. 66, № 5, стр. 631-640
Экстракционные свойства дифенилфосфорилмочевин с алифатическими ω-азотсодержащими радикалами
А. М. Сафиулина a, *, А. В. Лизунов a, Н. Е. Борисова b, Т. В. Баулина c, Е. И. Горюнов c, И. Б. Горюнова c, В. К. Брель c
a Акционерное общество “Высокотехнологический научно-исследовательский институт неорганических материалов им. академика А.А. Бочвара”
123098 Москва, ул. Рогова, 5а, Россия
b Московский государственный университет им. М.В. Ломоносова
119991 Москва, Ленинские горы, 1, Россия
c Институт элементоорганических соединений им. А.Н. Несмеянова РАН
119991 Москва, ул. Вавилова, 28, Россия
* E-mail: AMSafiulina@bochvar.ru
Поступила в редакцию 11.11.2020
После доработки 29.12.2020
Принята к публикации 30.12.2020
Аннотация
Исследована экстракция лантанидов и актинидов из азотнокислых сред растворами N-(дифенилфосфорил)-N'-н-пропилмочевин, содержащими в ω-положении алкильного заместителя имидазолильный, диэтиламиновый, пирид-2-ил, 2-оксопирролидиновый фрагменты. Показано, что относящиеся к иттриевой подгруппе лантаниды Ho(III) и Yb(III) экстрагируются значительно лучше La(III) и Nd(III), принадлежащих цериевой подгруппе. Лучшими экстракционными свойствами обладает N-(дифенилфосфорил)мочевина, содержащая ω-(2-оксопирролидино)пропильный радикал у терминального атома азота. Данная зависимость получила свое теоретическое обоснование при моделировании комплексообразования, поскольку координация иона f-элемента с амидным атомом кислорода оказывается предпочтительной.
ВВЕДЕНИЕ
Полидентатные фосфорорганические соединения привлекают повышенное внимание, учитывая их широкое использование в экстракционной практике. Вследствие того, что фосфорильная группа легко поляризуется и обладает высокой координирующей способностью по отношению к ряду d- и f-элементов, дизайн новых фосфорилсодержащих полидентатных лигандов, обладающих высокой синтетической доступностью, представляет фундаментальный и практический интерес [1–6]. Дополнительным преимуществом фосфорсодержащих экстрагентов является возможность изменять координирующие свойства фосфорильной группы путем варьирования заместителей у атома фосфора [7–11]. Кроме того, методы органической химии позволяют конструировать соединения, различающиеся количеством донорных центров, что открывает широкие возможности для направленной модификации их экстракционных свойств [12–19].
Ранее нами исследованы оригинальные фосфоразотсодержащие лиганды: N-(диорганилфосфорил)мочевины R2P(O)NHC(O)NHR' (I), в особенности N-дифенилфосфорилированные производные (Ia, R = Ph), которые обладают высокой экстракционной способностью к актинидам и лантанидам в азотнокислых средах [20–22]. В [23] мы показали, что природа заместителей у атома фосфора N-органофосфорилмочевин R(R'O)P(O)NHC(O)NHC8H17-н – фосфонатных аналогов мочевин (Ib) – оказывает большое влияние на их экстракционную способность. Замена одной фенильной группы у атома фосфора на алкоксильный или ароксильный радикал, как оказалось, приводит к значительному снижению экстракционной способности по отношению к 4f- и 5f-элементам.

На экстракционную способность соединений класса фосфорилмочевин существенное влияние оказывает также природа заместителей у терминального атома азота. В работе [20] были исследованы N-(дифенилфосфорил)-N'-н-алкил(С6-С10)мочевины Ph2P(O)NHC(O)NHCnH2n+ 1 (Iа, n = 6–10) и установлено, что наибольшую эффективность по отношению к урану(VI), торию(IV), америцию(III) и европию(III) проявляет N'-н-октильное производное.
Известно, что фосфориламиды на основе 2-амино-5,7-диметил-1,8-нафтиридина способны извлекать трехвалентные лантаниды из карбонатных сред [24, 25]. Подобный необычный эффект обусловлен наличием дополнительных центров координации, находящихся в азотсодержащем гетероциклическом фрагменте. В случае мочевин Ia следует предположить, что введение разнообразных азотсодержащих фрагментов в алкильный радикал, связанный с терминальным атомом азота, будет способствовать увеличению экстракционных свойств соответствующих лигандов.
В настоящей работе исследовано влияние алифатических ω-азотсодержащих радикалов у атома азота N' дифенилфосфорилмочевин (Ia) на их экстракционную способность по отношению к лантанидам и актинидам.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
N-(Дифенилфосфорил)-N'-(н-пропил)мочевины, содержащие в ω-положении пропильного радикала гетероарильные и алкильные азотсодержащие заместители (II–V), были получены исходя из коммерчески доступных дифенилхлорфосфина и RN(CH2)3NH2 (RN = имидазол-1-ил, диэтиламино, пирид-2-ил, 2-оксопирролидино) соответственно с применением оригинальных трехстадийных “one-pot” процессов, ключевой стадией которых является каталитический синтез дифенилфосфорилизоцианата (VI) (cхема). Все стадии этих процессов протекают с достаточно высокой скоростью при комнатной температуре, а выходы аналитически и спектрально чистых целевых соединений приближаются к количественному.
Схема

Применявшиеся ω-RN-н-пропиламины (Aldrich или Acros, 95–99+%) очищали перегонкой над твердым КОН. Дифенилхлорфосфин (Aldrich, 98%) перегоняли в вакууме непосредственно перед реакцией. Безводный MgCl2 (Aldrich, ≥98%) использовали без дополнительной очистки. Циановокислый натрий (Aldrich, 96%) сушили 4 ч при 120°С в вакууме (1 Торр) над P2O5. Хлористый сульфурил (Acros, 98.5%) перегоняли непосредственно перед реакцией. Четыреххлористый углерод и ацетонитрил абсолютировали перегонкой над P2O5. Все операции проводили в атмосфере аргона.
Общая методика синтеза N-дифенилфосфорил-N'-(ω-RN-н-пропил)мочевин II–V
К раствору 3.64 г (0.0165 моль) Ph2PCl в 7 мл абсолютированного CCl4 при перемешивании при комнатной температуре на магнитной мешалке в течение 30 мин добавляли по каплям раствор 2.67 г (0.0199 моль) SO2Cl2 в 5 мл абсолютированного CCl4 и перемешивали еще 1 ч при этой температуре. Растворитель и другие летучие компоненты реакционной смеси удаляли в вакууме. Остаток растворяли в 30 мл абсолютированного MeCN. К полученному раствору добавляли 0.040 г (0.420 ммоль) мелко растертого безводного MgCl2, перемешивали до полного растворения последнего, прибавляли 2.16 г (0.033 моль) NaOCN и перемешивали 1 ч при комнатной температуре. К полученной суспензии добавляли 0.0165 моль RN(CH2)3NH2 и перемешивали 1 ч при комнатной температуре, удаляли растворитель и к сухому остатку добавляли 60 мл дистиллированной воды, перемешивали в течение 1 ч при комнатной температуре. Осадок отфильтровывали, последовательно промывали смесью 17 мл дистиллированной воды и 3 мл MeCN, дистиллированной водой (2 × 20 мл) и сушили на воздухе.
N-Дифенилфосфорил-N'-[3-(имидазол-1-ил)пропил]-мочевина (II).
Выход 99.6%. tпл = 184–185°С.
| С | Н | N | P | |
|---|---|---|---|---|
| Найдено, %: | 61.84; | 5.74; | 15.09; | 8.27. |
| Для С19Н21N4О2Р | ||||
| вычислено, %: | 61.95; | 5.75; | 15.21; | 8.41. |
Спектр ЯМР 1Н (δН, м.д.): 1.80 квинтет (2Н, СН2СН2СН2, 3JH–H = 6.9 Гц); 2.96 дт (2Н, NHCH2, 3JH–CH = 6.7 Гц, 3JH–NH = 6.1 Гц); 3.91 т (2Н, СН2-имидазолил, 3JH–H = 7.0 Гц); 6.65 т (1Н, NHCH2, 3JH–H = 5.7 Гц); 6.88 т (1Н, 4Н-имидазолил, 3JH–H ~ ~ 4JH–H = 0.9 Гц); 7.13 т (1Н, 5Н-имидазолил, 3JH–H ~ ~ 4JH–H = 1.1 Гц); 7.48–7.54 м (4Н, m-C6H5); 7.54–7.59 м (3Н, p-C6H5 + 2H-имидазолил); 7.72–7.80 м (4Н, o-C6H5); 8.45 шс [1Н, NHP(O)].
Спектр ЯМР 13С{1H} (δC, м.д.): 31.52 с (СН2СН2СН2); 36.87 с (NHCH2); 43.92 c (CH2-имидазолил); 119.75 с (5С-имидазолил); 128.87 с (4С-имидазолил); 129.01 д (m-C6H5, 3JC–P = 12.7 Гц); 131.56 д (o-C6H5, 2JC–P = 10.0 Гц); 132.28 д (p-C6H5, 4JC–P = 2.7 Гц); 133.38 д (ипсо-C6H5, 1JC–P = 129.0 Гц); 137.69 с (2С-имидазолил); 155.53 c (C=O).
Спектр ЯМР 31P{1H} (δP, м.д.): 15.48 с.
N-Дифенилфосфорил-N'-(3-диэтиламинопропил)-мочевина (III).
Выход 90.9%. tпл = 157–157.5ºС (хлороформ–гексан).
| C | H | N | P | |
|---|---|---|---|---|
| Найдено, %: | 64.39; | 7.69; | 11.21; | 8.35. |
| Для C20H28N3O2P | ||||
| вычислено, %: | 64.33; | 7.56; | 11.25; | 8.29. |
Спектр ЯМР 1Н (δН, м.д.): 0.92 т (6Н, СН3, 3JH–H = = 7.0 Гц); 1.47 квинтет (2Н, СН2СН2СН2, 3JH–H = = 6.8 Гц); 2.32 т (2Н, СН2NEt2, 3JH–H = 7.1 Гц); 2.41 кв (4Н, СН2Ме, 3JH–H = 7.1 Гц); 3.01 дт (2Н, NHCH2, 3JH–CH = 6.4 Гц, 3JH–NH = 6.0 Гц); 6.55 т (1Н, NHCH2, 3JH–H = 5.3 Гц); 7.45–7.62 м (6Н, m- + p-C6H5); 7.75 дд (4Н, o-C6H5, 3JH–H = 7.1 Гц, 3JH–P = 12.4 Гц); 8.34 шс [1Н, NHP(O)].
Спектр ЯМР 13С{1H} (δC, м.д.): 12.14 с (СН3); 27.57 с (СН2СН2СН2); 38.06 с (NHCH2); 46.72 c (CH2Me); 50.11 c (CH2NEt2); 128.97 д (m-C6H5, 3JC–P = 12.2 Гц); 131.54 д (o-C6H5, 2JC–P = 9.9 Гц); 132.22 c (p-C6H5); 133.50 д (ипсо-C6H5, 1JC–P = = 128.2 Гц); 155.35 c (C=O).
Спектр ЯМР 31P{1H} (δP, м.д.): 15.53 с.
N-Дифенилфосфорил-N'-[3-(пирид-2-ил)пропил]-мочевина (IV).
Выход 98.4%. tпл = 177–178°С.
| C | H | N | P | |
|---|---|---|---|---|
| Найдено, %: | 66.40; | 5.72; | 11.50; | 8.38. |
| Для С21Н22N3О2Р | ||||
| вычислено, %: | 66.48; | 5.84; | 11.08; | 8.16. |
Спектр ЯМР 1Н (δН, м.д.): 1.76 квинтет (2Н, СН2СН2СН2, 3JH–H = 7.2 Гц); 2.69 т (2Н, СН2С5H4N, 3JH–H = 7.7 Гц); 3.02 дт (2Н, NHCH2, 3JH–CH = 6.5 Гц, 3JH–NH = 6.2 Гц); 6.65 т (1Н, NHCH2, 3JH–H = 5.5 Гц); 7.19 дд (1Н, 5Н-C5H4N, 3JH–H = 7.3 Гц, 3JH–H = 5.0 Гц); 7.21 д (1Н, 3Н-C5H4N, 3JH–H = 7.8 Гц), 7.51 дт (4Н, m-C6H5, 3JH–H = 7.4 Гц, 4JH–Р = 3.0 Гц); 7.56 т (2Н, p-C6H5, 3JH–H = 6.8 Гц); 7.68 дт (1Н, 4Н-C5H4N, 3JH–H = 7.7 Гц, 4JH–Н = = 1.7 Гц); 7.75 дд (4Н, o-C6H5, 3JH–H = 7.1 Гц, 3JH–Р = = 12.3 Гц); 8.41 шс [1Н, NHP(O)], 8.47 шд (1Н, 6Н-C5H4N, 3JH–H = 4.1 Гц).
Спектр ЯМР 13С{1H} (δC, м.д.): 29.90 с (СН2СН2СН2); 35.10 с (СН2С5H4N); 39.16 с (NHCH2); 121.70 с (5С-C5H4N); 123.17 с (3С-C5H4N); 129.00 д (m-C6H5, 3JC–P = 13.3 Гц); 131.55 д (o-C6H5, 2JC–P = 10.0 Гц); 132.24 с (p-C6H5); 133.47 д (ипсо-C6H5, 1JC–P = 129.4 Гц); 136.92 с (4С-C5H4N); 149.45 с (6С-C5H4N); 155.38 c (C=O); 161.40 с (2С-C5H4N).
Спектр ЯМР-31Р{1H} (δР, м.д.): 15.24 с.
N-Дифенилфосфорил-N'-[3-(2-оксопирролидино)пропил]мочевина (V).
Выход 97.2%. tпл = 175–176°С.
| C | H | N | P | |
|---|---|---|---|---|
| Найдено, %: | 60.98; | 6.45; | 10.60; | 7.53. |
| Для С20Н24N3О3Р · 0.5 Н2О | ||||
| вычислено, %: | 60.91; | 6.39; | 10.65; | 7.85. |
Спектр ЯМР 1Н (δН, м.д.): 1.53 квинтет (2Н, СН2СН2СН2, 3JH–H = 6.8 Гц); 1.89 квинтет (2Н, 4Н-пирролидино, 3JH–H = 7.5 Гц); 2.21 т (2Н, 3Н-пирролидино, 3JH–H = 8.0 Гц); 2.95 дт (2Н, NHCH2, 3JH–CH = 6.4 Гц, 3JH–NH = 6.1 Гц); 3.15 т (2Н, СН2-пирролидино, 3JH–H = 6.9 Гц); 3.28 т (2Н, 5Н-пирролидино, 3JH–H = 7.0 Гц); 6.59 т (1Н, NHCH2, 3JH–H = 5.7 Гц); 7.51 дт (4Н, m-C6H5, 3JH–H = = 7.4 Гц, 4JH–P = 2.8 Гц); 7.56 т (2Н, p-C6H5, 3JH–H = = 7.0 Гц); 7.74 дд (4Н, o-C6H5, 3JH–H = 7.1 Гц, 3JH–P = = 12.4 Гц); 8.50 шс [1Н, NHP(O)].
Спектр ЯМР 13С{1H} (δC, м.д.): 17.96 с (4С-пирролидино); 27.69 с (СН2СН2СН2); 30.90 с (3С-пирролидино); 37.21 с (СН2NH); 39.71 c (CH2-пирролидино); 46.73 с (5С-пирролидино); 128.99 д (m-C6H5, 3JC–P = 12.7 Гц); 131.55 д (o-C6H5, 2JC–P = 10.0 Гц); 132.20 д (p-C6H5, 4JC–P = 1.9 Гц); 133.48 д (ипсо-C6H5, 1JC–P = 128.9 Гц); 155.40 с [C(O)(NH)2]; 174.43 c (2С-пирролидино).
Спектр ЯМР 31Р{1H} (δР, м.д.): 15.06 c.
Спектры ЯМР 1H и 13С{1H} мочевин II–V регистрировали на приборе Bruker AV-600 (рабочая частота 600.22 MГц (1H) и 150.925 MГц (13C{1H})), а спектры ЯМР 31P{1H} – на приборе Bruker AV-400 (рабочая частота 161.98 MГц) в растворе (CD3)2SO (с = 0.1 моль/л). Внутренний эталон для спектров ЯМР 1H – сигналы остаточных протонов дейтерированного растворителя, а для спектров ЯМР 13С{1H} – сигналы ядер атомов углерода дейтерированного растворителя; внешний эталон для спектров ЯМР 31Р{1H} – 85%-ная H3PO4. Отнесение сигналов в спектрах ЯМР 1H и 13С{1H} было проведено с использованием корреляций COSY, HMQC и HMBC.
Исследование экстракции металлов. В работе использовали хлороформ (х. ч.), арсеназо III (ч. д. а.), HNO3 (ос. ч.), ГСО 8363-2003 закись-окись урана (аттестовано на содержание урана 84.784 ± ± 0.016%), Th(NO3)4 · 4H2O (х. ч.), La(NO3)3 ⋅ 6H2O (х. ч.), Nd(NO3)3 · 6H2O (х. ч.), Ho(NO3)3 ⋅ 6H2O (х. ч.), Yb(NO3)3 · 6H2O (х. ч.). Растворы готовили объемно-весовым методом, водные растворы – в бидистиллированной воде. Растворы нитратов исследуемых элементов готовили растворением навески соответствующего нитрата в 0.01 моль/л растворе HNO3. Концентрацию растворов нитратов металлов (0.1 ммоль/л) уточняли спектрофотометрически по методике [26] с использованием спектрофотометра Cary 5000 Scan (Varian). Концентрацию растворов HNO3 определяли потенциометрическим титрованием 0.1 моль/л NaOH с использованием рН-метра/кондуктометра S470 SevenExcellence™ (Mettler Toledo) с точностью ±0.01 ед. рН. Электродную пару калибровали по стандартным буферным растворам с рН 1.68, 4.01 и 9.21 (Mettler Toledo) (значения при 20°С). Концентрацию раствора NaOH уточняли потенциометрическим титрованием с 0.1 моль/л HCl (фиксанал).
Исследование экстракции катионов металлов выполняли по следующей методике. В пробирку с притертой пробкой вносили 1.5 мл раствора азотной кислоты, концентрацию которой варьировали от 0.052 до 5.0 моль/л; 0.5 мл 0.1 ммоль/л раствора нитрата металла, 2 мл 0.01 моль/л раствора лиганда в хлороформе. Фазы перемешивали в течение 20 мин в ротаторе. Время установления равновесия экстракции проверяли, увеличивая время контакта фаз до 120 мин, коэффициенты распределения при этом не изменялись. Расслаивание фаз осуществляли центрифугированием. После разделения фаз концентрацию катионов металлов в водной фазе определяли спектрофотометрическим методом [26]. Для каждой концентрации проводили не менее трех независимых опытов. Погрешность результатов составила ~20%, учитывая не исключенную и случайную составляющие. Соответственно доверительный интервал определяемых концентраций металлов в эксперименте составляет 0.002 ммоль/л.
Все эксперименты проводили при температуре 20 ± 1°С.
Коэффициенты распределения при экстракции (D = [M]орг/[M]водн) определяли при постоянных концентрациях экстрагента (0.01 моль/л в хлороформе) и исходных концентрациях металла в эксперименте (0.025 ммоль/л в водной фазе).
Расчеты выполняли на суперкомпьютере MVS-50K Межведомственного суперкомпьютерного центра РАН (www.jscc.ru). Все расчеты проводили с использованием программы Природа-9 [27, 28] (функционал PBE [29, 30]). Комплексы с f-элементами изучали с использованием cc-pVDZ-подобных базисов [27], для тяжелых атомов использовали скалярно-релятивистские базисные наборы, описанные в той же работе. Используемые одноэлектронные базисные наборы, представляющие собой сжатые наборы гауссовых функций с угловой частью, представленной действительными сферическими гармониками, являются орбитальными базисными наборами, которые применяли для решения уравнений Кона–Шэма. Вспомогательные базисные наборы использовали для расчета электронной плотности для быстрой оценки обменно-корреляционного вклада. Такой подход ускоряет вычисления без заметной потери точности [28]. Геометрию лиганда и его комплексов с f-элементами оптимизировали без ограничений по симметрии системы. Анализ колебательных спектров использовали для идентификации стационарных точек. Применение этой методики ранее позволило удовлетворительно объяснить исчезновение экстракционной способности в тетраалкилзамещенных диамидах 2,2'-бипиридил-6,6'-дикарбоновой кислоты [31].
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Соединения II–V входят в состав элементоорганических лигандов класса фосфорилмочевин. Способность лигандов II–V к комплексообразованию с актинидами и лантанидами определяется не только природой заместителей у фосфорильной группы, но и у терминального атома азота. Представляло интерес изучить влияние этого фактора на экстракционные свойства лигандов II–V по отношению к ряду актинидов и лантанидов.
Введение в структуру фосфорилмочевины алкилимидазольного фрагмента приводит к выявлению кислотного характера взаимодействия при экстракции урана(VI) лигандом II. Как следует из рис. 1, при экстракции урана(VI) 0.01 моль/л раствором лиганда II в хлороформ значения коэффициентов распределения (DU) уменьшаются с ростом концентрации HNO3. В диапазоне концентраций азотной кислоты от 0.01 до 2 моль/л экстракционная способность лиганда II к лантану(III), так же как и к урану(VI), снижается. Однако в области концентраций до 1 моль/л HNO3 уран(VI) и лантан(III) извлекаются почти одинаково (на ~15%), с ростом концентрации азотной кислоты уран(VI) практически не извлекается в хлороформ, а зависимость DLa проходит через минимум в области ~2 моль/л азотной кислоты. В аналогичных условиях эксперимента экстракционная способность лиганда II по отношению к Th(IV), Nd(III), Ho(III) и Yb(III), напротив, растет. Причем эти металлы по сравнению с U(VI) и La(III) экстрагируются значительно лучше. Так, Th(IV) и Nd(III) извлекаются в органическую фазу на ~35%, а Ho(III), Yb(III) – на ~40%.
Рис. 1.
Зависимость логарифмов коэффициентов распределения Th(IV), U(VI), La(III), Nd(III), Ho(III) и Yb(III) при экстракции 0.01 моль/л раствором лиганда II в хлороформе.

Следует отметить, что поведение урана(VI) кардинально отличается от поведения тория или лантанидов при извлечении лигандом II. При общем сохранении координации иона уранила с атомами кислорода фосфиноксида наблюдается ряд различий в типе связывания лиганда c образованием более энергетически выгодного гидратированного комплекса UO2(НII)2NO3 · H2O [15]. Что касается тория и лантанидов, то они экстрагируются с образованием в органической фазе комплексов типа M(HII)2(NO3)x [15].
При экстракции ряда исследуемых актинидов и лантанидов 0.01 моль/л раствором соединения III в хлороформе коэффициенты распределения растут уже во всем диапазоне концентраций азотной кислоты (рис. 2). Соединение III, в молекуле которого у терминального атома азота содержится радикал с ω-диэтиламинной группировкой, при экстракции U(VI), Th(IV) и лантанидов проявляет свойства нейтрального соединения.
Рис. 2.
Зависимость логарифмов коэффициентов распределения Th(IV), U(VI), La(III), Nd(III), Ho(III) и Yb(III) при экстракции 0.01 моль/л раствором лиганда III в хлороформе.

В области концентраций HNO3 от 2 до 4 моль/л исследуемые лантаниды экстрагируются в раствор лиганда III в хлороформе немного лучше, чем U(VI), Th(IV) (рис. 2). Так, степень извлечения лантанидов в области 4 моль/л HNO3 в одну стадию составляет ~30%, в то время как экстракционная способность лиганда III по отношению к U(VI), Th(IV) – ~15%.
Соединение IV, в терминальную часть молекулы которого встроен ω-(пирид-2-ил)пропильный радикал, проявляет основные свойства. При экстракции трехвалентных лантанидов 0.01 моль/л раствором лиганда IV в хлороформе коэффициенты распределения растут с повышением кислотности среды более резко по сравнению с лигандом III. При этом, как и в случае с лигандом III, экстракционная способность фосфорилмочевины IV по отношению к лантанидам несколько выше, чем к актинидам (рис. 3). La(III), Nd(III), Ho(III) и Yb(III) в области концентраций от 2 до 4 моль/л HNO3 извлекаются в одну стадию на ~30%; Th(IV) в этих условиях экстрагируется лишь незначительно. Коэффициенты распределения урана(VI) (DU) растут с увеличением кислотности среды с разной скоростью. Так, до 1.5 моль/л HNO3 рост DU незначителен, но с дальнейшим повышением концентрации HNO3 коэффициенты распределения урана сравнимы с D лантанидов.
Рис. 3.
Зависимость логарифмов коэффициентов распределения Th(IV), U(VI), La(III), Nd(III), Ho(III) и Yb(III) при экстракции 0.01 моль/л раствором лиганда IV в хлороформе.

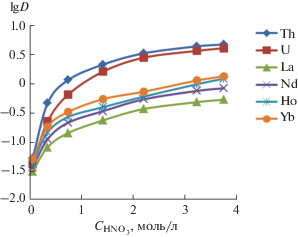
Иная картина наблюдается при экстракции актинидов и лантанидов 0.01 моль/л раствором соединения V в CHCl3 (рис. 4). Экстракционная способность лиганда V, содержащего у терминального атома азота ω-(2-оксопирролидино)пропильный фрагмент, по отношению и к актинидам, и к лантанидам оказалась значительно выше, чем у других описанных в данной работе производных N-(дифенилфосфорил)мочевин, причем U(VI) и Th(IV) извлекаются эффективнее лантанидов. Лантаниды, входящие в иттриевую подгруппу (Ho(III) и Yb(III)), экстрагируются значительно лучше (~60% извлечения в раствор соединения V в одну стадию), чем La(III) и Nd(III), принадлежащие цериевой подгруппе (~45% извлечения). Торий(IV) и уран(VI) извлекаются в органическую фазу с коэффициентами распределения 4.5 и 3.9 соответственно, что составляет ~80% экстракции в одну стадию. Наблюдается также симбатный рост коэффициентов распределения и актинидов, и лантанидов при повышении концентрации азотной кислоты.
Рис. 4.
Зависимость логарифмов коэффициентов распределения Th(IV), U(VI), La(III), Nd(III), Ho(III) и Yb(III) при экстракции 0.01 моль/л раствором лиганда V в хлороформе.
Методом сдвига равновесия (рис. 5) был определен состав экстрагируемых комплексов урана(VI), тория(IV) и неодима(III) с лигандом V, содержащим ω-(2-оксопирролидино)пропильный радикал.
Рис. 5.
Логарифмическая зависимость коэффициентов распределения урана(VI), тория(IV) и неодима(III) от логарифма концентрации лиганда V.

Сольватное число для урана(VI) близко к 2, для тория – 1.5, а для неодима – 1. Следовательно, в указанных условиях неодим(III) экстрагируется в виде моносольвата, торий(IV) – в виде моно- и дисольватов, а уран(VI) – в виде дисольвата:
(1)
${\text{N}}{{{\text{d}}}^{{{\text{3 + }}}}}{\text{ + 3NO}}_{3}^{ - }\,{\text{ + L = Nd}}{{\left( {{\text{N}}{{{\text{O}}}_{{\text{3}}}}} \right)}_{{\text{3}}}}{{\cdot L,}}$(2)
${\text{T}}{{{\text{h}}}^{{{\text{4 + }}}}}{\text{ + 4N}}{{{\text{O}}}_{{\text{3}}}}\,{\text{ + L = Th}}{{\left( {{\text{N}}{{{\text{O}}}_{{\text{3}}}}} \right)}_{{\text{4}}}}{{\cdot L,}}$(3)
${\text{T}}{{{\text{h}}}^{{{\text{4 + }}}}}{\text{ + 4N}}{{{\text{O}}}_{{\text{3}}}}\,{\text{ + 2L = Th}}{{\left( {{\text{N}}{{{\text{O}}}_{{\text{3}}}}} \right)}_{{\text{4}}}}{{\cdot}}\,{\text{2L,}}$(4)
${\text{UO}}_{2}^{{2 + }}\,{\text{ + 2}}\left( {{\text{NO}}_{3}^{ - }} \right){\text{ + 2L = U}}{{{\text{O}}}_{{\text{2}}}}\left( {{\text{N}}{{{\text{O}}}_{{\text{3}}}}} \right){{ \cdot 2L}}{\text{.}}$Для объяснения разницы в экстракционной способности в серии лигандов проведено квантово-химическое моделирование строения и энергии координации ионов f-элементов с лигандом V методом теории функционала плотности. Предварительное исследование лиганда показало, что наибольший отрицательный заряд (табл. 1) наблюдается для атома кислорода фосфиноксидной группы, тогда как для атомов азота заряды систематически ниже. Атомы кислороды двух карбонильных групп, хотя и несут близкие отрицательные заряды, неравноценны. Атом кислорода карбоксамидной группы по сравнению с атомом кислорода карбамидного фрагмента несет немного больший по величине отрицательный заряд, также являясь привлекательной позицией для дополнительной координации иона металла.
Таблица 1.
Заряды по Хиршфелду и Малликену на некоторых гетероатомах (q)
| Способ | P=O | Карбамидный CO | Амидный CO* | Амидный N | Карбамидный NHR | Карбамидный NHP |
|---|---|---|---|---|---|---|
| Хиршфелд | –0.3346 | –0.2610 | –0.2694 | –0.0204 | –0.0819 | –0.1318 |
| Малликен | –0.4818 | –0.3837 | –0.3848 | –0.3032 | –0.4514 | –0.3405 |
Координация лиганда V с ионом уранила может проходить по монодентатному, бидентатному и тридентатному типу. Все три типа координации могут быть реализованы при образовании комплексов с ионом уранила (табл. 2). Энергия взаимодействия лиганда V с ионом уранила, катионом UO2(NO3)+ и молекулой UO2(NO3)2 сильно зависит от типа координации лиганда. Координация атома кислорода карбоксамидной группы энергетически менее выгодна, чем фосфиноксидного атома кислорода, но существенно выигрышнее по энергии по сравнению с координацией атома кислорода карбамидного фрагмента. При сравнении энергетического выигрыша (уравнение (7)) бидентатной координации лиганда с карбамидным и фосфиноксидным атомами кислорода и монодентатной координации с амидным и фосфиноксидным атомами кислорода монодентатная координация оказывается даже более выигрышной, но всего на 0.88 ккал/моль. В случае стерически не нагруженного катионного комплекса выигрыш в энергии при монодентатной координации минимален, однако включение в координацию амидного атома кислорода незначительно проигрывает включению в координацию атома кислорода фосфиноксидной группы (уравнение (6)). Для свободного уранил-катиона глобальному минимуму на поверхности потенциальной энергии (ППЭ) отвечает структура, в которой в координацию включены амидные атомы кислорода обоих лигандов.
Таблица 2.
Электронные энергии взаимодействия лиганда V с катионом уранила, моно- и динитратами уранила, ккал/моль
| Номер | Реакция | Энергия взаимодействия, ккал/моль | |||
|---|---|---|---|---|---|
| 5 | ${\text{UO}}_{2}^{{2 + }}\xrightarrow{{{\text{ + 2L}}}}{\text{U}}{{{\text{O}}}_{2}}\left( {\text{L}} \right)_{2}^{{2 + }}$ | CO*CO CO*COPO |
COPO COPO |
CO* CO* |
|
| –368.21 | –349.60 | –276.00 | |||
| 6 | ${\text{U}}{{{\text{O}}}_{2}}{{\left( {{\text{N}}{{{\text{O}}}_{3}}} \right)}^{ + }}\xrightarrow{{{\text{ + 2L}}}}{\text{U}}{{{\text{O}}}_{2}}{{\left( {\text{L}} \right)}_{2}}{{\left( {{\text{N}}{{{\text{O}}}_{3}}} \right)}^{ + }}$ | CO*PO CO* |
CO* CO* |
COPO COPO |
COPO PO |
| –158.70 | –129.44 | –151.67 | –160.34 | ||
| 7 | ${\text{U}}{{{\text{O}}}_{2}}{{\left( {{\text{N}}{{{\text{O}}}_{3}}} \right)}_{2}}\xrightarrow{{{\text{ + 2L}}}}{\text{U}}{{{\text{O}}}_{2}}{{\left( {\text{L}} \right)}_{2}}{{\left( {{\text{N}}{{{\text{O}}}_{3}}} \right)}_{2}}$ | COPO PO |
CO* CO* |
||
| –54.00 | –54.88 | ||||
В отличие от стерически не нагруженного свободного иона уранила, на ППЭ комплекса нитрата тория с реагентом V не наблюдается тридентатной координации лиганда (табл. 3); для указанной системы наблюдается только моно- и бидентатная координация. Для комплексов, содержащих два лиганда, наблюдается только монодентатная координация последнего, и на ППЭ не существует комплексов с координацией по атому кислорода карбамидного фрагмента. Хотя координация амидного атома кислорода приводит к меньшему выигрышу в энергии, чем фосфиноксидного атома кислорода, эта разница невелика (уравнение (9)). Для бидентатной координации также наблюдается существенно больший выигрыш при включении в координацию амидного атома кислорода (рис. 6а), а не атома кислорода карбамидной группы, несмотря на возникающий в последнем случае устойчивый шестичленный хелатный металлоцикл (рис. 6б).
Таблица 3.
Энергии взаимодействия лиганда V с нитратом тория в стехиометрических соотношениях 1 : 1 и 2 : 1, ккал/моль
| Номер | Реакция | Энергия взаимодействия, ккал/моль | ||
|---|---|---|---|---|
| 8 | ${\text{Th}}{{\left( {{\text{N}}{{{\text{O}}}_{{\text{3}}}}} \right)}_{{\text{4}}}}\xrightarrow{{{\text{ + L}}}}{\text{Th}}\left( {\text{L}} \right){{\left( {{\text{N}}{{{\text{O}}}_{3}}} \right)}_{4}}$ | CO* | COPO | CO*PO |
| –32.41 | –40.67 | –52.99 | ||
| 9 | ${\text{Th}}{{\left( {{\text{N}}{{{\text{O}}}_{{\text{3}}}}} \right)}_{{\text{4}}}}\xrightarrow{{{\text{ + 2L}}}}{\text{Th}}{{\left( {\text{L}} \right)}_{2}}{{\left( {{\text{N}}{{{\text{O}}}_{3}}} \right)}_{4}}$ | CO* CO* |
PO PO |
|
| –62.28 | –68.84 | |||
Рис. 6.
Строение комплексов нитрата тория с лигандом V, содержащих координированную с металлом амидную (а) и карбамидную (б) группы.

На ППЭ комплекса реагента V с нитратом неодима наблюдаются три минимума, которые отвечают тридентатной и бидентатной координации лиганда (табл. 4). Глобальному минимуму отвечает тридентатный тип координации, при котором все три донорных атома включены в координацию с металлом. В отличие от комплекса тория, образование шестичленного хелатного металлоцикла при бидентатной координации неодима с атомами кислорода карбамидной и фосфиноксидной групп энергетически более выгодно, чем бидентатная координация металла с двумя атомами кислорода карбонильных групп. Вероятно, это вызвано меньшими стерическими затруднениями вокруг иона металла, окруженного тремя противоионами.
Таблица 4.
Энергии взаимодействия лиганда V с нитратом неодима, ккал/моль
| Номер | Реакция | Энергия взаимодействия, ккал/моль | ||
|---|---|---|---|---|
| 10 | ${\text{Nd}}{{\left( {{\text{N}}{{{\text{O}}}_{3}}} \right)}_{3}}\xrightarrow{{{\text{ + L}}}}{\text{Nd}}\left( {\text{L}} \right){{\left( {{\text{N}}{{{\text{O}}}_{3}}} \right)}_{3}}$ | CO*CO | CO*COPO | COPO |
| –39.54 | –51.75 | –43.36 | ||
Таким образом, можно однозначно установить, что именно присутствие в молекуле экстрагента амидной группы приводит к значительному повышению экстракционных характеристик за счет энергетически выгодной координации иона металла с атомом кислорода карбоксамидной группы.
ЗАКЛЮЧЕНИЕ
В ряду исследованных соединений наилучшими экстракционными свойствами обладает N-(дифенилфосфорил)мочевина V, содержащая ω-(2-оксопирролидино)пропильный радикал у терминального атома азота, что может быть предпосылкой для создания технологии переработки техногенных отходов различного происхождения на базе этого лиганда.
Список литературы
Розен А.М., Крупнов Б.В. // Успехи химии. 1996. Т. 65. № 11. С. 1052. [Rozen A.M., Krupnov B.V. // Russ. Chem. Rev. 1996. V. 65. № 11. P. 973. https://doi.org/10.1070/RC1996v065n11ABEH000241]
Leoncini A., Huskens J., Verboom W. // Chem. Soc. Rev. 2017. V. 46. № 23. https://doi.org/10.1039/c7cs00574a
Horwitz E.P., Kalina D.G., Kaplan L. et al. // Separation Sci. Technol. 1982. V. 17. № 10. P. 1261. https://doi.org/10.1080/01496398208060649
Jensen M., Chiarizia R., Ulicki J.S. et al. // Solvent Extr. Ion Exch. 2015. V. 33. № 4. P. 329. https://doi.org/10.1080/07366299.2015.1046292
Ta A.T., Hegde G.A., Etz B.D. et al. // J. Phys. Chem. B. 2018. V. 122. № 22. P. 5999. https://doi.org/10.1021/acs.jpcb.8b03165
Mahanty B., Mohapatra P.K., Leoncini A. et al. // Separ. Purif. Technol. 2019. V. 229. № 15. P. 115846. https://doi.org/10.1016/j.seppur.2019.115846
Mohapatra P.K., Kandwal P., Iqbal M. et al. // Dalton Trans. 2013. V. 42. P. 4343. https://doi.org/10.1039/c3dt32967d
Sengupta A., Mohapatra P.K., Pathak P. et al. // New J. Chem. 2017. V. 41. P. 836. https://doi.org/10.1039/C6NJ03102A
Braley J.C., Lumetta G.J., Carter J.C. et al. // Solvent Extr. Ion Exch. 2013. V. 31. № 6. P. 567. https://doi.org/10.1080/07366299.2013.785912
Sasaki Y., Umetani S. // J. Nucl. Sci. Technol. 2006. V. 43. № 7. P. 794. https://doi.org/10.1080/18811248.2006.9711161
Sharova E.V., Artyushin O.I., Odinets I.L. // Russ. Chem. Rev. 2014. V. 83. № 2. P. 95. https://doi.org/10.1070/RC2014v083n02ABEH004384
Туранов А.Н., Карандашев В.К., Матвеева А.Г. и др. // Радиохимия. 2017. Т. 59. № 5. С. 430. [Turanov A.N., Kadandashev V.K., Matveeva A.G. et al. // Radiochemistry. 2017. V. 59. № 5. P. 490.] https://doi.org/10.1134/S1066362217050083
Dam H.H., Reinhoudt D.N., Verboom W. // Chem. Soc. Rev. 2007. V. 36. P. 367. https://doi.org/10.1039/B603847F
Tatarinov D.A., Mironov V.F., Kostin A.A. et al. // Phosphorus, Sulfur, and Silicon. 2011. V. 186. P. 694. https://doi.org/10.1080/10426507.2010.515955
Борисова Н.Е., Сафиулина А.М., Лизунов А.В. и др. // Журн. неорган. химии. 2019. Т. 64. № 3. С. 330. [Borisova N.E., Safiullina A.M., Lizunov A.V. et al. // Russ. J. Inorg. Chem. 2019. V. 64. № 3. P. 414.] https://doi.org/10.1134/S0036023619030057
Turanov A.N., Karandashev V.K., Baulin V.E. et al. // Solvent Extr. Ion Exch. 2009. V. 27. № 4. P. 551. https://doi.org/10.1080/07366290903044683
Туранов А.Н., Карандашев В.К., Артюшин О.И. и др. // Журн. неорган. химии. 2020. Т. 65. № 8. С. 1099. [Turanov A.N., Karandashev V.K., Artyushin O.I. et al. // Russ. J. Inorg. Chem. 2020. V. 65. № 8. P. 1226. https://doi.org/10.1134/S0036023620080185
Демин С.В., Жилов В.И., Нефедов С.Е. и др.// Журн. неорган. химии. 2012. Т. 57. № 6. С. 970. [Demin S.V., Nefedov S.E., Zhilov V.I. et al. // Russ. J. Inorg. Chem. 2012. V. 57. № 6. P. 897.] https://doi.org/10.1134/S0036023612060095
Туранов А.Н., Карандашев В.К., Артюшин О.И. и др. // Журн. неорган. химии. 2020. Т. 65. № 6. С. 837. [Turanov A.N., Karandashev V.K., Artyushin O.I. et al. // Russ. J. Inorg. Chem. 2020. V. 65. № 6. P. 905.] https://doi.org/10.31857/S0044457X20060240
Тананаев И.Г., Летюшов А.А., Сафиулина А.М. и др. // Докл. АН. 2008. Т. 422. № 6. С. 762. [Tananaev I.G., Letyushov A.A., Safiulina A.M. et al. // Dokl. Chem. 2008. Т. 422. № 2. P. 260.] https://doi.org/10.1134/S0012500808100054
Горюнов Е.И., Шипов А.Э., Горюнова И.Б. и др. // Докл. АН. 2011. Т. 438. № 4. С. 480. [Goryunov E.I., Shipov A.E., Goryunova I.B. et al. // Dokl. Chem. 2011. V. 438. № 2. Р. 151.] https://doi.org/10.1134/S0012500811060012
Safiulina A.M., Goryunov E.I., Letyushov A.A. et al. // Mendeleev Commun. 2009. V. 19. P. 263. https://doi.org/10.1016/j.mencom.2009.09.010
Горюнов Е.И., Баулина Т.В., Горюнова И.Б. и др. // Изв. АН. Сер. химическая. 2014. № 1. С. 141. [Goryunov E.I., Baulina T.V., Goryunova I.B. et al. // Russ. Chem. Bull. 2014. V. 63. № 1. P. 141.] https://doi.org/10.1007/s11172-014-0408-y
Лемпорт П.С., Горюнов Е.И., Горюнова И.Б. и др. // Докл. АН. 2009. Т. 425. № 6. С. 773. [Lemport P.S., Goryunov E.I., Goryunova I.B. et al. // Dokl. Chem. 2009. V. 425. № 2. P. 84.] https://doi.org/10.1134/S0012500809040053
Сафиулина А.М., Синегрибова О.А., Матвеева А.Г. и др. // Журн. неорган. химии. 2012. Т. 57. № 1. С. 115. [Safiulina A.M., Grigoriev M.S., Nifant’ev E.E. et al. // Russ. J. Inorg. Chem. 2012. V. 57. № 1. P. 108.] https://doi.org/10.1134/S0036023612010196
Саввин С.Б. Органические реагенты группы арсеназо III. М.: Атомиздат, 1971. 352 с.
Laikov D.N. // Chem. Phys. Lett. 2005. V. 416. № 1–3. P. 116. https://doi.org/10.1016/j.cplett.2005.09.046
Laikov D.N. // Chem. Phys. Lett. 1997. V. 281. № 1–2. P. 151. https://doi.org/10.1016/S0009-2614(97)01206-2
Perdew J.P., Burke K., Ernzerhof M. // Phys. Rev. Lett. 1996. V. 77. P. 3865. https://doi.org/10.1103/PhysRevLett.77.3865
Perdew J.P., Burke K., Ernzerhof M. // Phys. Rev. Lett. 1997. V. 78. P. 1396. https://doi.org/10.1103/PhysRevLett.78.1396
Borisova N.E., Kostin A.A., Eroshkina E.A. et al. // Eur. J. Inorg. Chem. 2014. P. 2219. https://doi.org/10.1002/ejic.201301271
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии