

Журнал неорганической химии, 2022, T. 67, № 10, стр. 1359-1366
Особенности гликоль-цитратного синтеза высокодисперсного оксида состава La0.6Sr0.4Co0.2Fe0.8O3 – δ
Т. Л. Симоненко a, *, Н. П. Симоненко a, Е. П. Симоненко a, Н. Т. Кузнецов a
a Институт общей и неорганической химии им. Н.С. Курнакова РАН
119991 Москва, Ленинский пр-т, 31, Россия
* E-mail: egorova.offver@gmail.com
Поступила в редакцию 16.04.2022
После доработки 11.05.2022
Принята к публикации 12.05.2022
- EDN: BNOOQZ
- DOI: 10.31857/S0044457X22600736
Аннотация
Исследованы особенности процесса гликоль-цитратного синтеза высокодисперсного оксида состава La0.6Sr0.4Co0.2Fe0.8O3 – δ при варьировании концентрации окислителя (нитрогруппы) и восстановителя (этиленгликоля) в составе реакционной системы, а также определено влияние данных параметров на термическое поведение, спектральные характеристики, фазовый состав и микроструктуру полученных нанопорошков. Показано, что предложенные условия синтеза позволяют снизить температуру формирования однофазных высокодисперсных оксидов заданного состава, перспективных в качестве катодных материалов среднетемпературных твердооксидных топливных элементов.
ВВЕДЕНИЕ
Как известно, важным аспектом декарбонизации экономики является внедрение новых систем генерации “зеленой” энергии. На сегодняшний день все больше внимания уделяется развитию сектора энергетики, основанного на возобновляемых источниках энергии, а общемировое количество вырабатываемой ими электроэнергии неуклонно растет. В данном контексте необходимо выделить твердооксидные топливные элементы (ТОТЭ), преобразующие химическую энергию топлива в электрическую посредством электрохимических реакций с высокой эффективностью и низким уровнем выбросов загрязняющих веществ [1–3]. ТОТЭ имеют перспективы применения как в условиях, требующих обеспечения электропитания портативных приборов [4–6], так и при создании мощных (рассчитанных на десятки мегаватт) стационарных установок выработки электроэнергии, способных обеспечивать электро- и теплоснабжение жилых строений и других объектов [7–11]. Твердооксидные топливные элементы характеризуются наиболее высокими рабочими температурами по сравнению с другими типами топливных элементов, что обусловливает их наибольший коэффициент преобразования топлива без потребности в дорогостоящих катализаторах платиновой группы [12, 13]. Однако высокие рабочие температуры ускоряют процессы деградации конструкционных и функциональных элементов топливной ячейки, снижая тем самым ее мощность и рабочий ресурс. Обозначенная проблема приводит к увеличению стоимости итоговых устройств, а также к удорожанию производимой ими электроэнергии, что является основным фактором, сдерживающим процесс широкой коммерциализации технологий твердооксидных топливных элементов [14, 15]. В связи с этим сегодня значительные усилия ученых и исследователей направлены на снижение рабочей температуры топливных ячеек (до 400–650°С) и переход к среднетемпературным ТОТЭ, что в первую очередь требует создания материалов, которые по своим функциональным характеристикам (значениям электропроводности, уровню каталитической активности, химической устойчивости и т.д.) не уступали бы классическим высокотемпературным материалам [16–19]. Необходимо также учитывать, что снижение рабочих температур топливных ячеек может повлечь за собой возникновение поляризационных потерь со стороны катода, что негативно отразится на мощностных показателях итогового устройства [20–22]. Снизить влияние описанного эффекта можно посредством поиска новых и модификации существующих катодных материалов с высокой ионной и электронной проводимостью, а также электрокаталитической активностью в среднетемпературном интервале. Одними из наиболее перспективных в данном ключе представляются материалы со структурой типа перовскита (с общей формулой АВО3), которые благодаря возможности тонкого регулирования взаимосвязанных факторов в ряду состав–структура–свойства находят широкое применение не только в области альтернативной энергетики (при создании топливных элементов, литий-воздушных аккумуляторов, суперконденсаторов и фотовольтаических солнечных элементов), но также в области электро- и фотокатализа [23–27]. В настоящее время в качестве катодных материалов для среднетемпературных ТОТЭ активно исследуются смешанные проводники со структурой типа перовскита состава Ln1 –xAxMO3 – δ (Ln = La, Sm, Nd, Nd; A = Ca, Sr, Ba; M = Co, Fe, Ni), которые представляют собой альтернативу высокотемпературному катоду состава La1 –xSrxMnO3 – δ (LSM), который демонстрирует требуемый уровень проводимости при 1000°С [28–31]. Среди перечисленных среднетемпературных оксидов La1 –xSrxCo1 –yFeyO3 – δ (LSCF) считается одним из наиболее многообещающих кандидатов на роль катода ТОТЭ, что обусловлено его высокой электронной/ионной проводимостью и каталитической активностью по отношению к реакции восстановления кислорода на его поверхности в широком диапазоне температур [32–35]. Среди наиболее распространенных подходов к синтезу оксидов сложного состава со структурой перовскита необходимо отметить группу методов сжигания: глицин-нитратный [36], цитрат-нитратный [37], а также гликоль-цитратный синтез [38]. Особенность данных подходов состоит в том, что они позволяют эффективно получать однофазные нанокристаллические материалы, в состав которых входят разные по своей химической природе элементы [39, 40]. Кроме того, важным преимуществом данных методов является возможность контроля интенсивности окислительно-восстановительной реакции, в ходе которой формируются целевые продукты, за счет варьирования соотношения между органическими восстановителями (этиленгликоль, лимонная кислота) и окислителем (нитрогруппы). В свою очередь, это позволяет снизить температуры формирования оксидов заданного состава, сохраняя их высокую дисперсность, что, как правило, положительно сказывается на функциональных характеристиках итоговых материалов.
Цель работы – исследование процесса гликоль-цитратного синтеза оксида состава La0.6Sr0.4Co0.2Fe0.8O3 – δ, а также изучение влияния концентрации окислителя и восстановителя в реакционной системе на физико-химические характеристики получаемых нанопорошков.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Синтез оксида состава La0.6Sr0.4Co0.2Fe0.8O3 – δ проводили с помощью гликоль-цитратного метода в соответствии со следующей методикой: необходимые навески нитратов лантана, стронция и кобальта, а также хлорида железа растворяли в минимальном количестве дистиллированной воды. Затем с целью замещения хлорид-ионов на нитрогруппы к полученному раствору солей добавляли концентрированную азотную кислоту и проводили термообработку реакционной системы при кипении в течение 30 мин. После этого при перемешивании по каплям добавляли концентрированный водный раствор гидрата аммиака (ω = 25%), доводя pH раствора до 5.5. Далее к полученной реакционной системе добавляли лимонную кислоту, этиленгликоль и нитрат аммония, а затем упаривали раствор до вязкого состояния. На следующем этапе с целью инициирования окислительно-восстановительной реакции и воспламенения реакционные системы подвергали термообработке при 300°С в течение 1 ч, а для удаления остаточного углерода формируемые порошки дополнительно прокаливали при 600°С (2 ч).
Для установления влияния концентрации окислителя и восстановителей на процесс синтеза, а также на фазовый состав и микроструктуру получаемого оксида была проведена серия экспериментов с различным соотношением лимонной кислоты, этиленгликолем и нитратом аммония в составе реакционных систем (табл. 1).
Таблица 1.
Отношения этиленгликоля и нитрата аммония к лимонной кислоте, используемые при синтезе порошков состава La0.6Sr0.4Co0.2Fe0.8O3 – δ
| Номер эксперимента | n(C2H6O2)/ n(C6H8O7) | n(NH4NO3)/ n(C6H8O7) |
|---|---|---|
| 1 | 2 | 0.33 |
| 2 | 2 | 0.50 |
| 3 | 3 | 0.33 |
| 4 | 3 | 0.50 |
Синхронный (ТГА/ДСК) термический анализ использовали для изучения процесса инициирования и окислительно-восстановительных реакций, протекающих при нагревании реакционных систем, а также для их трансформации с образованием оксидов заданного состава. Термический анализ проводили с помощью термоанализатора SDT Q-600 (TA Instruments), контролируемый нагрев осуществляли в Al2O3-микротиглях в диапазоне температур 25–1000°С со скоростью 10 град./мин в токе воздуха, скорость потока газа составляла 250 мл/мин. Масса исследуемых реакционных систем составляла 26–28 мг, а порошков, термообработанных при 300°С в течение 1 ч, – 6–9 мг.
С целью регистрации ИК-спектров пропускания порошков готовили соответствующие суспензии в вазелиновом масле, которые далее в виде пленки помещали между стеклами из бромида калия. Спектральный анализ выполняли в интервале волновых чисел 350–4000 см–1 (время накопления сигнала 15 с, разрешение 1 см–1) с использованием ИК-фурье-спектрометра ИнфраЛЮМ ФТ-08.
Рентгенофазовый анализ (РФА) полученных порошков проводили с помощью дифрактометра Bruker D8 Advance (излучение CuKα, λ = 1.5418 Å, Ni-фильтр, E = 40 кэВ, I = 40 мА, диапазон 2θ = = 15°–80°, разрешение 0.02°, время накопления сигнала в точке 0.3 с).
Микроструктуру полученных оксидных порошков исследовали методами растровой (РЭМ; трехлучевая рабочая станция Carl Zeiss NVision 40, оснащенная энергодисперсионным микрозондовым анализатором Oxford Instruments X-MAX 80) и просвечивающей электронной микроскопии (ПЭМ; JEOL JEM-1011 с цифровой фотокамерой ORIUS SC1000W).
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
С помощью синхронного (ТГА/ДСК) термического анализа изучен процесс инициирования окислительно-восстановительной реакции при нагревании реакционных систем в токе воздуха (рис. 1а, 1б). Как видно из соответствующих термограмм, для исследуемых систем наблюдаются две существенные ступени потери массы в интервалах температур 25–150 и 150–250°С. Третья ступень уменьшения массы в диапазоне 250–600°С является гораздо менее интенсивной. При этом низкотемпературная стадия снижения массы (Δm составляет ~50%) во всех случаях сопровождается эндотермическим эффектом с максимумом в интервале 92–105°С и относится к испарению растворителя. Следует отметить, что для реакционной системы 2, характеризующейся наибольшей концентрацией окислителя (нитрат аммония) по отношению к восстановителям (лимонная кислота и этиленгликоль), скорость уменьшения массы на данном этапе является максимальной, а максимум теплового эффекта наиболее смещен в низкотемпературную область, находясь при 92°С. При последующем повышении температуры до 180°С для систем 1, 2 и 3 имеет место практически линейное падение массы, а в случае реакционной системы 4, которая содержит максимальное количество нитрата аммония и этиленгликоля, наблюдается более медленное снижение массы. При достижении температуры ~180°С во всех случаях происходит инициирование окислительно-восстановительной реакции и активное газовыделение, в результате чего для систем 1, 3 и 4 наблюдается резкая потеря массы (Δm находится в диапазоне 25–37%) практически с одинаковой скоростью. Термическое поведение реакционной системы 2 на данном этапе отличается меньшей скоростью снижения массы и минимальной величиной Δm. Из соответствующих кривых ДСК видно (рис. 1б), что воспламенение сопровождается выделением энергии с максимумом экзоэффекта для систем 1, 3 и 4 в узком интервале температур 199–201°С. Для реакционной системы 2 наблюдается смещение данного теплового эффекта в область более высоких температур (210°С), следует так же отметить существенное увеличение его интенсивности по сравнению с остальными системами. Последующий рост температуры до 600°С для систем 1, 3 и 4 сопровождается малоинтенсивным снижением массы, а для системы 2 характерна более заметная величина Δm (~3%). На данном этапе происходит окисление образовавшегося углерода, что подтверждается наличием слабого экзотермического эффекта с максимумом в области 400–450°С. Величина итоговой потери массы при нагревании реакционных систем до 1000°С находится в диапазоне 97.9–98.4%. Таким образом, результаты термического анализа исследуемых реакционных систем свидетельствуют о том, что основные химические превращения протекают при их нагревании до температуры ~250°С, а окисление образующегося углерода завершается до 600°С.
Рис. 1.
Кривые ТГА (слева) и ДСК (справа) реакционных систем 1–4 после упаривания (а, б), а также порошков, образовавшихся после их термообработки при 300°С (в, г).
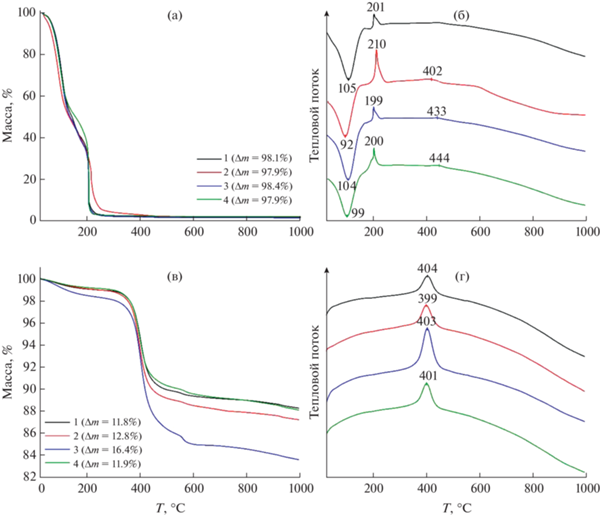
С использованием синхронного (ТГА/ДСК) термического анализа изучено термическое поведение на воздухе порошков, полученных в результате термообработки реакционных систем при 300°С в течение 1 ч (рис. 1в, 1г). Так, при нагревании данных порошков до 1000°С наблюдаются четыре ступени потери массы, первая из которых (Δm составляет 1–2%) находится в интервале температур 25–300°С и связана с удалением сорбированных атмосферных газов и воды с поверхности порошков. Наиболее интенсивная ступень уменьшения массы (9–12%) находится в диапазоне 300–550°С и сопровождается экзотермическим эффектом, что свидетельствует об окислении остаточного углерода. Для порошка, полученного с использованием реакционной системы 3, на данном и других этапах наблюдается наибольшая величина Δm, что объясняется недостаточной концентрацией окислителя (в частности, нитрата аммония) для более полного окисления органических компонентов на стадии инициирования окислительно-восстановительной реакции и воспламенения. Дальнейшее нагревание приводит к потере массы порошками в узком интервале температур (550–580°С), сопровождающейся малоинтенсивным экзоэффектом, что, как и последующее постепенное уменьшение массы до температуры 1000°С, относится к окислению более устойчивых фракций остаточного углерода. По результатам термического анализа был определен режим дополнительной термообработки (600°С, 2 ч) соответствующих порошков с целью удаления остаточного углерода.
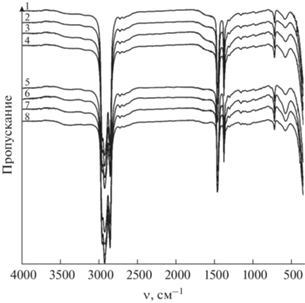
По данным ИК-фурье-спектроскопии (рис. 2), порошки, полученные в результате гликоль-цитратного синтеза и последующей дополнительной термообработки, имеют идентичный набор функциональных групп. Так, в спектрах порошков во всех случаях имеются интенсивные характеристические полосы поглощения с максимумами при 660 и 575 см–1, относящиеся к колебаниям связей M–O, свидетельствующие о формировании целевых оксидов [41, 42]. Кроме того, видно, что после проведения дополнительной термообработки (600°С, 2 ч) практически исчезают полосы поглощения в интервале волновых чисел 1700–1530 см–1, обусловленные колебаниями С–О-, С=О- и ОН-групп, что свидетельствует об удалении остаточного углерода. Результаты ИК-спектроскопии также подтверждают отсутствие в составе полученных оксидных порошков примесей реагентов и побочных продуктов.
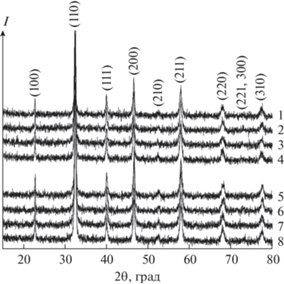
Исследование кристаллической структуры порошков, полученных в результате термообработки реакционных систем при 300°С (1 ч) и после удаления остаточного углерода (600°С, 2 ч), проводили с помощью РФА (рис. 3). Как видно из рентгенограмм, уже после инициирования окислительно-восстановительной реакции и термообработки при 300°С были получены целевые продукты, характеризующиеся кубической кристаллической структурой типа перовскита (пр. гр. $~Pm\bar {3}m$), которые не содержат каких-либо кристаллических примесей. Тем не менее, учитывая данные термического анализа, можно предположить наличие аморфного углерода в составе порошков после проведения первой стадии термообработки. Образец 3, содержащий максимальное в исследуемом концентрационном ряду количество восстановителя (этиленгликоля), характеризуется минимальным средним размером области когерентного рассеяния (ОКР) ~26 нм. Данный факт может быть связан с образованием наибольшего количества остаточного углерода в ходе окислительно-восстановительной реакции, препятствующего активному укрупнению оксидных частиц. Образцы 1 и 4 характеризуются средним размером ОКР порядка 27 и 29 нм, тогда как в случае образца 2, содержащего максимальное количество окислителя, наблюдается наибольшее значение среднего размера ОКР (~30 нм), что, вероятно, обусловлено наиболее интенсивным выделением энергии в ходе воспламенения реакционной системы. Проведение дополнительной термообработки при 600°С не привело к изменению типа кристаллической структуры полученных порошков, а также практически не повлияло на средний размер ОКР.
С использованием растровой электронной микроскопии изучена микроструктура полученных оксидных порошков состава La0.6Sr0.4Co0.2Fe0.8O3 – δ (рис. 4). Как видно из микрофотографий, порошки имеют традиционную для продуктов гликоль-цитратного синтеза вспененную морфологию и состоят из агломератов в виде пленок сложной формы толщиной от десятков нанометров до нескольких микрометров. При этом внутренний объем оксидных пленок представляет собой высокопористый материал, состоящий из частиц размером 20–30 нм, а их внешняя поверхность заметно отличается по своей микроструктуре и характеризуется высокой плотностью. Толщина плотной приповерхностной области агломератов, как правило, не зависит от особенностей их внутренней микроструктуры и варьируется в диапазоне 15–20 нм. Полученные данные позволили определить, что в результате вспенивания и воспламенения реакционной системы 1 с минимальной концентрацией нитрата аммония и этиленгликоля формируются оксидные пленки с наименьшей толщиной. В случае одновременного увеличения содержания окислителя (NH4NO3) и одного из восстановителей (C2H6O2) продукт гликоль-цитратного синтеза состоит из оксидных пленок наибольшей толщины. Таким образом, характер микроструктуры оксидного порошка существенно зависит от состава реакционной системы, что позволяет путем варьирования концентрации окислителя и восстановителя контролировать и прогнозировать особенности морфологии, дисперсность, удельную площадь поверхности и, соответственно, функциональные свойства получаемого материала. Результаты проведенного рентгеноспектрального элементного микроанализа подтвердили соответствие химического состава исследуемых порошков заданному.
Рис. 4.
Микроструктура оксидов состава La0.6Sr0.4Co0.2Fe0.8O3 – δ (а – 1, б – 2, в – 3, г – 4) после термообработки на воздухе при 600°С (по данным РЭМ).

Для более детального определения характера морфологии агломератов, размера и формы оксидных частиц полученные материалы были изучены с помощью просвечивающей электронной микроскопии (рис. 5). Как видно из микрофотографий, для всех порошков оксидные агломераты представляют собой пленки сложной формы, что согласуется с данными РЭМ. При этом подтверждено, что агломераты состоят из частиц размером около 20–30 нм. С учетом ранее приведенных результатов рентгенофазового анализа можно сделать вывод о том, что наблюдаемые оксидные частицы в основном монокристаллические. Кроме того, исследуемые оксидные пленки состава La0.6Sr0.4Co0.2Fe0.8O3 – δ являются пористыми, а размер пор находится на уровне до 10 нм. По данным электронной микроскопии, оксидные порошки не содержат примесей, отличающихся от основного продукта по своей микроструктуре.

ЗАКЛЮЧЕНИЕ
Проведено исследование процесса синтеза высокодисперсного оксида состава La0.6Sr0.4Co0.2Fe0.8O3 – δ с помощью гликоль-цитратного метода, в том числе определено влияние концентрации окислителя и восстановителей на интенсивность окислительно-восстановительной реакции при нагревании реакционной системы, дисперсность и микроструктурные особенности получаемых продуктов. Показано, что предложенные условия синтеза позволяют получать высокодисперсные порошки (средний размер ОКР 26–30 нм) заданного состава при более низкой температуре по сравнению с указанной в литературных источниках. Установлено, что оксидные порошки после удаления остаточного углерода (600°С, 2 ч) являются однофазными и не содержат примесных включений. С применением синхронного (ТГА/ДСК) термического анализа изучены особенности протекания окислительно-восстановительной реакции формирования оксидов состава La0.6Sr0.4Co0.2Fe0.8O3 – δ в зависимости от концентрации нитрата аммония и этиленгликоля, в частности, повышение концентрации окислителя приводит к значительному росту интенсивности выделения энергии в ходе реакции, что ведет к увеличению среднего размера ОКР, тогда как увеличение содержания этиленгликоля способствует повышению количества остаточного углерода, сдерживающего рост данного параметра. Полученные с помощью комплекса взаимодополняющих методов физико-химического анализа результаты свидетельствуют о высокой перспективности предложенного метода синтеза при формировании компонентов функциональных чернил для аддитивного формирования планарных катодных материалов в качестве компонентов среднетемпературных ТОТЭ.
Список литературы
Sazali N., Salleh W.N.W., Jamaludin A.S. et al. // Membranes (Basel). 2020. V. 10. № 5.
Kuterbekov K.A., Nikonov A.V., Bekmyrza K.Z. et al. // Nanomaterials. 2022. V. 12. № 7. P. 1059. https://doi.org/10.3390/nano12071059
Ponomareva A.A., Ivanova A.G., Shilova O.A. et al. // Glass Phys. Chem. 2016. V. 42. № 1. P. 1. https://doi.org/10.1134/S1087659616010120
Zhou J., Wang Z., Han M. et al. // Int. J. Hydrogen Energy. 2022. V. 47. № 6. P. 4109. https://doi.org/10.1016/j.ijhydene.2021.11.049
Cheekatamarla P.K., Finnerty C.M., Robinson C.R. et al. // J. Power Sources. 2009. V. 193. № 2. P. 797. https://doi.org/10.1016/j.jpowsour.2009.04.060
Ding J., Liu J., Feng Y. et al. // Int. J. Hydrogen Energy. 2011. V. 36. № 13. P. 7649. https://doi.org/10.1016/j.ijhydene.2011.03.145
Minh N.Q. // J. Korean Ceram. Soc. 2010. V. 47. № 1. P. 1. https://doi.org/10.4191/KCERS.2010.47.1.001
Al-Khori K., Bicer Y., Boulfrad S. et al. // Int. J. Hydrogen Energy. 2019. V. 44. № 56. P. 29604. https://doi.org/10.1016/j.ijhydene.2019.07.016
McConnell V.P. // Fuel Cells Bull. 2007. V. 2007. № 9. P. 12. https://doi.org/10.1016/S1464-2859(07)70368-1
Sghaier S.F., Khir T., Ben Brahim A. // Int. J. Hydrogen Energy. 2018. V. 43. № 6. P. 3542. https://doi.org/10.1016/j.ijhydene.2017.08.216
Mehrpooya M., Ghorbani B., Abedi H. // Energy Convers. Manag. 2020. V. 206. P. 112467. https://doi.org/10.1016/j.enconman.2020.112467
Mahato N., Banerjee A., Gupta A. et al. // Prog. Mater. Sci. 2015. V. 72. P. 141. https://doi.org/10.1016/j.pmatsci.2015.01.001
Kalinina M.V., Morozova L.V., Egorova T.L. et al. // Glass Phys. Chem. 2016. V. 42. № 5. P. 505. https://doi.org/10.1134/S1087659616050060
Yokokawa H., Tu H., Iwanschitz B. et al. // J. Power Sources. 2008. V. 182. № 2. P. 400. https://doi.org/10.1016/j.jpowsour.2008.02.016
Jang I., Kim S., Kim C. et al. // J. Power Sources. 2018. V. 392. P. 123. https://doi.org/10.1016/j.jpowsour.2018.04.106
Lee Y.H., Ren H., Wu E.A. et al. // Nano Lett. 2020. V. 20. № 5. P. 2943. https://doi.org/10.1021/acs.nanolett.9b02344
Ni N., Cooper S.J., Williams R. et al. // ACS Appl. Mater. Interfaces. 2016. V. 8. № 27. P. 17360. https://doi.org/10.1021/acsami.6b05290
Simonenko T.L., Kalinina M.V., Simonenko N.P. et al. // Glass Phys. Chem. 2018. V. 44. № 4. P. 314. https://doi.org/10.1134/S1087659618040144
Simonenko T.L., Simonenko N.P., Gorobtsov P.Y. et al. // J. Colloid Interface Sci. 2021. V. 588. P. 209. https://doi.org/10.1016/j.jcis.2020.12.052
Zhang X., Zhang W., Zhang L. et al. // Electrochim. Acta. 2017. V. 258. P. 1096. https://doi.org/10.1016/j.electacta.2017.11.163
Simner S., Bonnett J., Canfield N. et al. // J. Power Sources. 2003. V. 113. № 1. P. 1. https://doi.org/10.1016/S0378-7753(02)00455-X
Venezia E., Viviani M., Presto S. et al. // Nanomaterials. 2019. V. 9. № 4. P. 654. https://doi.org/10.3390/nano9040654
Cheng J., Jiang Y., Zhang M. et al. // Phys. Chem. Chem. Phys. 2017. V. 19. № 16. P. 10227. https://doi.org/10.1039/C7CP00110J
Cheng J., Jiang Y., Zou L. et al. // ChemistryOpen. 2019. V. 8. № 2. P. 206. https://doi.org/10.1002/open.201800247
Tezsevin I., Van De Sanden M.C.M., Er S. // J. Phys. Chem. Lett. 2021. V. 12. № 17. P. 4160. https://doi.org/10.1021/acs.jpclett.1c00827
Monama G.R., Ramohlola K.E., Iwuoha E.I. et al. // Results Chem. 2022. V. 4. P. 100321. https://doi.org/10.1016/j.rechem.2022.100321
Yang Q., Niu X., Zhu Y. et al. // J. Hazard. Mater. 2022. V. 432. P. 128686. https://doi.org/10.1016/j.jhazmat.2022.128686
Simonenko T.L., Simonenko N.P., Simonenko E.P. et al. // Russ. J. Inorg. Chem. 2021. V. 66. № 5. P. 662. https://doi.org/10.1134/S0036023621050193
Li Z., Toshiyuki M., Auchterlonie G.J. et al. // Appl. Mater. Interfaces. 2011. P. 2772.
Develos-Bagarinao K., Ishiyama T., Kishimoto H. et al. // Nat. Commun. 2021. V. 12. № 1. P. 3979. https://doi.org/10.1038/s41467-021-24255-w
Zuo X., Chen Z., Guan C. et al. // Materials (Basel). 2020. V. 13. № 10. P. 2267. https://doi.org/10.3390/ma13102267
Wang C.C., Gholizadeh M., Hou B. et al. // RSC Adv. 2021. V. 11. № 1. P. 7. https://doi.org/10.1039/D0RA09239H
Qiu P., Yang X., Zou L. et al. // ACS Appl. Mater. Interfaces. 2020. V. 12. № 26. P. Acsami.0c01962. https://doi.org/10.1021/acsami.0c01962
Marinha D., Dessemond L., Cronin J.S. et al. // Chem. Mater. 2011. V. 23. № 24. P. 5340. https://doi.org/10.1021/cm2016998
Zhang W., Wang H., Guan K. et al. // ACS Appl. Mater. Interfaces. 2019. V. 11. № 30. P. 26830. https://doi.org/10.1021/acsami.9b06668
Muhammed Ali S.A., Anwar M., Somalu M.R. et al. // Ceram. Int. 2017. V. 43. № 5. P. 4647. https://doi.org/10.1016/j.ceramint.2016.12.136
Zhou G.-H., Fu X.-Z., Luo J.-L. et al. // Mater. Technol. 2013. V. 28. № 1–2. P. 3. https://doi.org/10.1179/1753555712Y.0000000035
Chrzan A., Karczewski J., Gazda M. et al. // J. Solid State Electrochem. 2015. V. 19. № 6. P. 1807. https://doi.org/10.1007/s10008-015-2815-x
Simonenko N.P., Sakharov K.A., Simonenko E.P. et al. // Russ. J. Inorg. Chem. 2015. V. 60. № 12. P. 1452. https://doi.org/10.1134/S0036023615120232
Simonenko T.L., Simonenko N.P., Simonenko E.P. et al. // Russ. J. Inorg. Chem. 2021. V. 66. № 4. P. 477. https://doi.org/10.1134/S0036023621040203
Thuy N.T., Minh D. Le // Adv. Mater. Sci. Eng. 2012. V. 2012. P. 1. https://doi.org/10.1155/2012/380306
Hussain M., Muneer M., Abbas G. et al. // Ceram. Int. 2020. V. 46. № 11. P. 18208. https://doi.org/10.1016/j.ceramint.2020.04.143
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии