

Журнал неорганической химии, 2023, T. 68, № 7, стр. 885-895
Синтез и электронная структура биметаллических AuFe нанокомпозитов
А. Ю. Васильков a, А. А. Воронова a, *, А. В. Наумкин a, И. Е. Бутенко a, Я. В. Зубавичус b
a Институт элементоорганических соединений им. А.Н. Несмеянова РАН
119334 Москва, ул. Вавилова, 28, Россия
b ЦКП “СКИФ”, Институт катализа им. Г.К. Борескова СО РАН
630559 Кольцово, Россия
* E-mail: voronova.anastasiia.a@mail.ru
Поступила в редакцию 27.01.2023
После доработки 02.03.2023
Принята к публикации 02.03.2023
- EDN: RIJQUX
- DOI: 10.31857/S0044457X23600147
Аннотация
Наночастицы Au, Fe и AuFe получены методом металло-парового синтеза с использованием ацетона в качестве дисперсионной среды. Состав и электронная структура частиц исследованы методами ПЭМ, СЭМ, РФЭС, XANES и EXAFS. Получены частицы Au и Fe со средним диаметром 5.3 и 1.8 нм соответственно. Методы рентгеновской диагностики показали, что золото в основном находится в состоянии Au0, а состояния Au+ и Au3+ присутствуют в небольших количествах, железо представляет собой смесь нестехиометрических оксидов с состояниями, близкими к Fe2+ и Fe3+. Биметаллические наночастицы являются твердым раствором с неупорядоченной структурой и связями Au–Fe–O и Au–O–Fe. Зарегистрировано наличие углеродсодержащей оболочки на всех типах металлических частиц. Полученные материалы могут быть перспективны для создания улучшенных противомикробных средств и новых методов лечения онкологических заболеваний.
ВВЕДЕНИЕ
Онкология является серьезной проблемой здравоохранения и причиной смертности и заболеваемости во всем мире [1, 2]. Химиотерапия, один из основных способов лечения, предусматривает ряд ограничений, связанных с тем, что неспецифичность препаратов приводит к отрицательным клиническим результатам.
Химиотерапия в первую очередь поражает быстро делящиеся клетки, в том числе и здоровые, так как химиотерапевтические средства токсичны и для них, включая клетки костного мозга, пищеварительного тракта, макрофаги и волосяные фолликулы [3, 4].
Металлические наночастицы (НЧ) могут обладать потенциалом для решения проблем, связанных с обычной химиотерапией. Известно, что металлические НЧ весьма эффективны в терапии рака, обеспечивая специфическую избирательность в воздействии на клетки и доставку лекарств [5–7].
Использование наночастиц оксида железа (FeOx НЧ) имеет ряд преимуществ: селективное поглощение НЧ раковыми клетками посредством неспецифического эндоцитоза, возможность модифицирования поверхности НЧ лекарственными препаратами и генерации тепла под воздействием переменного магнитного поля (магнитная гипертермия) [8].
Уникальные оптические свойства наночастиц золота (Au НЧ) позволяют использовать их в биологических и фармацевтических областях. Они могут применяться в терапевтических методах, основанных на визуализации, и для сверхчувствительного обнаружения раковых клеток [9–11].
Дальнейшее развитие этого направления связано с исследованиями гетерометаллических наночастиц, при использовании которых в качестве биологически активных систем наблюдается синергетический эффект [12, 13]. Биметаллические железосодержащие наносистемы являются одними из наиболее исследуемых объектов, так как существует возможность управления их свойствами путем модифицирования Fe наночастицами другого металла [14–16]. Системы, содержащие наночастицы Au и Fe, эффективны в катализе [17–20], мультимодальной магнитно-резонансной томографии/компьютерной томографии (МРТ/КТ) [21–23], радиосенсибилизации под контролем МРТ [24], усиленной гипертермии [25, 26] и других применениях [15, 27–29]. Показано, что введение золота в FeOx НЧ не снижает намагниченность системы [30], что позволяет использовать внешнее магнитное воздействие для системы Au–Fe в целом [31]. Структура и электронное состояние наночастиц AuFe существенно меняется в зависимости от способа их получения [32–34], что может приводить к синергизму [35]. Это стимулирует разработку новых методов синтеза систем Au–Fe и всестороннее исследование физико-химических свойств этих материалов.
В известных в настоящее время способах получения металлических наночастиц в матрицах различной природы, как правило, используется химическое восстановление солей металлов [36–40]. В большинстве случаев эти методы имеют ряд существенных ограничений, усложняющих биомедицинское применение полученных материалов: наличие значительного количества примесей поверхностно-активных веществ и остатков продуктов синтеза и сложность контроля полноты восстановления металла.
Свою эффективность в данной области показал экологически допустимый метод металло-парового синтеза (МПС) [41–44]. Метод позволяет получать моно- и биметаллические наночастицы металлов, что существенно расширяет круг доступных биологически активных гетерометаллических систем. При использовании МПС удается избежать или значительно снизить загрязнения, вносимые в материалы биомедицинского назначения в процессе их синтеза.
Первые исследования биологической активности, проведенные с наночастицами золота и железа, полученными методом МПС, показали, что наночастицы Au проявляют противоопухолевый потенциал [45], воздействуя на клетки цервикальной и колоректальной аденокарциномы человека. Наночастицы Fe проявляли цитотоксическую активность в отношении линии раковых клеток А549 [46].
Целью настоящей работы является синтез монометаллических наночастиц золота, железа и биметаллических наночастиц золота–железа методом МПС как потенциальных противораковых препаратов, а также исследование структуры полученных материалов и электронного состояния металла в нанокомпозитах.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Наночастицы Au, Fe и AuFe получали методом МПС [47, 48]. В типовом эксперименте испаряли 0.2–0.4 г металла и 120–140 мл ацетона. Совместную конденсацию паров металла и органического лиганда проводили в статическом кварцевом пятилитровом реакторе. Пары металлов генерировали в вакууме 10–2 Па путем резистивного нагрева молибденовой лодочки для испарения Au и вольфрамового прутка для Fe и AuFe. Затем проводили совместную конденсацию паров металла с ацетоном на охлаждаемых жидким азотом стенках реактора (схема 1 ). Продолжительность синтеза 40–60 мин. После его окончания охлаждение снимали, а матрицу соконденсата нагревали до комнатной температуры. Органозоль металлов in situ сифонировали из реактора в вакуумированную колбу, из которой далее отгоняли ацетон, а полученные черни металлов исследовали.
Наночастицы металлов анализировали методом просвечивающей электронной микроскопии (ПЭМ) с помощью прибора LEO 912AB OMEGA (Zeiss, Германия) при ускоряющем напряжении 100 кВ. Для анализа образцы предварительно измельчали в агатовой ступке, суспендировали в этаноле и диспергировали ультразвуком в течение 10–15 мин. Далее небольшое количество помещали на формвар/углеродную сетку. Распределение частиц образцов по размерам было рассчитано путем измерения размера 200 отображаемых частиц с помощью программного обеспечения SigmaScan Pro.
Морфологию поверхности нанокомпозитов изучали методом сканирующей электронной микроскопии (СЭМ) на микроскопе Hitachi TM4000Plus (Япония) при ускоряющем напряжении 15 кВ в режиме вторичных электронов. Энергодисперсионные рентгеновские исследования (EDX) проводили с использованием спектрометра Quantax 75 (Bruker, Германия).
Термогравиметрический анализ был проведен на приборе Derivatograph-С (МОМ, Венгрия) на образцах массой ~15 мг при скорости нагревания 5 град/мин в атмосфере аргона.
Рентгеновские фотоэлектронные спектры регистрировали на спектрометре Axis Ultra DLD (Kratos) с использованием монохроматического AlKα-излучения при мощности рентгеновской пушки 150 Вт. Обзорные спектры и спектры высокого разрешения регистрировали при энергии пропускания 160 и 40 эВ соответственно. Обзорный спектр записывали с шагом 1 эВ, спектры высокого разрешения – с шагом 0.1 эВ. Размер анализируемой области составлял около 300 × × 700 мкм2. Образцы закрепляли на держателе с помощью двухсторонней адгезионной ленты и исследовали при комнатной температуре при остаточном давлении в камере спектрометра ≤10–8 Торр. Для устранения эффекта зарядки образцов съемку спектров проводили с использованием нейтрализатора. Калибровку спектров образца AuAc осуществляли по состоянию С–С/C–Н, выделенному в спектре C1s, которому была приписана энергия 285.0 эВ, а спектров образцов FeAc и AuFeAc – по низкоэнергетическому пику в спектрах O1s, которому была приписана энергия связи 530.0 эВ. Вычитание фона неупругих потерь энергии электронов проводили по методу Ширли.
Порошковые дифрактограммы были измерены на автоматизированном дифрактометре ДРОН-3 в геометрии Брегга–Брентано, параметры съемки: CuKα-излучение (λ = 1.5418 Å), графитовый монохроматор на вторичном пучке, режим генератора 36 кВ × 20 мА, непрерывное θ/2θ-сканирование, шаг по углу 0.05°, скорость сканирования 1 град/мин. Теоретические дифрактограммы для нанокристаллических моделей были рассчитаны по формуле Дебая с использованием процедуры, описанной в [49].
Измерения рентгеноабсорбционных спектров на K-крае Fe и L3-крае Au для синтезированных наноматериалов проводили на экспериментальной станции “Структурное материаловедение” Курчатовского источника синхротронного излучения [50]. Энергию волны падающего на образец рентгеновского излучения выбирали с помощью моноблочного прорезного монохроматора Si (111). Спектры XANES и EXAFS измеряли в режиме пропускания с использованием двух ионизационных камер, расположенных до и после образца. В области XANES шаг сканирования составлял 0.3–0.5 эВ. В области EXAFS шаг по энергии был переменным, но постоянным по значениям волновых чисел фотоэлектрона Δk = 0.05 Å–1. Время съемки спектра составляло 30–40 мин.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Свойства материалов на основе наночастиц металлов определяются условиями получения нанокомпозита, составом и размером частиц, а также природой их специфического взаимодействия со стабилизирующим носителем. В отличие от Au, наночастицы Fe, получаемые МПС, экстремально активны и легко окисляются при контакте с воздухом или следами влаги и кислорода в органическом реагенте. Поэтому все эксперименты по получению биметаллических наноматериалов AuFe были выполнены in situ в вакууме или инертной атмосфере. Варьируя природу органического реагента можно менять как размер частиц, так и электронное состояние металла и, как следствие, биологическую активность системы в целом.
На рис. 1 представлены микрофотографии ПЭМ системы Fe–ацетон и гистограмма распределения частиц железа (FeAc) по размерам. Были зарегистрированы частицы с диаметром в диапазоне от 1 до 2.8 нм. Частицы с диаметром 1.8 нм составляют 25% от общего количества. Наночастицы FeAc имеют размытый профиль, что может свидетельствовать о рыхлой поверхности наночастиц, обусловленной, по-видимому, окислением.
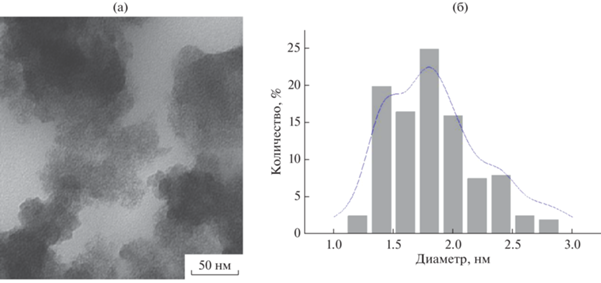
Анализ микрофотографий показал, что частицы золота имеют сферическую форму и диаметр от 2.3 до 24 нм. 25% наночастиц AuAc имеют диаметр 5.3 нм. На рис. 2 представлены микрофотографии ПЭМ системы Au–ацетон и гистограмма распределения частиц золота (AuAc) по размерам.
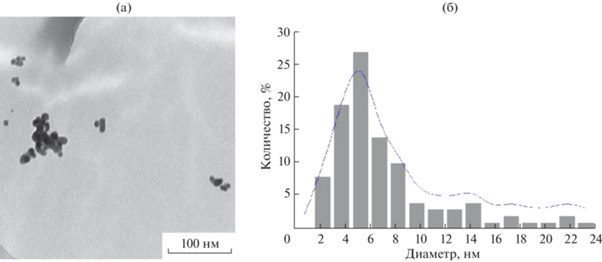
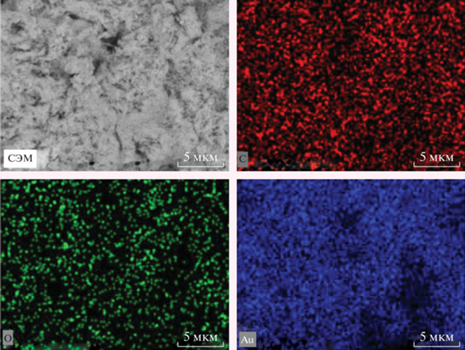
Анализ результатов СЭМ показал, что наночастицы металла после удаления дисперсионной среды – ацетона – образуют агрегаты с широким распределением частиц по размерам. Эти образования имеют форму “виноградной грозди”, которая состоит из более мелких металлических наночастиц (рис. 3).
Спектр EDX, характеризующий элементный состав НЧ AuAc, представлен на рис. 4. Полученные результаты свидетельствуют о том, что образующиеся в синтезе наночастицы Au существенно отличаются по реакционной способности от “инертного” компактного металла. Значительное количество углеродного материала указывает на активную хемосорбцию ацетона или его фрагментов на поверхности металла. В табл. 1 представлены данные о концентрациях элементов.
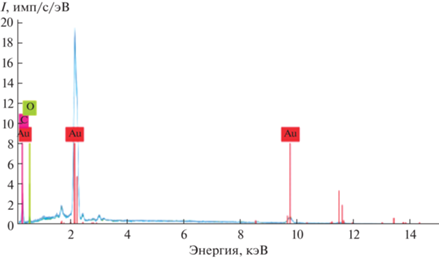
Таблица 1.
Концентрации элементов в образцах (ат. %), рассчитанные по энергодисперсионным рентгеновским спектрам
| Образец | Au | Fe | O | C |
|---|---|---|---|---|
| FeAc | – | 19.3 | 24.0 | 56.7 |
| AuAc | 32.9 | – | 7.1 | 60.0 |
| AuFeAc | 12.6 | 6.0 | 11.2 | 70.1 |
Полученные материалы были исследованы методами термогравиметрического анализа (ТГА) и дифференциальной сканирующей калориметрии (ДСК). Анализ кривых ТГА и ДСК показал, что наночастицы FeAc характеризуются низкотемпературной потерей массы из-за удаления сорбированного растворителя в отличие от образца AuAc. Окисление железа начинается при температуре 350°C. Для образцов золота вес на воздухе не меняется, но в аргоне прослеживается потеря веса на 2%.
Для биметаллических наночастиц также характерна низкотемпературная потеря массы за счет удаления сорбированного растворителя. Экзоэффект при 250°С может быть связан с процессами, протекающими в гетерометаллической частице AuFeAc. Этот факт требует дополнительного исследования и анализа продуктов эволюции биметаллических частиц.
Поверхность наночастиц является одним из важных компонентов, определяющих физико-химические характеристики наночастиц; образцы FeAc, AuAc и AuFeAc были исследованы с использованием РФЭС.
Фотоэлектронная спектроскопия является ведущим аналитическим методом для характеризации различных химических/физических форм элементов в поверхностных структурах. На рис. 5 представлены спектры Au4f образцов AuAc и AuFeAc, а в табл. 2 – их характеристики. Исходя из подобия пиков и спин-орбитального расщепления металлического [51] и окисленного [52] золота, можно сделать вывод, что основным состоянием золота является Au0, но также наблюдается присутствие Au+ и Au3+. Тот факт, что энергии связи пиков Au4f7/2 превышают величину 84.0, характерную для фольги Au, может быть проявлением размерного эффекта, тогда как различия в полуширине (AuAc – 0.84 эВ и AuFeAc – 0.97 эВ) могут быть связаны с возможным образованием связей Au–Fe, Au–C–O–Fe и Au–O–Fe.
Рис. 5.
Фотоэлектронные спектры Au4f образцов AuAc и AuFeAc (а), спектр Au4f образца AuAc совмещен со спектром образца AuFeAc (б), совмещенные фотоэлектронные спектры Au4f7/2 образцов AuAc и AuFeAc (в).
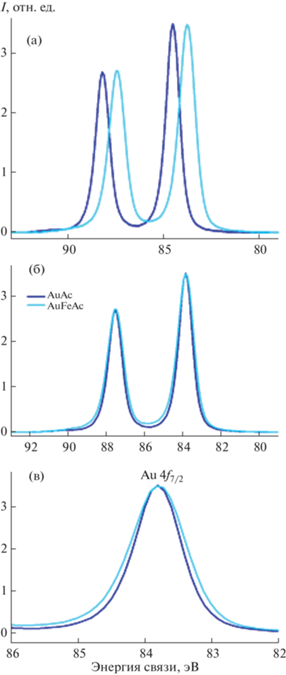
Таблица 2.
Характеристики фотоэлектронных спектров Au4f и Fe2p, эВ
| Образец | Au4f7/2 | Au4f5/2 | Au4f5/2–Au4f7/2 | Fe2p3/2 | Fe2p1/2 | Fe2p1/2–Fe2p3/2 | сат | сат–Fe2p3/2 |
|---|---|---|---|---|---|---|---|---|
| FeAc | – | – | – | 711.1 | 724.5 | 13.4 | 718.9 | 7.8 |
| AuAc | 84.7 | 88.4 | 3.7 | – | – | – | – | – |
| AuFeAc | 84.4 | 88.0 | 3.6 | 711.4 | 724.6 | 13.2 | 718.4 | 7.0 |
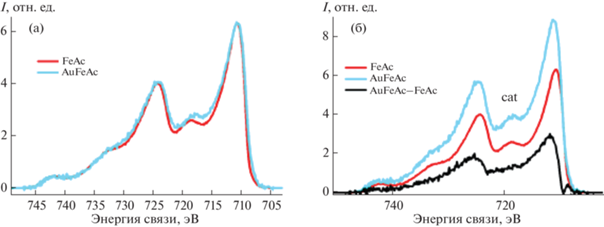
Наблюдаемые сателлиты с энергиями связи 718.4 и 718.9 эВ являются идентификаторами состояния Fe3+. Различие в величинах энергетических интервалов сат–Fe2p3/2 (0.8 эВ), наиболее вероятно, связано с дифференциальной зарядкой, обусловленной присутствием нескольких фаз, содержащих железо, или, другими словами, областей, различающихся по зарядовым состояниям атомов Fe. Для определения их долей в спектрах Fe2p была проведена перекалибровка спектров посредством совмещения сателлитных пиков для достижения их наилучшего совпадения, нормировка по их интенсивности и вычитание одного спектра из другого (рис. 6б). Из энергий связи пиков разностного спектра и отсутствия сателлитного пика следует вывод о присутствии состояния Fe3O4, относительная доля которого ~32%.
Рис. 6.
Фотоэлектронные спектры Fe2p образцов FeAc и AuFeAc: исходный (а), нормированный по сателлиту (б).
Сравнение энергетических интервалов для монометаллических и биметаллических систем Fe2p3/2–Au4f7/2:
Дополнительная информация об электронном состоянии и локальном окружении атомов золота и железа в композитах получена методом рентгеноабсорбционной спектроскопии XANES/EXAFS. Спектры XANES исследованного образца на основе наночастиц AuFeAc приведены на рис. 7. В случае K-края железа спектр исследуемого образца существенно отличается от спектра металлического железа энергетическим сдвигом края поглощения на 7–8 эВ, но близок к спектрам оксидов железа в степени окисления +2…+3. В предкраевой области спектра ⁓7115 эВ наблюдается выраженный пик, предполагающий, что часть ионов железа находится в тетраэдрическом окружении атомов кислорода. Среди распространенных кислородных соединений железа к спектру исследуемого соединения по положению и форме ключевых спектральных особенностей максимально близок спектр магнетита Fe3O4. Основное отличие спектра образца AuFeAc от магнетита заключается в существенном уширении и размытии спектров, что характерно для разупорядоченных и наноразмерных систем. По всей видимости, атомы железа в исследуемом образце образуют оксидные наночастицы, близкие по структуре к магнетиту.
Рис. 7.
Спектры XANES образца AuFeAc на K-крае Fe (слева) и L3-крае Au (справа). Для сравнения приведены спектры реперных материалов.
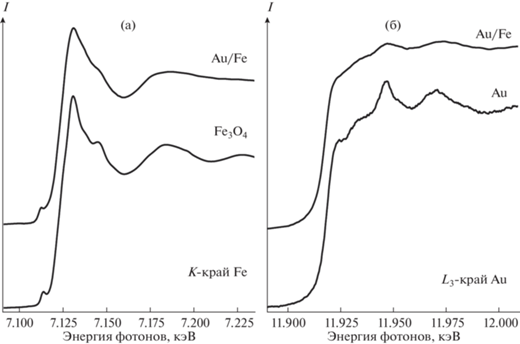
Спектры околокраевой тонкой структуры рентгеновского поглощения XANES вблизи L3-края Au для композитов сопоставлены со спектром монометаллического образца, полученного по аналогичной методике (рис. 7). В случае L3-края золота в спектре исследуемого образца нет значимого энергетического сдвига края поглощения относительно металлического золота: в нем просматриваются все спектральные особенности, характерные для Au, только очень сильно размытые. Этот факт предполагает, что основная часть атомов золота находится в металлическом окружении.
Фурье-трансформанты спектров EXAFS для образца AuFeAc на двух краях поглощения изображены на рис. 8. В соответствии с результатами XANES, обсужденными выше, локальное окружение атомов железа близко к характерному для Fe3O4. В Фурье-трансформанте наблюдаются два доминирующих максимума, надежно относимых к контактам Fe–O и Fe…Fe. Наилучшее соответствие между экспериментальной и расчетной кривыми достигнуто для модели с двумя наборами расстояний Fe–O и Fe…Fe (всего 4 пути рассеяния). Оптимизированные параметры приведены в табл. 3. Для идеальной шпинельной структуры магнетита характерные расстояния Fe–O (Fe…Fe) составляют 1.89 и 2.06 Å (2.97 и 3.48 Å).
Рис. 8.
Фурье-трансформанты спектров EXAFS для образца AuFeAc на K-крае Fe (слева) и L3-крае Au (справа): экспериментальные данные (сплошные линии) и оптимизированные модельные кривые (точки). Для сравнения приведены ФТ реперных материалов.
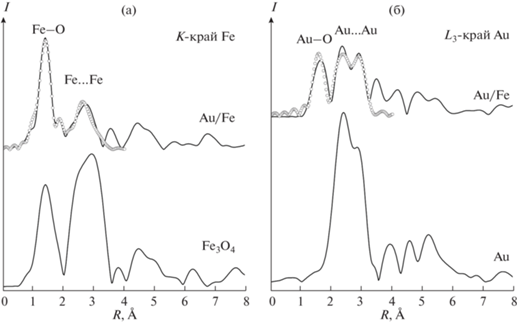
Таблица 3.
Параметры локального окружения атомов железа и золота (N – эффективное координационное число, R – межатомное расстояние, σ2 – параметр Дебая–Валлера) по результатам уточнения спектров EXAFS на K-крае Fe
| Сфера | N | R, Å | σ2, Å2 |
|---|---|---|---|
| Железо | |||
| Fe–O | 2.8 | 1.93 | 0.0034 |
| Fe–O | 2.1 | 2.12 | 0.0034 |
| Fe…Fe | 5.8 | 3.08 | 0.0191 |
| Fe…Fe | 2.9 | 3.43 | 0.0191 |
| Золото | |||
| Au–O | 0.5 | 2.00 | 0.0024 |
| Au–Au | 4.4 | 2.80 | 0.0127 |
| Au…Au | 2.4 | 3.15 | 0.0169 |
В Фурье-трансформанте спектра Au наблюдается сложная структура из большого числа максимумов сопоставимой интенсивности. По сравнению с Фурье-трансформантой стандарта, содержащего относительно крупные чисто золотые наночастицы, образец AuFeAc выявляет дополнительный максимум на коротких межатомных расстояниях и более сильное расщепление доминирующего максимума контактов Au–Au. Наилучшая подгонка была достигнута с использованием трех путей рассеяния: Au–O, Au–Au и Au…Au, соответствующие параметры приведены в табл. 3. Введение путей рассеяния Au–Fe не приводит к улучшению качества подгонки. Таким образом, EXAFS не показывает образования сплавных частиц AuFeAc, хотя и длина связи, по результатам уточнения, оказывается существенно короче, чем для объемного золота (2.80 вместо 2.89 Å). С другой стороны, результаты EXAFS предполагают, что часть атомов золота оказывается в оксидоподобном окружении. Нельзя исключать, что они входят непосредственно в структуру железо-оксидных наночастиц.
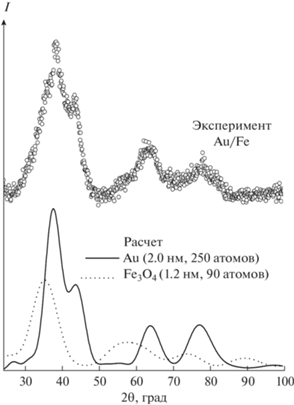
Порошковая дифрактограмма образца AuFeAc, приведенная на рис. 9, содержит очень широкие брэгговские пики, что позволяет классифицировать данный материал как практически аморфный. Тем не менее, положения основных максимумов оказываются близкими к рефлексам в ГЦК-структуре объемного золота. Проведенный расчет по формуле Дебая для модели наночастицы ГЦК-золота диаметром 2 нм (250 атомов в кластере) дает кривую, в общих чертах воспроизводящую экспериментальную кривую. Однако форма доминирующего максимума вблизи 40° и, в особенности, второго максимума ⁓65° указывает на дополнительный вклад другой фазы. На роль второй фазы может претендовать магнетит с еще меньшей областью когерентного рассеяния (на рис. 9 приведен результат расчета для наночастицы из 90 атомов диаметром 1.2 нм).
Рис. 9.
Дифрактограмма образца AuFeAc по сравнению с результатами моделирования для наноразмерных кластеров Au и Fe3O4.
Анализ полученных результатов позволяет предложить структуру синтезированных наночастиц AuFeAc. Образец AuFeAc представляет собой сложный по составу твердый раствор наночастиц гетерометаллического оксида, который находится в матрице, сформированной из наночастиц оксида железа c переменной зарядовой плотностью. Золото находится в состоянии Au0 и в оксидоподобном окружении, из чего можно сделать вывод, что биметаллические частицы представляют собой неупорядоченную структуру твердого раствора [55]. Происходит образование связи Au–Fe–O, это доказывают данные РФЭС, XANES и Фурье-трансформанты спектров EXAFS. Следует отметить наличие углеродсодержащей оболочки на всех типах металлических частиц, образующих этот многокомпонентный материал. Присутствие значительного количества углеводородных фрагментов может играть важную роль в стабилизации металлических наночастиц и формировании биологических свойств материала.
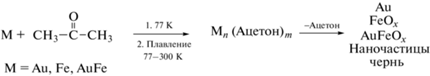
Схема 1. Синтез наночастиц Au, Fe и AuFe с использованием ацетона в качестве дисперсионной среды.
ЗАКЛЮЧЕНИЕ
Полученные результаты демонстрируют перспективность использования метода металло-парового синтеза для получения наночастиц золота, железа и биметаллических структур золото–железо.
Просвечивающая электронная микроскопия показала, что использование в синтезе в качестве органического реагента ацетона приводит к получению наночастиц Au и Fe со средними размерами 5.3 и 1.8 нм соответственно. Как наночастицы, так и нанокомозит содержит значительное количество углеводородных соединений, которые могут играть важную роль в стабилизации моно- и биметаллических наночастиц и определять свойства материалов.
Биметаллические нанокомпозиты обладают неупорядоченной структурой со связями Au–Fe–O и Au–O–Fe, в которых золото, в основном, находится в состояниях Au0, Au+ и Au3+, а железо представляет собой смесь нестехиометрических оксидов, степени окисления которых близки к Fe2+ и Fe3+, со значительной долей магнетита.
Полученные результаты обеспечивают основу для использования наночастиц металлов в исследованиях in vivo для получения эффективных средств для лечения онкологических заболеваний.
Список литературы
Conde J., Doria G., Baptista P. // J. Drug Delivery. 2012. V. 2012. P. 1. https://doi.org/10.1155/2012/751075
Vinardell M.P., Mitjans M. // Nanomaterials. 2015. V. 5. № 2. P. 1004. https://doi.org/10.3390/nano5021004
Sutradhar K.B., Amin M.L. // ISRN Nanotechnology. 2014. V. 2014. P. 1. https://doi.org/10.1155/2014/939378
Zhao G., Rodriguez B.L. // Int. J. Nanomedicine. 2013. V. 8. P. 61. https://doi.org/10.1371/journal.pone.0065896
Akhtar M.J., Alhadlaq H.A., Kumar S. et al. // Arch Toxicol. 2015. V. 89. № 11. P. 1895. https://doi.org/10.1007/s00204-015-1570-1
Ahmad M.Z., Akhter S., Jain G.K. et al. // Expert Opin Drug Deliv. 2010. V. 7. № 8. P. 927. https://doi.org/10.1517/17425247.2010.498473
Гилевская К.С., Машкин М.Е., Красковский А.Н. и др. // Журн. неорган. химии. 2021. Т. 66. № 8. С. 1017. https://doi.org/10.31857/S0044457X21080067
Attari E., Nosrati H., Danafar H. et al. // J. Biomed. Mater. Res. 2019. V. 107. P. 2492. https://doi.org/10.1002/jbm.a.36755
Rostami M., Aghajanzadeh M., Zamani M. et al. // Res. Chem. Intermed. 2018. V. 44. P. 1889. https://doi.org/10.1007/s11164-017-3204-0
Ершов A.Ю., Мартыненков A.A., Якиманский A.В. и др. // Журн. общ. химии. 2022. Т. 92. № 5. С. 788. https://doi.org/10.31857/S0044460X22050146
Zamani M., Rostami M., Aghajanzadeh M. et al. // J. Mater Sci. 2018. V. 53. P. 1634. https://doi.org/10.1007/s10853-017-1673-6
Zain N.M., Stapley A.G.F., Shama G. // Carbohydrate Polymers. 2014. V. 112. P. 195. https://doi.org/10.1016/j.carbpol.2014.05.081
Valodkar M., Modi S., Pal A. et al. // Mater. Res. Bull. 2011. V. 46. P. 384. https://doi.org/10.1016/j.materresbull.2010.12.001
Dong Q., Yang H., Wan C. et al. // Nanoscale Res. Lett. 2019. V. 14. P. 235. https://doi.org/10.1186/s11671-019-3053-4
Efremova M.V., Veselov M.M., Barulin A.V. et al. // ACS Nano. 2018. V. 12. P. 3190. https://doi.org/10.1021/acsnano.7b06439
Efremova M.V., Naumenko V.A., Spasova M. et al. // Sci. Rep. 2018. V. 8. P. 11295. https://doi.org/10.1038/s41598-018-29618-w
Hao Zh., Cheng D., Guo Y. et al. // Appl. Catal. B: Environ. 2001. V. 33. P. 217. https://doi.org/10.1016/S0926-3373(01)00172-2
Seeburg D., Liu D., Radnik J. et al. // Catalysts. 2018. V. 8. P. 42. https://doi.org/10.3390/catal8020042
Naumkin A.V., Budnikov A.V., Buzin M.I. et al. // Ineos Open. 2022. V. 1. P. 1. https://doi.org/10.32931/io2126a
Finch R.M., Hodge N.A., Hutchings G.J. et al. // Phys. Chem. Chem. Phys. 1999. V. 1. P. 485. https://doi.org/10.1039/A808208A
Tomitaka A., Arami H., Raymond A. et al. // RSC Nanoscale. 2017. V. 9. P. 764. https://doi.org/10.1039/C6NR07520G
Kozenkova E., Levada K., Efremova M.V. et al. // Nanomaterials. 2020. V. 10. P. 1646. https://doi.org/10.3390/nano10091646
Cai H., Li K., Li J. et al. // Small. 2015. V. 11. P. 4584. https://doi.org/10.1002/smll.201500856
Maniglio D., Benetti F., Minati L. et al. // Nanotechnology. 2018. V. 29. P. 315101. https://doi.org/10.1088/1361-6528/aac4ce
Brennan G., Thorat N.D., Pescio M. et al. // RSC Nanoscale. 2020. V. 12. P. 12632. https://doi.org/10.1039/D0NR01463J
Guardia P., Nitti S., Materia M.E. et al. // RSC J. Mater. Chem. B. 2017. V. 5. P. 4587. https://doi.org/10.1039/C7TB00968B
Leung K.C.-F., Xuan S., Zhu X. et al. // RSC Chem. Soc. Rev. 2012. V. 41. P. 1911. https://doi.org/10.1039/C1CS15213K
Tomitaka A., Ota S., Nishimoto K. et al. // RSC Nanoscale. 2019. V. 11. P. 6489. https://doi.org/10.1039/C9NR00242A
Majouga A., Sokolsky-Papkov M., Kuznetsov A. et al. // Colloids Surf. B. 2015. V. 125. P. 104. https://doi.org/10.1016/j.colsurfb.2015.11.009
Kinoshita T., Seino S., Okitsu K. et al. // J. Alloys Compounds. 2003. V. 359. P. 46. https://doi.org/10.1016/S0925-8388(03)00198-1
Lin J., Zhou W., Kumbhar A. et al. // J. Solid State Chem. 2001. V. 159. P. 26. https://doi.org/10.1006/jssc.2001.9117
Roduner E. // Chem. Soc. Rev. 2006. V. 35. P. 583. https://doi.org/10.1039/B502142C
Love J.C., Estroff L.A., Kriebel J.K. et al. // Chem. Rev. 2005. V. 105. P. 1103. https://doi.org/10.1021/cr0300789
Zeng H., Du X.W., Singh S.C. et al. // Adv. Funct. Mater. 2012. V. 22. P. 1333. https://doi.org/10.1002/adfm.201102295
Lyon J.L., Fleming D.A., Stone M.B. et al. // Nano Lett. 2004. V. 4. № 4. P. 719. https://doi.org/10.1021/nl035253f
Chen W., Cai W., Zhang L. et al. // J. Colloid Interface Sci. 2001. V. 238. № 2. P. 291. https://doi.org/10.1006/jcis.2001.7525
Geethalakshmi R., Sarada D.V. // Int. J. Nanomed. 2012. V. 7. P. 5375. https://doi.org/10.2147/IJN.S36516
Popov V.V., Menushenkov A.P., Yastrebtsev A.A. et al. // J. Alloys Compd. 2022. V. 910. 164922. https://doi.org/10.1016/j.jallcom.2022.164922
Рашидова С.Ш., Вохидова Н.Р., Алексеева О.В. и др. // Журн. неорган. химии. 2022. Т. 67. № 12. С. 1851. https://doi.org/10.31857/S0044457X22601146
Агафонов А.В., Сироткин Н.А., Титов В.А. и др.// Журн. неорган. химии. 2022. Т. 67. № 3. С. 271. https://doi.org/10.31857/S0044457X22030023
Vasil’kov A., Dovnar R., Smotryn S. et al. // Antibiotics. 2018. V. 7. P. 80. https://doi.org/10.3390/antibiotics7030080
Abd-Elsalam K.A., Alghuthaymi M.A., Shami A. et al. // J. Fungi. 2020. V. 6. P. 112. https://doi.org/10.3390/jof6030112
Cardenas-Trivino G., Cruzat-Contreras C. // J. Cluster Sci. 2018. V. 29. P. 1081. https://doi.org/10.1007/s10876-018-1419-x
Vasil’kov A., Rubina M., Naumkin A. et al. // Gels 2021. V. 7. P. 1. https://doi.org/10.3390/gels7030082
Sztandera K., Gorzkiewicz M., Klajnert-Maculewicz B. // Mol. Pharmaceutics. 2019. V. 16. № 1. P. 1. https://doi.org/10.1021/acs.molpharmaceut.8b00810
Miri A., Najafzadeh H., Darroudi M. et al. // Chem. Open. 2021. V. 10. P. 327. https://doi.org/10.1002/open.202000186
Vasil’kov A., Batsalova T., Dzhambazov B. et al. // Surf. Interface Anal. 2021. V. 53. P. 1. https://doi.org/10.1002/sia.7038
Vasil’kov A.Yu., Migulin D.A., Muzalevskiy V.M. et al. // Mend. Commun. 2022. V. 32. P. 478. https://doi.org/10.1016/j.mencom.2022.07.016
Belyakova O.A., Zubavichus Y.V., Neretin I.S. et al. // J. Alloys Comps. 2004. V. 382. P. 46. https://doi.org/10.1016/j.jallcom.2004.05.047
Chernyshov A.A., Veligzhanin A.A., Zubavichus Y.V. // Nucl. Instr. Meth. Phys. Res. A. 2009. V. 603. P. 95. https://doi.org/10.1016/j.nima.2008.12.167
Moulder J.F., Chastain J., King R.C. et al. Handbook of X-Ray Photoelectron Spectroscopy: A Reference Book of Standard Spectra for Identification and Interpretation of XPS Data. PerkinElmer, Eden Prairie. 1995. 261 p.
Grosvenor A.P., Kobe B.A., Biesinger M.C. et al. // Surface and Interface Analysis. 2004. V. 36. № 12. P. 1564. https://doi.org/10.1002/sia.1984
Mansour A.N., Brizzolara R.A. // Surface Sci. Spectra. 1996. V. 4. P. 345. https://doi.org/10.1116/1.1247831
Anderson J.F., Kuhn M., Diebold U. // Surface Sci. Spectra. 1996. V. 4. P. 266. https://doi.org/10.1116/1.1247796
Tymoczko A., Kamp M., Prymak O. et al. // RSC Nanoscale. 2018. V. 10. P. 16434. https://doi.org/10.1039/C8NR03962C
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии