

Журнал неорганической химии, 2021, T. 66, № 5, стр. 641-647
Получение наночастиц ε-Fe2O3 в матрицах, образованных плотной упаковкой сфер диоксида кремния
А. И. Шарапаев a, *, С. А. Кузнецова a, А. Н. Норенко a, А. Г. Мурадова a, Н. П. Симоненко b, Е. В. Юртов a
a Российский химико-технологический университет им. Д.И. Менделеева
125047 Москва, Миусская пл., 9, Россия
b Институт общей и неорганической химии им. Н.С. Курнакова РАН
119071 Москва, Ленинский пр-т, 31, Россия
* E-mail: a.sharapaev@gmail.com
Поступила в редакцию 04.12.2020
После доработки 23.12.2020
Принята к публикации 24.12.2020
Аннотация
Разложением нитрата железа(III) в пустотах плотной упаковки сфер диоксида кремния получены смеси полиморфных модификаций оксида железа(III) с высоким содержанием ε-Fe2O3. Показана возможность управления фазовым составом нанопорошков Fe2O3 за счет использования сфер диоксида кремния различного размера. Определены критические размеры наночастиц Fe2O3, соответствующие переходам γ-Fe2O3 → ε-Fe2O3 и ε-Fe2O3 → α-Fe2O3, которые составляют 10 ± 2 и 28 ± 3 нм соответственно. Максимальное содержание ε-Fe2O3 достигается при размере сфер диоксида кремния 110 нм и составляет 83%.
ВВЕДЕНИЕ
Наноматериалы на основе оксидов железа находятся в центре внимания исследователей благодаря возможности их применения при создании новых биомедицинских препаратов, магнитных адсорбентов, катализаторов, в электронике, космической технике [1–5]. Среди разнообразных оксидов железа особое место занимает модификация ε-Fe2O3 [6, 7]. Высокая коэрцитивная сила (~20 кЭ) [8, 9], умеренная намагниченность насыщения и ферроэлектрические свойства делают ε-Fe2O3 перспективным материалом для поглощения и аттенюации электромагнитного излучения в диапазоне 50–200 ГГц, а также для хранения информации [9–11].
Термодинамически наиболее устойчивой формой оксида железа(III) является α-Fe2O3 [7], однако в случае наночастиц возможна стабилизация других модификаций за счет минимизации поверхностной энергии. В связи с этим фазовый состав нанопорошков Fe2O3 сильно зависит от размера образующих их наночастиц. Известно, что при размере наночастиц менее 20 нм преобладает γ-Fe2O3, размер частиц более 60–100 нм благоприятствует образованию α-Fe2O3, а в области размеров 20–60 нм возможно преобладание β- и ε-модификаций Fe2O3 [6, 7, 12].
Кристаллизация наночастиц и протекание полиморфных превращений в системах на основе Fe2O3 требуют воздействия высоких температур (более 600°C) [12–14]. Действие высокой температуры приводит к укрупнению наночастиц, поэтому целенаправленное получение ε-Fe2O3 практически всегда включает ограничение агрегации и роста наночастиц. Имеются сведения о получении ε-Fe2O3 без использования матрицы диоксида кремния путем термического разложения железосодержащих минералов [7], гидротермального [15] и плазмодинамического синтеза [16, 17] и даже с использованием магнетотактических бактерий [18], однако подобные методы либо не обеспечивают высокого содержания целевой модификации, либо неявно включают процесс разделения модификаций. Поэтому задача получения материалов с высоким содержанием ε-Fe2O3 по-прежнему требует решения.
Наиболее простым и распространенным способом получения ε-Fe2O3 является использование аморфного геля диоксида кремния с включениями гидроксидов железа [19, 20] либо сочетание золь-гель и микроэмульсионного методов [21, 22]. При этом для получения нанопорошков с высоким содержанием ε-Fe2O3 требуется введение небольших количеств ионов щелочноземельных металлов (Ba2+, Sr2+) для ограничения роста нанокристаллитов. Использование такого подхода не позволяет точно контролировать размер получаемых наночастиц и, как следствие, достигать высокого содержания ε-Fe2O3.
С целью более точного контроля размера получаемых наночастиц и увеличения доли ε-Fe2O3 используется термическая обработка наночастиц-предшественников заданного размера (α-Fe2O3 [23], γ-Fe2O3 [24], FeO [25], β-FeOOH [26, 27] или Fe3O4 [28]) в оболочке диоксида кремния или пропитка мезопористых силикагелей соединениями железа (FeSO4 [12, 29], Fe(NO3)3 [30–32], Fe(C10H9CHO) [12, 33]). Максимальное содержание ε‑Fe2O3 при этом ограничивается распределением по размерам исходных наночастиц или пор силикагеля и не превышает 70%. Причиной этого является широкое распределение по размерам наночастиц-предшественников в первом случае и малый размер пор силикагелей – во втором. Существенным недостатком использования наночастиц-предшественников является и то, что в большинстве случаев не удается добиться изолированного покрытия отдельных наночастиц оболочкой диоксида кремния.
В качестве альтернативы мезопористым силикагелям в роли темплата могут выступать плотноупакованные опалоподобные структуры из сферических частиц диоксида кремния. Размер пустот в таких матрицах определяется размерами частиц SiO2 [34, 35], что позволяет достаточно точно управлять размером формируемых наночастиц.
Подобные структуры широко используются для создания новых каталитических и сенсорных материалов на основе оксидов-перовскитов и ферритов-шпинелей [36, 37]. Известно, что размер пустот таких матриц оказывает влияние на фазовый состав синтезируемых в них наночастиц (например, титанатов висмута [38], оксида титана [39]). Получение оксидов железа в подобных матрицах также исследовалось. Так, в работе [40] прокаливанием при 450°C опалоподобной матрицы с размером исходных частиц 280 нм, пропитанной нитратом железа, была получена смесь наночастиц α- и γ-Fe2O3. Несмотря на широкое использование, темплатный синтез в пустотах плотноупакованных и опалоподобных структур диоксида кремния для получения наночастиц ε-Fe2O3 ранее не применялся.
В настоящей работе для контроля размера наночастиц Fe2O3 и направленного получения ε-Fe2O3 был использован темплатный синтез в матрицах, образованных плотной упаковкой монодисперсных частиц SiO2 размером 80–140 нм.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Для получения монодисперсных частиц SiO2 использовали тетраэтоксисилан (99+%, Across), 25%-ный раствор аммиака (Лаверна) и изопропиловый спирт (х. ч., ЭКОС-1). Для пропитки матриц применяли Fe(NO3)3 ∙ 9H2O (99+%, Across). Реактивы использовали без дополнительной очистки. Бидистиллированная вода была получена в лаборатории.
Исследование пленок диоксида кремния методом оптической микроскопии в проходящем свете выполняли на микроскопе Axiostar Plus (Zeiss, Германия) с цифровой фотокамерой Canon, при этом использовали стандартную процедуру настройки по Келлеру.
Для определения размера, формы и микроструктуры частиц SiO2 и структур на их основе применяли сканирующий электронный микроскоп JEOL JEM-6510LV. Исследование наночастиц методом просвечивающей электронной микроскопии (ПЭМ) выполняли на микроскопе JEOL JEM-1011.
Рентгенофазовый анализ образцов осуществляли на рентгеновском дифрактометре D8 Advance (Bruker-AXS, Германия) с использованием CuKα-излучения, измерения проводили в режиме 2θ–θ с шагом 0.01°, время накопления в точке 0.5 с.
Сферические частицы диоксида кремния размером <100 нм получали в ходе щелочного гидролиза тетраэтоксисилана в среде изопропилового спирта (метод Штобера) [41], наночастицы большего размера – путем доращивания слоя диоксида кремния на поверхности наночастиц-затравок [42]. В качестве наночастиц-затравок использовали наночастицы диоксида кремния, полученные методом Штобера.
Матрицы для создания наночастиц ε-Fe2O3 были получены путем естественного осаждения сферических частиц SiO2 из водно-спиртовой дисперсии в неоднородном поле температур. В качестве подложек использовали чашки Петри. Объем дисперсии частиц SiO2 выбирали исходя из необходимой расчетной толщины матрицы. Градиент температуры создавался за счет нагрева нижней части чашек Петри до 70°C. Испарение растворителя происходило в течение 30 мин.
После высушивания матрицы пропитывали раствором нитрата железа(III). Избыток раствора удаляли вакуумной фильтрацией, после чего матрицы высушивали при 120°C и прокаливали при температурах 900–1100°C в течение 2–16 ч. Для исследования наночастиц оксида железа методом ПЭМ диоксид кремния растворяли в 5 М растворе NaOH в течение 72 ч.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Исследовано влияние состава растворителя на однородность получаемых пленок. Для этого использовали водно-спиртовые дисперсии с концентрацией воды от 25 до 50 об. %, полученные разбавлением исходной дисперсии. Установлено, что увеличение содержания воды в дисперсии приводит к снижению однородности получаемых пленок, что можно объяснить повышением поверхностного натяжения раствора и соответствующим увеличением внутренних напряжений при его высыхании. На рис. 1 приведена оптическая микрофотография и результаты сканирующей электронной микроскопии пленки.

При средней расчетной толщине <10–15 мкм получаемые пленки обладают высокой макроскопической однородностью. Механическое разрушение таких пленок приводит к образованию относительно правильных ромбических фрагментов с углами, близкими к 60° и 120°, что объясняется гексагональной симметрией пленки, характерной для большинства коллоидных кристаллов (рис. 1а).
Как видно на изображении, полученном методом сканирующей электронной микроскопии (рис. 1б), пленки характеризуются плотной упаковкой сфер SiO2. Увеличение средней толщины >25 мкм приводит к формированию оптически неоднородных пленок, имеющих многочисленные макроскопические дефекты. Механическое разрушение таких пленок способствует образованию фрагментов произвольной формы. Для дальнейших исследований средняя толщина пленок была ограничена уровнем 10–15 мкм.
Для проверки формирования упорядоченных структур дополнительно были получены пленки из дисперсий с размером частиц 200–300 нм. При нормальном падении света на поверхность пленки наблюдается яркая окраска, цвет которой соответствует дифракции на плоскостях {111} структуры, образованной сферическими частицами SiO2 соответствующего размера. На рис. S1 а представлено фотоизображение пленки, состоящей из наночастиц размером 230 нм. Структурная окраска при нормальном падении света сохраняется после пропитки матриц раствором нитрата железа(III) и прокаливания, что свидетельствует о сохранении структуры. При этом окраска изменяется в более длинноволновую область вследствие увеличения эффективного показателя преломления среды (рис. S1 б).
На рис. 2 представлены ПЭМ-изображения наночастиц диоксида кремния, использованных для получения матриц. Как видно из рис. 2, наночастицы обладают узким распределением по размерам и склонны к самопроизвольному образованию слоев гексагональной симметрии с плотнейшей упаковкой наночастиц.

Для изучения влияния продолжительности прокаливания на кристаллизацию наночастиц оксида железа(III) прокаливание проводили при 1000°C от 2 до 16 ч. Средний размер частиц диоксида кремния, образующих матрицы, составлял 120 ± 4 нм. Как видно из рис. 3, увеличение времени прокаливания сопровождается ростом контрастности наночастиц на электронно-микроскопических снимках, что свидетельствует об увеличении их кристалличности. Следует отметить наличие частиц округлой и продолговатой формы, что может свидетельствовать о различии их кристаллической структуры. Увеличение продолжительности прокаливания не оказывает значительного влияния на средний размер наночастиц, но приводит к увеличению количества наблюдаемых стержнеобразных частиц.
Рис. 3.
ПЭМ-изображения наночастиц Fe2O3, полученных при прокаливании в течение 2 (а), 4 (б) и 8 ч (в).

Результаты исследования наночастиц оксида железа, полученных в матрицах с различным размером сфер SiO2, показывают, что размер наночастиц оксида железа пропорционален размеру частиц матрицы, что хорошо согласуется с образованием наночастиц Fe2O3 в пустотах плотнейшей упаковки (рис. 4). Полученные результаты свидетельствуют о том, что использованный подход позволяет управлять размером наночастиц оксида железа и получать наночастицы с размерами в области стабильности ε-Fe2O3.
Рис. 4.
Распределение наночастиц Fe2O3 по размерам для матриц, образованных частицами SiO2 размером 80 (а), 100 (б), 120 (в) и 140 нм (г).

На рис. 5 представлена характерная рентгеновская дифрактограмма нанопорошка. На дифрактограмме отчетливо видны рефлексы, соответствующие трем модификациям оксида железа(III): α-Fe2O3, γ-Fe2O3, ε-Fe2O3. Наиболее интенсивным является рефлекс, соответствующий плоскостям (122) ε-Fe2O3 и расположенный в области 32° (2θ). Присутствие β-Fe2O3 нельзя полностью исключить, однако отсутствие наиболее интенсивных рефлексов данной модификации говорит о ее малом содержании.
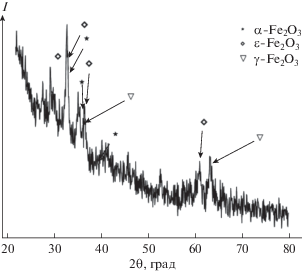
Анизотропия уширения дифракционных пиков свидетельствует о том, что частицы ε-Fe2O3 имеют форму эллипсоидов вращения с отношением больших осей 1 : 3 : 2 для осей кристаллической решетки a, b и c соответственно, что хорошо согласуется с результатами измерения анизометричных частиц по ПЭМ и кристаллографической симметрией ε-Fe2O3 (пр. гр. Pna21).
В табл. 1 дана оценка фазового состава полученных нанопорошков. Приведенные данные показывают, что увеличение размера сфер, образующих матрицу, сопровождается постепенным изменением фазового состава. При этом характер изменения согласуется с общепринятой гипотезой о влиянии размера наночастиц на устойчивость полиморфных модификаций Fe2O3 и порядке их следования.
Таблица 1.
Фазовый состав продуктов термообработки в зависимости от размера сфер, образующих матрицу, %
| Размер сфер SiO2, нм | α-Fe2O3 | γ-Fe2O3 | ε-Fe2O3 |
|---|---|---|---|
| 72 | 15 | 32 | 53 |
| 80 | 15 | 16 | 69 |
| 100 | 15 | 9 | 76 |
| 120 | 19 | 15 | 66 |
| 140 | 24 | 9 | 67 |
Для установления пороговых значений, соответствующих границам стабильности полиморфных модификаций, были рассчитаны квантили распределений объема наночастиц Fe2O3. Распределения объема наночастиц были построены по результатам просвечивающей электронной микроскопии. Критический размер, соответствующий переходу γ-Fe2O3 → ε-Fe2O3, составил 10 ± 2 нм. Размер, соответствующий переходу ε-Fe2O3 → → α-Fe2O3, составил 28 ± 3 нм.
Наблюдаемые распределения объемов наночастиц могут быть описаны логарифмически-нормальным распределением, параметры которого зависят от размера частиц матрицы. Установленные пороговые значения размеров, соответствующих стабилизации полиморфных модификаций Fe2O3, указывают на максимальное содержание ε-Fe2O3 при размере частиц матрицы 100–110 нм, что хорошо объясняет экспериментально наблюдаемые результаты.
Максимальное содержание ε-Fe2O3 и размер наночастиц матрицы, при котором оно достигается, сильно зависят от размеров, соответствующих переходам γ-Fe2O3 → ε-Fe2O3 и ε-Fe2O3 → α-Fe2O3. Максимальное содержание ε-Fe2O3 составляет от 70 до 80% для диапазонов переходов 8–12 и 25–30 нм соответственно.
Для подтверждения полученных закономерностей использовали плотноупакованные матрицы, полученные из дисперсий SiO2 со средним размером частиц 110 нм, прокаливание проводили при 1000°C. Полученные при этом образцы содержат до 83% ε-Fe2O3 (по сравнению с другими модификациями) с учетом большого количества аморфного диоксида кремния и возможного наложения рефлексов α- и γ-модификаций, снижающих точность количественного фазового анализа.
ЗАКЛЮЧЕНИЕ
Подтверждена возможность получения ε-Fe2O3 путем термического разложения нитрата железа(III) в пустотах матриц, образованных плотной упаковкой монодисперсных частиц SiO2.
Критические размеры наночастиц, соответствующие переходам γ-Fe2O3 → ε-Fe2O3 и ε-Fe2O3 → → α-Fe2O3, составляют 10 ± 2 и 28 ± 3 нм соответственно. Вследствие этого фазовый состав нанопорошков определяется размером наночастиц SiO2, образующих матрицу. Максимальное содержание ε-Fe2O3 достигается при размере частиц матрицы 100–110 нм, что было подтверждено экспериментально. Результаты работы могут быть использованы для контролируемого получения наночастиц ε-Fe2O3. Кроме того, предложенный подход можно применять для установления размерных границ стабильности полиморфных модификаций других неорганических соединений.
Список литературы
Strapolova V.N., Yurtov E.V., Muradova A.G. et al. // J. Spacecr. Rockets. 2017. V. 55. № 1. P. 1. https://doi.org/10.2514/1.A33805
Zarschler K., Rocks L., Licciardello N. et al. // Nanomedicine Nanotechnology, Biol. Med. 2016. V. 12. № 6. P. 1663. https://doi.org/10.1016/j.nano.2016.02.019
Ali A., Zafar H., Zia M. et al. // Nanotechnol. Sci. Appl. 2016. V. 9. P. 49. https://doi.org/10.2147/NSA.S99986
Mjakin S.V., Nikolaev A.M., Khamova T.V. et al. // Russ. J. Inorg. Chem. 2020. V. 65. № 4. P. 626. https://doi.org/10.1134/S0036023620040129
Naderi S., Morsali A., Bozorgmehr M.R. et al. // Russ. J. Inorg. Chem. 2019. V. 64. № 4. P. 503. https://doi.org/10.1134/S0036023619040156
MacHala L., Tuček J., Zbořil R. // Chem. Mater. 2011. V. 23. № 14. P. 3255. https://doi.org/10.1021/cm200397g
Lee S., Xu H. // J. Phys. Chem. C. 2016. V. 120. Acs. Jpcc.6b05287. https://doi.org/10.1021/acs.jpcc.6b05287
Jin J., Ohkoshi S., Hashimoto K. // Adv. Mater. 2004. V. 16. № 1. P. 48. https://doi.org/10.1002/adma.200305297
Tuček J., Zbořil R., Namai A. et al. // Chem. Mater. 2010. V. 22. № 24. P. 6483. https://doi.org/10.1021/cm101967h
Namai A., Sakurai S., Nakajima M. et al. // J. Am. Chem. Soc. 2009. V. 131. № 3. P. 1170. https://doi.org/10.1021/ja807943v
Ohkoshi S.I., Kuroki S., Sakurai S. et al. // Angew. Chem. Int. Ed. 2007. V. 46. № 44. P. 8392. https://doi.org/10.1002/anie.200703010
Sakurai S., Namai A., Hashimoto K. et al. // J. Am. Chem. Soc. 2009. V. 131. № 51. P. 18299. https://doi.org/10.1021/ja9046069
Brázda P., Večerníková E., Pližingrová E. et al. // J. Therm. Anal. Calorim. 2014. V. 117. № 1. P. 85. https://doi.org/10.1007/s10973-014-3711-9
Gich M., Roig A., Taboada E. et al. // Faraday Discuss. 2007. V. 136. P. 345. https://doi.org/10.1039/b616097b
Ma J., Chen K. // Ceram. Int. 2018. V. 44. № 16. P. 19 338. https://doi.org/10.1016/j.ceramint.2018.07.162
Shanenkov I., Sivkov A., Ivashutenko A. et al. // J. Alloys Compd. 2019. V. 774. P. 637. https://doi.org/10.1016/j.jallcom.2018.10.019
Sivkov A., Naiden E., Ivashutenko A. et al. // J. Magn. Magn. Mater. 2016. V. 405. P. 158. https://doi.org/10.1016/j.jmmm.2015.12.072
Wen T., Zhang Y., Geng Y. et al. // Biomater. Res. 2019. V. 23. № 1. P. 1. https://doi.org/10.1186/s40824-019-0162-1
Nikolic V.N., Spasojevic V., Panjan M. et al. // Ceram. Int. 2017. V. 43. № 10. P. 7497. https://doi.org/10.1016/j.ceramint.2017.03.030
Barick K.C., Varaprasad B.S.D.C.S., Bahadur D. // J. Non. Cryst. Solids 2010. V. 356. № 3. P. 153. https://doi.org/10.1016/j.jnoncrysol.2009.10.001
Jin J., Hashimoto K., Ohkoshi S. // J. Mater. Chem. 2005. V. 15. № 10. P. 1067. https://doi.org/10.1039/B416554C
López-Sánchez J., Muñoz-Noval A., Serrano A. et al. // RSC Adv. 2016. V. 6. № 52. P. 46 380. https://doi.org/10.1039/C6RA01912A
Tadić M., Spasojević V., Kusigerski V. et al. // Scr. Mater. 2008. V. 58. № 8. P. 703. https://doi.org/10.1016/j.scriptamat.2007.12.009
Taboada E., Gich M., Roig A. // ACS Nano 2009. V. 3. № 11. P. 3377. https://doi.org/10.1021/nn901022s
Nakaya M., Nishida R., Hosoda N. et al. // Cryst. Res. Technol. 2017. V. 52. № 11. P. 1700110. https://doi.org/10.1002/crat.201700110
Tadic M., Milosevic I., Kralj S. et al. // Nanoscale. 2017. V. 9. № 30. P. 10579. https://doi.org/10.1039/c7nr03639f
Tadic M., Milosevic I., Kralj S. et al. // Acta Mater. 2020. V. 188. P. 16. https://doi.org/10.1016/j.actamat.2020.01.058
Klekotka U., Satuła D., Kalska-Szostko B. // J. Magn. Magn. Mater. 2020. V. 497. At. 165 999. https://doi.org/10.1016/j.jmmm.2019.165999
Bukhtiyarova G.A., Shuvaeva M.A., Bayukov O.A. et al. // J. Nanoparticle Res. 2011. V. 13. № 10. P. 5527. https://doi.org/10.1007/s11051-011-0542-5
Delahaye E., Escax V., El Hassan N. et al. // J. Phys. Chem. B. 2006. V. 110. № 51. P. 26001. https://doi.org/10.1021/jp0647075
Kubíčková L., Kohout J., Brázda P. et al. // Hyperfine Interact. 2016. V. 237. № 1. https://doi.org/10.1007/s10751-016-1356-8
Brázda P., Kohout J., Bezdička P. et al. // Cryst. Growth Des. 2014. V. 14. P. 1039. https://doi.org/10.1021/cg4015114
Nakamura T., Yamada Y., Yano K. // J. Mater. Chem. 2006. V. 16. № 25. P. 2417. https://doi.org/10.1039/B604025J
Rouault Y., Assouline S. // Powder Technol. 1998. V. 96. № 1. P. 33. https://doi.org/10.1016/S0032-5910(97)03355-X
Aste T., Weaire D. // The pursuit of perfect packings. Taylor & Francis Group, 2008.
Sadakane M., Takahashi C., Kato N. et al. // Bull. Chem. Soc. Jpn. 2007. V. 80. № 4. P. 677. https://doi.org/10.1246/bcsj.80.677
Qin J., Cui Z., Yang X. et al. // Sens. Actuators, B: Chem. 2015. V. 209. P. 706. https://doi.org/10.1016/j.snb.2014.12.046
Ивичева С.Н., Каргин Ю.Ф., Куцев С.В. и др. // Журн. неорган. химии. 2015. Т. 60. № 11. С. 1439. https://doi.org/10.7868/s0044457x15110082
Ивичева С.Н., Каргин Ю.Ф., Ляпина О.А. и др. // Неорган. материалы. 2009. Т. 45. № 11. С. 1337.
Ивичева С.Н., Каргин Ю.Ф., Ашмарин А.А. и др. // Журн. неорган. химии. 2012. Т. 57. № 11. С. 1508.
Ibrahim I.A.M., Zikry A.A.F., Sharaf M.A. // J. Am. Sci. 2010. V. 6. № 11. P. 985. https://doi.org/10.7537/marsjas061110.133
Zaytseva M.P., Muradova A.G., Sharapaev A.I. et al. // Russ. J. Inorg. Chem. 2018. V. 63. № 12. P. 1684. https://doi.org/10.1134/S0036023618120239
Дополнительные материалы
- скачать ESM_1.jpg
- Рис. S1. Матрица SiO2 до (а) и после (б) пропитывания раствором Fe(NO)3.
{kind=link}
Инструменты
Журнал неорганической химии