

Журнал неорганической химии, 2023, T. 68, № 5, стр. 581-588
Новый подход к получению LuFeMgO4
М. Н. Смирнова a, *, О. Н. Кондратьева a, Г. Е. Никифорова a, А. В. Хорошилов a
a Институт общей и неорганической химии им. Н.С. Курнакова РАН
119991 Москва, Ленинский пр-т, 31, Россия
* E-mail: smirnovamn@igic.ras.ru
Поступила в редакцию 30.12.2022
После доработки 16.01.2023
Принята к публикации 17.01.2023
- EDN: SODSES
- DOI: 10.31857/S0044457X22602383
Аннотация
Предложен новый способ получения LuFeMgO4, основанный на реакции горения гелеобразного прекурсора, приготовленного из нитратов металлов и органического топлива. Исследована возможность получения этого оксида из стехиометрических композиций нитратов металлов с поливиниловым спиртом (ПВС) и глицином. Для рассматриваемых систем выполнена оценка адиабатических температур горения Tad. Продукты горения ПВС- и глицин-нитратных композиций до и после их термической обработки изучены с помощью РФА и ИК-спектроскопии. Установлено, что продукты реакции горения ПВС-нитратной композиции представляют собой рентгеноаморфный порошок, а глицин-нитратной – смесь нанокристаллических оксидов, содержащую 52.5 мас. % LuFeMgO4. По данным РФА и РЭМ, четырехчасовой отжиг этой смеси при 1300°C приводит к получению однофазного порошка LuFeMgO4 со слоистой микроструктурой и размером зерна около 1–2 мкм.
ВВЕДЕНИЕ
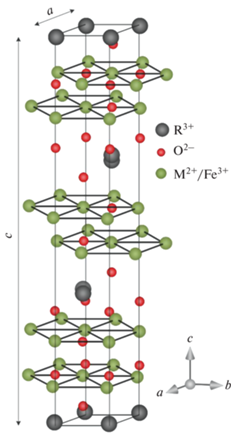
Исследование новых сложных оксидов редкоземельных металлов и железа, обладающих потенциально высокими магнитными и электрофизическими характеристиками, и поиск простых экономичных способов их получения являются неотъемлемой частью развития научно-технологического прогресса в области магнетоэлектроники и оптики. Среди таких соединений можно выделить ряд оксидов R(Fe3+M2+)O4 (R = Y, Er, Tm, Yb и Lu; M = Mg, Mn, Fe, Co, Cu и Zn), впервые синтезированных в 80-х гг. прошлого века [1]. Результаты нейтроно- и рентгенографических экспериментов [1, 2] показали, что при комнатной температуре эти соединения кристаллизуются в тригональной сингонии (пр. гр. R$\bar {3}$m) и имеют слоистую структуру, в которой двойные плоские гексагональные слои, образованные равным количеством статистически распределенных катионов M2+ и Fe3+, чередуются со слоями октаэдров RO6 (рис. 1). Авторами работ [3, 4] отмечено, что при низких температурах геометрические особенности этой структуры способствуют возникновению состояний, подобных состоянию спинового стекла.
Рис. 1.
Фрагмент кристаллической структуры оксидов R(Fe3+M2+)O4 (пр. гр. R$\bar {3}$m) при комнатной температуре.
Согласно литературным данным, в подавляющем числе работ для получения сложных оксидов R(Fe3+M2+)O4 использовали метод твердофазного взаимодействия [1, 5, 6] и зонной плавки [3, 4, 7–9]. При исследовании фазовых равновесий в системах R2O3–Fe2O3–MO (R = Y, Er, Tm, Yb и Lu; M = = Mg, Mn, Co, Cu и Zn), авторы [1] показали, что для получения однофазных порошков R(Fe3+M2+)O4 требуется длительный (до 120 ч) высокотемпературный отжиг (до 1500°C) смеси исходных простых оксидов. При более низких температурах отжига продуктами твердофазной реакции являются гексагональный ортоферрит RFeO3 и оксид MO. На примере серии R2O3–Fe2O3–MgO (R = Er, Tm, Yb и Lu) было установлено, что с ростом массы редкоземельного катиона скорость твердофазной реакции существенно возрастает. При этом было показано, что Lu-содержащие оксиды Lu(Fe3+M2+)O4, будучи наиболее стабильными в этом ряду [1], могут быть синтезированы при максимально низких для этих соединений температурах. Одним из наиболее подробно изученных оксидов в этом ряду является LuFeMgO4. Благодаря высокой диэлектрической проницаемости (ε(300 K) ⁓ 103–104) и удельному сопротивлению (ρ(300 K) ⁓ 107 Ом см) [6] этот материал считается перспективным для изготовления компонентов устройств долговременного хранения данных. С другой стороны, LuFeMgO4 относится к геометрически фрустрированным магнетикам [10], магнитные свойства которых определяются локальными магнитными взаимодействиями, возникающими вследствие хаотичного распределения катионов Mg2+ и Fe3+ в тригонально-бипирамидальных координационных полиэдрах. В этой связи очевидно, что функциональные свойства материалов на основе LuFeMgO4 будут зависеть от способа их синтеза и условий термической обработки. Примечательно, что систематическое исследование особенностей получения сложных оксидов R(Fe3+M2+)O4 методами “мягкой” химии ранее почти не проводилось и многие аспекты, включающие влияние условий синтеза на структурно-морфологические и физические свойства, а также перспективы практического применения, пока остаются слабоизученными. В связи с этим получение LuFeMgO4 в высокодисперсном состоянии открывает дополнительные возможности для модифицирования его функциональных свойств, а также для возникновения новых, ранее неизвестных.
Одним из простых и экономичных способов, позволяющих синтезировать высокодисперсные порошки сложных оксидов, является метод сжигания геля [11]. В его основе лежит реакция термического разложения гелеобразного прекурсора, приготовленного из водных растворов солей металлов (нитратов, ацетатов и др.) и органических соединений – топлива. Такой подход обеспечивает смешение исходных компонентов на молекулярном уровне, а подбор температуры и длительности термической обработки продуктов горения позволяет варьировать размер получаемых частиц и их чистоту. Можно отметить, что ранее этот метод был успешно применен для получения ряда сложных оксидов [12, 13], содержащих в том числе железо и РЗЭ [14–16].
При получении сложных оксидов методом сжигания геля важную роль играет выбор органического топлива [17]. От его природы и количества по отношению к нитратам металлов зависит режим реакции горения (адиабатическая температура горения Tад), а следовательно, состав и структура получаемых продуктов. Связано это с тем, что топливо может нести как функцию восстановителя, который, реагируя с нитратами металлов (окислителем), инициирует реакцию горения, так и выступать в качестве комплексообразующего агента. Формирование комплексных соединений с катионами металлов препятствует их раздельной кристаллизации в виде оксидов при испарении растворителя [18].
В данной работе исследуется возможность получения LuFeMgO4 методом сжигания геля с использованием глицина и ПВС в качестве топлива. Несмотря на разную химическую природу, оба этих соединения наиболее часто используют в качестве топлива из-за их способности к комплексообразованию с большим количеством катионов металлов [18, 19], а также умеренно высокой экзотермичности реакций с их участием [20, 21]. Методами РФА и ИК-спектроскопии было установлено, что продукты горения, полученные в результате термического разложения ПВС- и глицин-нитратных композиций, имеют принципиально разный фазовый состав, а формирование фазы LuFeMgO4 возможно при дополнительном отжиге при температурах выше 1300°С.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Водные растворы нитратов Mg(NO3)2, Fe(NO3)3 и Lu(NO3)3 готовили путем растворения магния (Мг-90, ω(Mg) = 99.90%), железа (ос. ч. 6–2, ω(Fe) = 99.96%) и лютеция (ЛюМ-1, ω(Lu) = = 99.99%) в разбавленной дистиллированной водой (V(HNO3)/V(H2O) = 1 : 3) азотной кислоте (ос. ч. 18–4, ω(HNO3) = 70%). Свежеприготовленные растворы солей смешивали в мольном соотношении Mg : Fe : Lu = 1 : 1 : 1. Полученный раствор делили на две части, к первой добавляли глицин (х. ч., ω(NH2CH2COOH) = 99.8%), а ко второй – поливиниловый спирт ((–СH2CH2OH)n, высший сорт, марка 20/1). В каждом случае соотношение нитратов металлов к топливу соответствовало стехиометрическому. Обе композиции постепенно нагревали до ⁓90°C и продолжали выпаривать при постоянном перемешивании до вязкого гелеобразного состояния. Реакция термического разложения композиции с добавлением глицина (глицин-нитратная композиция (ГНК)) сопровождалась пламенным горением, а с ПВС (ПВС-нитратная композиция (ПНК)) протекала в режиме тления. В обоих случаях был получен объемный темно-коричневый порошок.
Для определения температуры, достаточной для получения однофазного порошка LuFeMgO4, продукты горения обеих композиций отжигали на воздухе при различных температурах (от 500 до 1300°C). Время отжига при каждой температуре составляло 4 ч. Фазовый состав полученных порошков изучали методом РФА на дифрактометре Bruker D8 Advance (CuKα-излучение, λ = 1.5418 Å), оснащенном линейным детектором LynxEye и никелевым фильтром. Рентгенограммы регистрировали при комнатной температуре в диапазоне углов 2θ от 10° до 60°. Для обработки и интерпретации результатов РФА использовали программное обеспечение Bruker Topas v. 4.2 и электронную базу данных ICDD PDF-2. ИК-спектральное исследование образцов выполняли на инфракрасном Фурье-спектрометре Perkin Elmer Spectrum 65. Съемку спектров проводили в интервале 400–4000 см–1. Содержание остаточного углерода в порошке LuFeMgO4 определяли с помощью CHNS-элементного анализатора EA 3000 Eurovector. Особенности морфологии и размерные характеристики порошка LuFeMgO4 изучали методом растровой электронной микроскопии (РЭМ) на двухлучевом сканирующем электронно-ионном комплексе FIB-SEM TESCAN AMBER X. Тепловое поведение продуктов горения, подвергнутых промежуточной термообработке, изучали методом дифференциальной сканирующей калориметрии (ДСК) на установке Netzsch DSC 404 F1 Pegasus. Измерения проводили в Pt–Rh-тиглях в атмосфере сухого аргона марки 5.5 (99.9995 об. % Ar) в диапазоне температур от 40 до 1450°C.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Синтез LuFeMgO4 путем твердофазного взаимодействия между оксидами металлов требует длительного (48 ч) отжига при температуре 1440°C [1]. Снизить длительность высокотемпературного отжига можно за счет применения реакций горения композиций, приготовленных из нитратов металлов и органического топлива [22]. Высокая экзотермичность таких процессов обеспечивается за счет стехиометрического соотношения окислителя к восстановителю [22], а небольшое количество органических примесей, содержащееся в порошке-прекурсоре после реакции горения, полностью удаляется путем его дополнительной термической обработки [12, 13].
Гипотетические реакции взаимодействия нитратов металлов с глицином и ПВС (при условии полного превращения исходных реагентов в конечные продукты) выглядят следующим образом:
(1)
$\begin{gathered} {\text{Mg}}{{\left( {{\text{N}}{{{\text{O}}}_{{\text{3}}}}} \right)}_{{{\text{2(p - p)}}}}} + {\text{Fe}}{{\left( {{\text{N}}{{{\text{O}}}_{{\text{3}}}}} \right)}_{{{\text{3(p - p)}}}}}{\text{ + Lu}}{{\left( {{\text{N}}{{{\text{O}}}_{{\text{3}}}}} \right)}_{{{\text{3(p - p)}}}}}{\text{ + }} \\ {\text{ + }}\,\,{\text{4/}}n{{{\text{(C}}{{{\text{H}}}_{{\text{2}}}}{\text{CHOH)}}}_{{n({\text{тв}})}}} = {\text{LuFeMg}}{{{\text{O}}}_{{{\text{4}}({\text{тв}})}}} + \\ + \,\,{\text{8C}}{{{\text{O}}}_{{{\text{2(г)}}}}} + {\text{4 }}{{{\text{N}}}_{{{\text{2(г)}}}}} + {\text{8 }}{{{\text{H}}}_{{\text{2}}}}{{{\text{O}}}_{{{\text{(г)}}}}}{\text{,}} \\ \end{gathered} $(2)
$\begin{gathered} {\text{Mg}}{{\left( {{\text{N}}{{{\text{O}}}_{{\text{3}}}}} \right)}_{{{\text{2(р - р)}}}}} + {\text{Fe}}{{\left( {{\text{N}}{{{\text{O}}}_{{\text{3}}}}} \right)}_{{{\text{3(р - р)}}}}} + {\text{Lu}}{{\left( {{\text{N}}{{{\text{O}}}_{{\text{3}}}}} \right)}_{{{\text{3(р - р)}}}}}{\text{ + }} \\ {\text{ + }}\,\,{\text{4}}{\text{.44N}}{{{\text{H}}}_{{\text{2}}}}{\text{C}}{{{\text{Н}}}_{{\text{2}}}}{\text{СОО}}{{{\text{Н}}}_{{{\text{(тв)}}}}} = {\text{LuFeMg}}{{{\text{O}}}_{{{\text{4(тв)}}}}} + \\ {\text{ + }}\,\,{\text{8}}{\text{.88C}}{{{\text{O}}}_{{{\text{2(г)}}}}} + {\text{6}}{\text{.22}}{{{\text{N}}}_{{{\text{2(г)}}}}} + {\text{11}}{\text{.1}}{{{\text{H}}}_{{\text{2}}}}{{{\text{O}}}_{{{\text{(г)}}}}} + {\text{0}}{\text{.01 }}{{{\text{О}}}_{{{\text{2(г)}}}}}{\text{.}} \\ \end{gathered} $Максимальную температуру (Taд), до которой могут нагреться рассматриваемые реакционные системы (при условии протекания процесса в адиабатических условиях), можно оценить из уравнения:
(3)
$Q = - {{\Delta }_{r}}H_{m}^{^\circ } = \int\limits_{298.15\,\,{\text{K}}}^{{{T}_{{ad}}}} {{{n}_{j}}{{C}_{{p,j}}}{\text{d}}T,} $(4)
${{\Delta }_{r}}H_{m}^{^\circ } = \sum\limits_j {{{n}_{j}}{{\Delta }_{f}}H_{{m,j}}^{^\circ }} - \sum\limits_i {{{n}_{j}}{{\Delta }_{f}}H_{{m,i}}^{^\circ }} ,$Термодинамические характеристики продуктов реакции горения и исходных реагентов [23–27], необходимые для оценки Taд, приведены в табл. 1. Данные о величинах теплоемкости $C_{{p,m}}^{^\circ }$ (298.15 K) и стандартной энтальпии образования ${{\Delta }_{f}}H_{m}^{^\circ }$ (298.15 K) LuFeMgO4 в литературных источниках не найдены. Поэтому их приближенная оценка при T = 298.15 K была выполнена по правилу аддитивности с использованием данных для оксидов MgO, Fe2O3 и Lu2O3 из [27]. Значения ${{\Delta }_{r}}H_{m}^{^\circ }$ (298.15 K) для реакций горения ПНК и ГНК, рассчитанные по уравнениям (1) и (2), составляют ‒2129.5 и –2941.4 кДж/моль соответственно. Очевидно, что применение глицина увеличивает экзотермичность протекающего процесса приблизительно на одну треть. Эти данные коррелируют с экспериментальными наблюдениями, согласно которым интенсивность реакции горения ПНК (тление) заметно ниже, чем ГНК (пламенное горение). Как следует из результатов термодинамических расчетов, максимальные температуры Tад, теоретически достижимые в реакциях горения ПНК и ГНК, равны 1938 и 2110°C соответственно. Очевидно, что в обоих случаях они заметно выше по сравнению с температурой, необходимой для получения LuFeMgO4 твердофазным способом.
Таблица 1.
Термодинамические данные для оценки Tад и ${{\Delta }_{r}}H_{m}^{^\circ }$ для реакций (1) и (2)
| Соединение | Стандартная энтальпия образования ${{\Delta }_{f}}H_{m}^{^\circ }$(298.15 K), кДж/моль |
Теплоемкость $C_{{p,m}}^{^\circ }$, Дж/(K моль) |
Литература |
|---|---|---|---|
| Mg(NO3)2(р-р) | –882.87 | [23] | |
| Fe(NO3)3(р-р) | –668.56 | [23] | |
| Lu(NO3)3(р-р) | –1291.6 | [23] | |
| NH2CH2COOH(тв) | –528.5 | [24] | |
| (–СH2CH2OH)n(тв) | –515.64 | [25] | |
| LuFeMgO4(тв) | –1952.5a | 139.9b | Наст. работа |
| CO2(г) | –393.5 | 43 + 0.011T | [26] |
| H2O(г) | –241.8 | 30 + 0.015T | [26] |
| N2(г) | 0 | 27 + 0.004T | [26] |
| O2(г) | 0 | 25 + 0.015T | [26] |
a,bХарактеристики рассчитаны по правилу аддитивности с использованием величин $C_{{p,m}}^{^\circ }$ (298.15 K) и ${{\Delta }_{f}}H_{m}^{^\circ }$ (298.15 K) для MgO, Fe2O3 и Lu2O3 из [27].
Рассмотрим результаты рентгенодифракционных (рис. 2) и ИК-спектральных (рис. 3 и 4) исследований продуктов горения ПНК и ГНК, отожженных при 500, 700, 1000 и 1300°C. По данным РФА, продукты горения ПНК (рис. 2а, спектр 1) находятся в рентгеноаморфном состоянии. При этом их ИК-спектр (рис. 3, спектр 2) содержит широкие полосы поглощения, которые относятся к колебаниям –ОН $(\upsilon {{({\text{OH}})}_{{{{{\text{H}}}_{2}}{\text{O}}}}},$ $\upsilon {{({\text{OH}})}_{{{\text{COOH}}}}}$ ⁓3700–3100 см–1), ${\text{HCO}}_{3}^{ - }$ ($\upsilon {{({\text{OH}})}_{{{\text{COOH}}}}}$ 1634; δ(OCO)асим 1330 см–1) (асимметричные колебания C=O-групп) и групп ${\text{NO}}_{3}^{ - }$ (υ(NO) 1395; δ(ONO) 846 см–1) [28, 29]. Наличие таких полос характерно для колебаний связей в молекулах органических соединений, адсорбированных на поверхности образовавшегося порошка. Как видно из рис. 2а (спектр 3), полученный порошок остается рентгеноаморфным даже после четырехчасовой термообработки при 700°C, а в его ИК-спектре (рис. 3, спектр 4) сохраняются слабые полосы поглощения $(\upsilon {{({\text{COH}})}_{{{\text{C}}{{{\text{O}}}_{3}}}}}$ в области 1300–1500 см–1, характерные для карбонатных групп [29]. ДСК-кривая для этого порошка приведена на рис. 5. Ее анализ показал, что в интервале температур 40–700°C наблюдается серия слабых эндотермических эффектов с максимумами при 123, 358 и 583°C. Их наличие связано с термоокислительной деструкцией и удалением органического остатка из порошка.
Рис. 2.
Рентгенограммы продуктов термического разложения ПНК (а) и ГНК (б) до (спектр 1) и после их отжига (спектры 2–5) на воздухе при разных температурах.
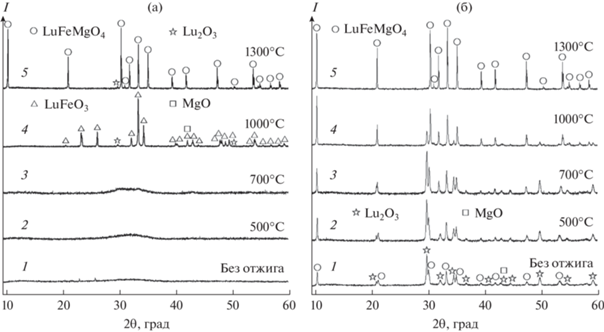
Рис. 3.
ИК-спектры ПВС (1) и продуктов термического разложения ПНК (2–6), отожженных на воздухе при разных температурах.
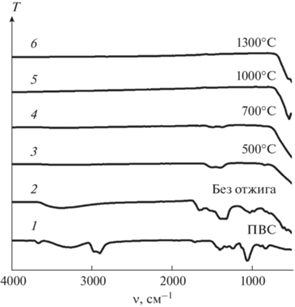
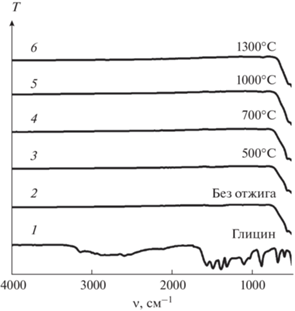
Рис. 4.
ИК-спектры глицина (1) и продуктов термического разложения ГНК (2–6), отожженных на воздухе при разных температурах.
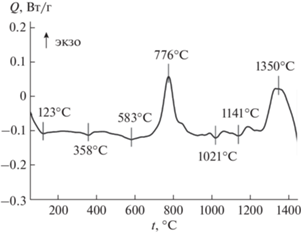
Экзотермический эффект, наблюдаемый при 726–816°C, связан, по-видимому, с образованием новой фазы. Это подтверждают результаты РФА продуктов горения ПНК, термообработанных при 1000°C (рис. 2а, спектр 4). На рентгенограмме появляются интенсивные рефлексы, относящиеся к ортоферриту лютеция LuFeO3 со структурой перовскита, а также наблюдается небольшое количество оксидов магния и лютеция (табл. 2). С увеличением температуры отжига до 1000°C полосы поглощения в высокочастотной области ИК-спектров (рис. 3, спектры 3–5) полностью исчезают, что свидетельствует о полном удалении углеродсодержащих остатков из этого порошка.
Таблица 2.
Фазовый состав продуктов реакции горения ПНК и ГНК, отожженных при разных температурах
| Температура отжига, °C | Содержание фаз в порошке, мас. % | |||
|---|---|---|---|---|
| LuFeMgO4 | LuFeO3 | Lu2O3 | MgO | |
| ПНК | ||||
| – | ||||
| 500 | ||||
| 700 | ||||
| 1000 | – | 84.0(7) | 1.9(2) | 14.1(7) |
| 1300 | 98.8(1) | – | 1.2(1) | – |
| ГНК | ||||
| – | 52.5(8) | 5.6(5) | 28.8(5) | 13.1(1) |
| 500 | 53.6(7) | 6.0(5) | 29.9(5) | 10.5(1) |
| 700 | 52.6(7) | 6.5(5) | 30.5(4) | 10.4(9) |
| 1000 | 72.4(1) | 9.2(5) | 9.3(2) | 9.1(1) |
| 1300 | 100.0 | – | – | – |
Наличие небольших эндоэффектов на кривой ДСК в интервалах 995–1048°C и 1088–1177°C (рис. 5) связано, по-видимому, с последовательным протеканием твердофазных реакций между оксидами LuFeO3, Lu2O3 и MgO. Образование LuFeMgO4 происходит при температурах 1270–1450°C и сопровождается экзотермическим эффектом с максимумом при ⁓1350°C. Появление этой фазы после термообработки продуктов горения ПНК при 1300°C подтверждается результатами РФА (табл. 2). Однако наряду с основной фазой в порошке присутствует небольшая примесь Lu2O3. Очевидно, применяющиеся условия отжига оказались недостаточными для получения однофазного LuFeMgO4.
Примечательно, что в случае ГНК продукты реакции горения уже на 52.5 мас. % состояли из LuFeMgO4. В качестве вторичных фаз в полученном порошке были обнаружены оксиды LuFeO3, Lu2O3 и MgO (табл. 2). При этом в ИК-спектре этой смеси фаз (рис. 4, спектр 2) отсутствовали какие-либо полосы, относящиеся к колебаниям связей органических соединений. Следовательно, реально достигнутой в системе температуры оказалось достаточно не только для полного выгорания органических остатков, но и для частичного образования наноразмерного оксида LuFeMgO4. Как видно из данных количественного РФА (табл. 2), термообработка полученного порошка при 500 и 700°C не влияет на фазовый состав смеси. Заметное увеличение содержания LuFeMgO4 наблюдается лишь после отжига при 1000°C, при этом суммарное количество вторичных фаз постепенно уменьшается в результате твердофазного взаимодействия между ними. Четырехчасовой отжиг порошка, полученного в результате горения ГНК, при 1300°C приводит к получению однофазного оксида LuFeMgO4 (пр. гр. R$\bar {3}$m, Z = 3) с параметрами элементарной ячейки a = 3.41313(4) Å, c = 25.2324(4) Å, V = 254.562(7) Å3. По данным CHNS-анализа, содержание остаточного углерода в этом порошке составляет 0.02 мас. %, что согласуется с данными ИК-спектроскопии (рис. 4, спектр 6). На рис. 6 показана характерная микроструктура однофазного порошка LuFeMgO4. Видно, что размер зерен варьируется в пределах 1–2 мкм, и на них отчетливо прослеживаются грани роста. Примечательно, что похожие морфологические особенности были обнаружены и у родственных слоистых оксидов InGaO3(ZnO)m (m = 1, …, n) [12, 30].

Сравнительный анализ данных РФА и ИК-спектроскопии для исследуемых ПНК- и ГНК-композиций показал, что фазовый состав и структура продуктов реакции зависят как от температуры горения, так и от природы используемого топлива. Как следует из данных РФА (табл. 2), термическое разложение обеих композиций не привело к образованию беспримесной кристаллической фазы LuFeMgO4. Это указывает на то, что реальные температуры горения Tад были ниже расчетных, и объясняется тем, что в протекающих процессах не удается избежать частичного рассеяния тепла в окружающую среду. В случае ПВС столь существенную разницу в значениях Tад можно объяснить тем, что реакция горения ПНК протекала в режиме тления, для которого характерны невысокие температуры Tад (<1000°C) [22]. Поскольку продуктом этой реакции является углеродсодержащий рентгеноаморфный порошок, а кристаллизация низкотемпературной фазы LuFeO3, как свидетельствуют данные РФА и ДСК, происходит при 726–816°C, можно предположить, что эта температура была ниже ⁓700°C. С другой стороны, термическое разложение ГНК, в результате которого частично образовался LuFeMgO4, протекало в режиме пламенного горения. В таком режиме температура достигает 1500°C [22], что заметно ниже Tад, рассчитанной для ГНК, но сопоставимо с температурой твердофазного синтеза LuFeMgO4. В совокупности с малой продолжительностью горения (⁓1 мин) и возникающим в реакционной зоне градиентом температур это не позволяет получить полностью однофазный порошок LuFeMgO4 при выбранных условиях, однако его содержание в полученной смеси оксидов достигает 52.5 мас. % (табл. 2).
Несмотря на то, что использование глицина и ПВС приводит к реализации различных режимов горения, оба этих топлива имеют склонность к образованию комплексных соединений с катионами металлов [18, 19, 31]. Анализируя состав продуктов горения (табл. 2), можно предположить, что в случае ГНК образование таких соединений произошло лишь частично. По-видимому, их совместное термическое разложение с нитратами металлов привело к раздельной кристаллизации в виде смеси LuFeMgO4, LuFeO3, Lu2O3 и MgO. В случае ПНК рентгеноаморфный продукт, содержащий органический остаток, мог образоваться из-за неполной термической деструкции полимерной сетки, образованной ПВС и распределенными в нем катионами металлов [18]. В этом случае отщепление углеродсодержащих фрагментов сетки прекращается при термообработке продуктов реакции только при температурах выше 700°C, а образование LuFeMgO4 (⁓1300°C) происходит через образование низкотемпературной фазы LuFeO3 (⁓800°C).
ЗАКЛЮЧЕНИЕ
Следует отметить, что оба топлива можно использовать в составе композиций для синтеза LuFeMgO4, однако в случае ПВС могут потребоваться более высокие температуры (>1300°C) и/или длительность термообработки. По сравнению с твердофазным способом [1] подход, основанный на методе сжигания геля, позволяет не только сократить длительность термообработки с 48 до 4 ч, но и снизить ее температуру с 1440 до 1300°C. Одним из направлений его дальнейшего совершенствования может стать поиск путей увеличения температуры реакции горения. Добиться этого можно при использовании композиций, обогащенных глицином, или за счет добавления сильных окислителей типа нитрата аммония.
Список литературы
Kimizuka N., Takayama E. // J. Solid State Chem. 1981. V. 40. P. 109. https://doi.org/10.1016/0022-4596(81)90368-6
Wiedenmann A., Gunsser W., Rossat-Mignod J. et al. // J. Magn. Magn. Mater. 1983. V. 31–34. P. 1442. https://doi.org/10.1016/0304-8853(83)90962-9
Ikeda N., Kohn K., Himoto E. et al. // J. Phys. Soc. Jpn. 1995. V. 64. P. 4371. https://doi.org/10.1143/JPSJ.64.4371
Todate Y., Kikuta C., Himoto E. et al. // J. Phys.: Condens. Matter. 1998. V. 10. P. 4057. https://doi.org/10.1088/0953-8984/10/18/015
Tanaka M., Siratori K., Kimizuka N. et al. // J. Phys. Soc. Jpn. 1984. V. 53. P. 4113. https://doi.org/10.1143/JPSJ.53.4113
Qin Y., Wang Z., Chen X.M. et al. // J. Appl. Phys. 2010. V. 108. P. 084111. https://doi.org/10.1063/1.3500309
Tanaka M., Himoto E., Todate Y. et al. // J. Phys. Soc. Jpn. 1995. V. 64. P. 2621. https://doi.org/10.1143/JPSJ.64.2621
Todate Y., Ohnishi N., Tanaka M. et al. // Hyperfine Interact. 1997. V. 104 P. 375. https://doi.org/10.1023/A:1012689507474
Iida J., Takekawa Sh., Kimizuka N. et al. // J. Cryst. Growth. 1990. V. 102. P. 398. https://doi.org/10.1016/0022-0248(90)90397-4
Todate Y., Himoto E., Kikuta C. et al. // Phys. Rev. B. 1998. V. 57. P. 485. https://doi.org/10.1103/PhysRevB.57.485
Lackner M. Combustion Synthesis: Novel Routes to Novel Materials. Bentham Science Publishers Ltd., 2010.
Kondrat'eva O.N., Smirnova M.N., Nikiforova G.E. et al. // J. Eur. Ceram. Soc. 2021. V. 41. P. 6559. https://doi.org/10.1016/j.jeurceramsoc.2021.05.063
Kondrat'eva O.N., Smirnova M.N., Nikiforova G.E. et al. // Ceram. Int. 2023. V. 49. P. 179. https://doi.org/10.1016/j.ceramint.2022.08.326
Kondrat'eva O.N., Nikiforova G.E., Shevchenko E.V. et al. // Ceram. Int. 2020. V. 46. № 4. P. 11390. https://doi.org/10.1016/j.ceramint.2020.01.169
Smirnova M.N., Glazkova I.S., Nikiforova G.E. et al. // Nanosyst.: Phys. Chem. Math. 2021. V. 12. P. 210. https://doi.org/10.17586/2220-8054-2021-12-2-210-217
Popkov V.I., Martinson K.D., Kondrashkova I.S. et al. // J. Alloys Compd. 2022. V. 859. Article 157812. https://doi.org/10.1016/j.jallcom.2020.157812
Carlos E., Martins R., Fortunato E. et al. // Chem. Eur. J. 2020. V. 26. P. 9099. https://doi.org/10.1002/chem.202000678
Khaliullin Sh.M., Zhuravlev V.D., Bamburov V.G. et al. // J. Sol-Gel Sci. Technol. 2020. V. 93. P. 251. https://doi.org/10.1007/s10971-019-05189-8
Chick L.A., Pederson L.R., Maupin G.D. et al. // Mater. Lett. 1990. V. 10. P. 6. https://doi.org/10.1016/0167-577X(90)90003-5
Smirnova M.N., Kop’eva M.A., Nipan G.D. et al. // Russ. J. Inorg. Chem. 2022. V. 67. P. 978. https://doi.org/10.1134/S0036023622070221
Zhuravlev V.D., Dmitriev A.V., Vladimirova E.V. et al. // Russ. J. Inorg. Chem. 2021. V. 66 P. 1895. https://doi.org/10.1134/S0036023621120226
Patil K.C., Hedge M.S., Rattan T., Aruna S.T. Chemistry of Nanocrystalline Oxide Materials: Combustion Synthesis, Properties and Applications, 1st ed., Singapore: World Scientific Publishing Co. Pte. Ltd., 2008.
http://www.chem.msu.ru/cgi-bin/tkv.pl?show=welcome.html
Dorofeeva O.V., Ryzhova O.N. // J. Chem. Thermodynamics. 2009. V. 41. P. 433. https://doi.org/10.1016/j.jct.2008.12.001
He Z., Xia Z., Hu J. et al. // J. Polym. Res. 2019. V. 26. Article 219. https://doi.org/10.1007/s10965-019-1894-2
Varma A., Mukasyan A.S., Rogachev A.S. et al. // Chem. Rev. 2016. V. 116. P. 14493. https://doi.org/10.1021/acs.chemrev.6b00279
Barin I. Thermochemical Data of Pure Substances, third ed., VCH Verlagsgesellschaft mbH, 1995.
Larkin P. Infrared and Raman Spectroscopy: Principles and Spectral Interpretation. Amsterdam: Elsevier, 2011.
Chukanov N.V., Chervonnyi A.D. Infrared Spectroscopy of Minerals and Related Compounds. Switzerland: Springer International Publishing, 2016.
Кондратьева О.Н., Смирнова М.Н., Никифорова Г.Е. // XI Конференция молодых ученых по общей и неорганической химии: Тез. докл. конф. М., 2021. 312 с.
Lisnevskaya I.V., Bobrova I.A., Lupeiko T.G. // J. Magn. Magn. Mater. 2016. V. 397. P. 86. https://doi.org/10.1016/j.jmmm.2015.08.084
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии