

Журнал неорганической химии, 2023, T. 68, № 7, стр. 896-903
Ce0.9(Mg,Ni)0.1O2: композит или твердый раствор?
М. Н. Смирнова a, *, Г. Д. Нипан a, М. А. Копьева a, Г. Е. Никифорова a, Г. А. Бузанов a, Е. И. Кожухова b, И. В. Козерожец a, А. Д. Япрынцев a, А. А. Архипенко a, М. С. Доронина a
a Институт общей и неорганической химии им. Н.С. Курнакова РАН
119991 Москва, Ленинский пр-т, 31, Россия
b НИЦ “Курчатовский институт” – ИРЕА
107076 Москва, ул. Богородский Вал, 3, Россия
* E-mail: smirnova_macha1989@mail.ru
Поступила в редакцию 30.01.2023
После доработки 16.02.2023
Принята к публикации 21.02.2023
- EDN: RIKAIU
- DOI: 10.31857/S0044457X23600159
Аннотация
Методом сжигания геля c последующей гидротермальной обработкой получены образцы состава Ce0.9(Mg1–xNix)0.1O2 (0 ≤ x ≤ 1, шаг x = 0.1). Рентгенофазовый анализ показал, что после сгорания геля и отжига при 1100°С образуется композит CeO2 (структура флюорита)/твердый раствор Mg1–xNixO (структура галита), а дополнительная гидротермальная обработка с последующим отжигом способствует образованию ограниченного твердого раствора Ce0.9(Mg1–xNix)0.1O2. Согласно результатам ИК-спектроскопии, композит CeO2–Mg1–xNixO не адсорбирует CO2 даже в присутствии паров воды, что также подтверждается спектрами диффузного отражения в УФ-видимой области. Напротив, твердый раствор Ce0.9(Mg1–xNix)0.1O2 поглощает CO2, о чем свидетельствуют результаты ИК-спектроскопии и термогравиметрического анализа.
ВВЕДЕНИЕ
В настоящее время значительно увеличилось число публикаций, посвященных исследованию сорбционных свойств оксида церия, в которых показано, что материалы на основе CeO2 могут быть использованы как для адсорбции углекислого газа (CO2) [1–3], так и в качестве сенсоров для определения концентрации угарного газа (CO) в атмосфере [4]. Поглощение CO2 (~5 вес. %) кристаллическим CeO2 со структурой флюорита [2, 3] увеличивается после введения катионов Cu или La в кристаллическую решетку [5]. Для композита 4 мол. % Mg–CeO2 адсорбция CO2 ниже, чем для CeO2 [5], однако с ростом концентрации Mg поглощение увеличивается и достигает 10.4 вес. % CO2 (Ce/Mg = 0.05, 30°C) [6]. Технологические процессы часто сопровождаются выделением угарного газа наряду с CO2. Диспропорционирование CO с образованием С и CO2 осуществляет композит Ni–MgO, адсорбирующий С на частицах Ni и CO2 в матрице MgO [7]. Создание материала, способного эффективно поглощать оба углеродных оксида (CO2 и CO), представляет особенный интерес для экологии. В качестве такого адсорбента может выступать композит Ni–MgO–CeO2 – катализатор сухого риформинга метана (CH4 + CO2 ↔ 2CO + 2H2) [8]. В зависимости от способа получения Ni–MgO–CeO2 (подобно тому, как это происходит в системе CoO–CeO2, где образуется многофазный композит (Co/CeO2 – метод пропитки) или ограниченный твердый раствор (Ce1–xCoxO2 – соосаждение) [9]) адсорбция CO2 может значительно варьироваться. Сведения о твердых растворах в системе MgO–CeO2 противоречивы. Незначительная растворимость CeO2 в MgO отмечается при твердофазном синтезе [10, 11] или соосаждении с последующим отжигом [12, 13]. При использовании органических гелей в процессе синтеза (Mg : Ce = 1 : 1 [14], Mg : Ce = = 1 : 3, 1 : 1, 3 : 1 [15]) и ионных жидкостей (Mg : Ce = = 4 : 1 [16]) после отжига при 500°C MgO не обнаруживается. В результате применения макромолекулярной поверхностно-активной методики синтеза (Mg : Ce = 1 : 9, 1 : 1, 9 : 1) и отжига при 650°C, по мнению авторов [17, 18], образуется протяженный твердый раствор на основе CeO2. Твердофазный синтез приводит к образованию ограниченного твердого раствора, в котором растворимость MgO достигает 8 мол. % [10]. Сведения о взаимодействии оксидов в системе NiO–CeO2, как и в системе MgO–CeO2, неоднозначны. Отсутствие растворимости CeO2 в NiO со структурой галита доказано в работах [19, 20], а вопрос о существовании ограниченного твердого раствора на основе CeO2 вызвал разногласия [21–23]. При твердофазном синтезе [19, 24] возможность образования Ce1–xNixO2 опровергается, однако применение микроволнового безводного золь-гель метода синтеза позволяет ввести 5 мол. % NiO в оксид церия CeO2 с кристаллической структурой флюорита [25], а после сжигания геля [26, 27] или гидроксидного золь-геля [28] образуется твердый раствор Ce1–xNixO2 для составов x < < 0.1 [26], x ≤ 0.15 [27] и x ≤ 0.20 [28]. В отличие от метастабильных ограниченных твердых растворов Ce1–xMgxO2 и Ce1–xNixO2, неограниченный твердый раствор Mg1–xNixO (0 ≤ x ≤ 1) существует стабильно в широком интервале температур [29, 30].
В настоящей работе исследовано фазовое состояние образцов, принадлежащих разрезу Ce0.9Mg0.1O2–Ce0.9Ni0.1O2 и полученных методом сжигания геля, а также влияние гидротермальной обработки на сорбционную способность изучаемых образцов. Для поликристаллов Ce0.9Mg0.05Ni0.05O2 определена возможность адсорбции CO2 при разной влажности и проанализировано поглощение в УФ-видимой части спектра излучений.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Образцы состава Ce0.9(Mg1–xNix)0.1O2 (0 ≤ x ≤ 1, шаг x = 0.1) получали методом сжигания геля [31, 32]. Прекурсоры Mg (металл, стружка, “тех.”), оксид никеля Ni2O3 (ч.) и кристаллогидрат нитрата церия Ce(NO3)3 · 6H2O (ч. д. а.), взятые в стехиометрических количествах, растворяли в разбавленной (1 : 1) азотной кислоте (ч. д. а.). Полученный раствор упаривали в конической колбе, переносили в керамическую чашку и добавляли поливиниловый спирт (C2H4O)x (ПВС) в виде порошка.
Упаривание проводили до образования геля, превращающегося постепенно, без возгорания в порошок, который дополнительно гомогенизировали, переносили в керамический тигель и отжигали при 600 и 1100°C не менее 4 ч с последующим охлаждением печи в инерционно-термическом режиме.
Синтезированные образцы после отжига при 600°C подвергали гидротермальной обработке в контейнерах из нержавеющей стали в кислотной (1.5 мас. % НСl), щелочной (1.5 мас. % NaOН) и нейтральной среде: к порошку Ce0.9Mg0.05Ni0.05O2(0.15, 0.12 и 0.1 г) добавляли 2.9 мл раствора НСl (коэффициент заполнения автоклава КЗА – 20%), 3.2 мл раствора NaOH (КЗА – 14%) и 3.4 мл дистиллированной воды (КЗА – 13%). Для предотвращения испарения воды из суспензии Ce0.9Mg0.05Ni0.05O2 в автоклав вне тефлонового контейнера заливали 0.5 мл дистиллированной воды. Герметизированный автоклав помещали в разогретую электрическую печь при температуре 400°С и выдерживали в течение 7 сут при постоянной температуре. Автоклавы охлаждали проточной водой, образцы промывали дистиллированной водой до рН 7 и выдерживали 24 ч в сушильном шкафу при температуре 80 ± 5°C.
Насыщение исследуемых образцов проводили как осушенным, так и содержащим пары воды углекислым газом (доля CO2 99.8 об. %, содержание H2O < 0.1%). В последнем случае CO2 барботировали через дистиллированную воду в склянке Дрекселя со стеклянным пористым дном и каплеуловителем, заполненным минеральной ватой, до стационарного содержания паров H2O при 25°С.
Рентгенофазовый анализ (РФА) выполняли на дифрактометре Bruker Advance D8 (CuKα-излучение) в интервале углов 2θ = 10°–70° с шагом сканирования 0.0133°. Обработку результатов проводили с помощью программного пакета DIFFRAC.EVA и базы данных ICDD PDF2.
Содержание Ni и Ce в образцах контролировали с помощью рентгенофлуоресцентной спектрометрии (РФС) на спектрометре Спектроскан Макс-GVM (Россия). Из-за физических особенностей метода определение магния представляло значительные трудности, поэтому использовали дуговую атомно-эмиссионную спектрометрию (ДАЭС) в соответствии с разработанной методикой [33].
ИК-спектры регистрировали на спектрометре Perkin Elmer Spectrum 65 FT-IR в области 4000–400 см–1 с разрешением 2 см–1.
Спектры диффузного отражения в диапазоне 200–1000 нм регистрировали с помощью модульной оптической системы Ocean Optics (дейтериево-галогеновый источник DH-2000-BAL, интегрирующая сфера ISP-80-8-R диаметром 80 мм, детектор QE650000). В качестве образца сравнения использовали стандарт WS-1 (Ocean Optics) из политетрафторэтилена. Ширину запрещенной зоны рассчитывали из данных диффузного отражения (R) через построение Таука в координатах:
где F(R) – функция Кубелки–Мунка, равная (1 ‒ R2)/2R, R – диффузное отражение, h – постоянная Планка, ν – частота падающего излучения.Термический анализ проводили в атмосфере воздуха (газовый поток 150 мл/мин) в алундовых тиглях на термоанализаторе SDT Q600 (TA Instruments, США).
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
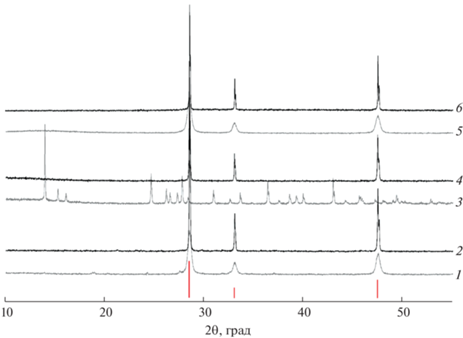
При синтезе образцов методом сжигания геля с поливиниловым спиртом и отжиге при 1100°C ограниченные твердые растворы состава Ce0.9(Mg1–xNix)O2 (0 ≤ x ≤ 1) не образуются. На рис. 1 приведены результаты рентгеновской дифракции образцов граничных составов Ce0.9Mg0.1O2 и Ce0.9Ni0.1O2, где наряду с CeO2 со структурой флюорита присутствуют рефлексы, относящиеся к структурам периклаза MgO и бурсенита NiO. По данным полнопрофильного анализа дифрактограмм, процентное содержание этих оксидов соответствует исходным количествам Mg и Ni в образцах.
Рис. 1.
Дифрактограммы образцов Ce0.9Mg0.1О2 (1) и Ce0.9Ni0.1О2 (2) (синтез методом сжигания геля с ПВС и отжиг при 1100°С). Внизу представлена штрих-диаграмма CeO2 (ICDD PDF2#01-071-4199).
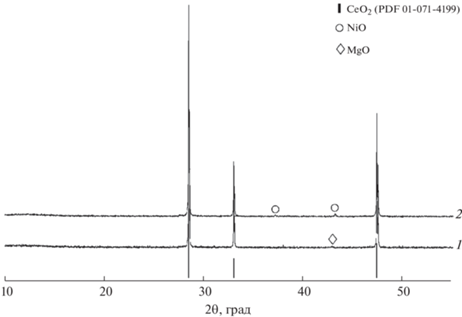
Аналогичная ситуация наблюдается для всех остальных образцов в системе Ce0.9(Mg1–xNix)O2 (0 < x < 1), в которых CeO2 образуется совместно с твердым раствором Mg1–xNixO, обладающим структурой галита.
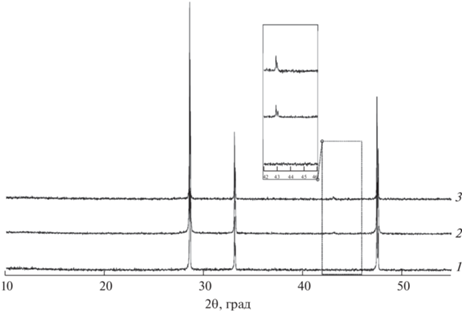
Для исследования способности сорбции углекислого газа образцы Ce0.9(Mg1–xNix)O2, отожженные при 1100°C, охлаждали в атмосфере сухого или влажного CO2. Насыщение образцов углекислым газом не привело к изменению фазового состава (рис. 2).
Рис. 2.
Дифрактограммы образцов CeО2 (1) и Ce0.9Mg0.0.5Ni0. 05О2 (2) (синтез методом сжигания геля с ПВС, отжиг при 1100°С, охлаждение в атмосфере влажного СО2), а также образца Mg0.05Ni0.05Ce0.9О2, охлажденного в атмосфере сухого СО2 (3).
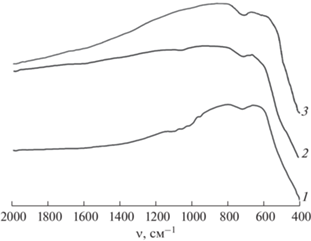
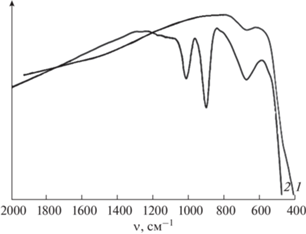
В ИК-спектрах образцов Ce0.9(Mg1 – xNix)O2, отожженных как на воздухе, так и в атмосфере углекислого газа, присутствуют только полосы поглощения, соответствующие валентным колебаниям Ce–O при 420 [34, 35], 720 [35] и 1010 см–1 [35] (рис. 3). Насыщение потока CO2 парами воды не привело к активации адсорбции CO2 композитами Ce0.9(Mg1–xNix)O2, в отличие, например, от оксида магния, для которого повышение относительной влажности до 50% повышает адсорбцию CO2 в пять раз [36].
Рис. 3.
ИК-спектры образцов СeO2 (1) и Ce0.9Mg0.05Ni0.05О2 (2, 3); синтез методом сжигания геля с ПВС, отжиг при 1100°С, охлаждение в атмосфере сухого СО2 (1, 2) и влажного CO2 (3).
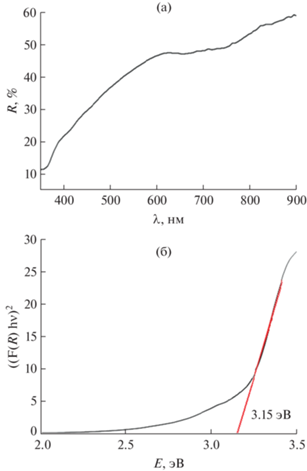
Отсутствие растворимости MgO и NiO в кубическом CeO2 подтверждает анализ спектров диффузного отражения в УФ-видимой области (рис. 4), согласно которому ширина запрещенной зоны (3.15 эВ) образца Ce0.9(Mg0.05Ni0.05)O2 (синтез методом сжигания геля с ПВС, отжиг при 1100°С, охлаждение в атмосфере влажного CO2) соответствует ширине запрещенной зоны для кристаллического CeO2, варьирующей в интервале 3.1–3.35 эВ [37–40].
Рис. 4.
Спектр диффузного отражения Ce0.9Mg0.05Ni0.05О (синтез методом сжигания геля с ПВС, отжиг при 1100°С, охлаждение в атмосфере влажного CO2) (а) и построение Таука для определения величины ширины запрещенной зоны CeO2 (б).
Для образца состава Ce0.9Mg0.05Ni0.05О2, полученного методом сжигания геля ПВС и отожженного при 600°C, была проведена дополнительная гидротермальная обработка в щелочной, кислотной или нейтральной среде с последующим отжигом при 1100°C. Гидротермальная обработка в щелочной и нейтральной среде приводит к аморфизации оксида церия, которая проявляется значительным уширением дифракционных рефлексов на рентгенограмме (рис. 5, кривые 1 и 5). В кислой среде происходит химическое взаимодействие оксида церия с соляной кислотой с образованием целого ряда кристаллогидратов хлорида церия (рис. 5, кривая 3). На дифрактограммах образцов после конечного отжига при 1000°C присутствуют только рефлексы, относящиеся к кристаллической структуре CeO2. Никакого присутствия Mg1–xNixO с кристаллической структурой галита в пределах чувствительности метода РФА не обнаружено (рис. 5, кривые 2, 4 и 6).
Рис. 5.
Дифрактограммы образцов Ce0.9Mg0.05Ni0.05О2; синтез методом сжигания геля с ПВС, отжиг при 600°С, гидротермальная обработка при 400°C с NaOH (1), HCl (3) и H2O (5); отжиг при 1100°С (2, 4, 6 соответственно). Внизу представлена штрих-диаграмма CeO2 (ICDD PDF2#01-071-4199).
С помощью элементного анализа (РФС и ДАЭС) было показано, что мольное соотношение металлов (Ni, Ce и Mg) в процессе гидротермальной обработки осталось неизменным в пределах погрешности определения (рис. 6).
Рис. 6.
Спектры РФС (а) и ДАЭС (б) образца Ce0.9Mg0.05Ni0.05О2; синтез методом сжигания геля с ПВС, отжиг при 600°С, гидротермальная обработка с Н2О при 400°C.
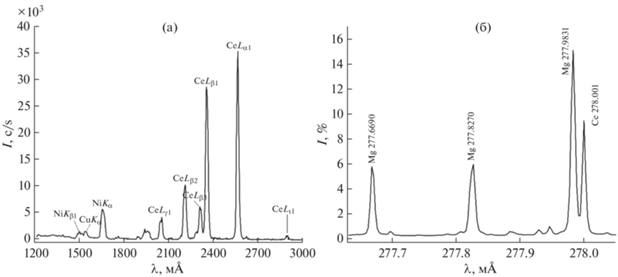
По данным количественного анализа (РФС), массовое соотношение Ce : Ni составило 43 : 1, что соответствует стехиометрическому составу. Результаты ДАЭС подтвердили ожидаемое содержание магния, которое составило 0.75 мас. %. Полученные результаты свидетельствуют о том, что для образца Ce0.9Mg0.05Ni0.05O2 после его гидротермальной обработки и дополнительного отжига сохранился исходный состав.
В ИК-спектрах образцов, полученных в результате гидротермальной обработки в водной среде и отжига при 1100°С с охлаждением в атмосфере влажного CO2, появляются две дополнительные полосы поглощения в диапазонах частот 880–900 и 1020–1050 см–1 (рис. 7), которые могут быть отнесены к колебаниям C–OH-группы [41], в частности, к валентным-колебаниям ν1(C–O).
Рис. 7.
ИК-спектры образца Ce0.9Mg0.05Ni0.05О2, синтезированного методом сжигания геля с ПВС с последующей обработкой: отжиг при 1100°С, охлаждение в атмосфере влажного CO2 (1); отжиг при 600°С, гидротермальная обработка с Н2О при 400°С; отжиг при 1100°С, охлаждение в атмосфере влажного CO2 (2).
Вода играет определяющую роль в механизме адсорбции углекислого газа на поверхности оксидов металлов. В отсутствие воды CO2 адсорбируется, как правило, в виде бикарбоната и/или карбоната [42]. Увеличение влажности повышает вероятность взаимодействия воды с поверхностью оксида и образования барьера для прямой адсорбции CO2, поэтому во влажной среде углекислый газ сорбируется преимущественно в виде сольватированного карбоната [43].
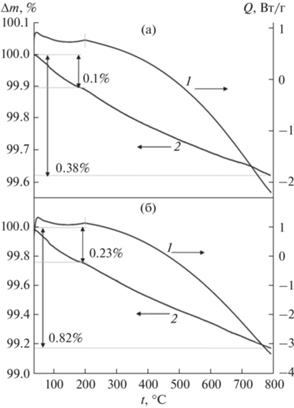
На рис. 8 представлены результаты анализа ДСК-ТГ двух образцов Ce0.9Mg0.05Ni0.05О2, отожженных при 1100°С и охлажденных в атмосфере влажного CO2, при этом второй образец (нижний рисунок) перед отжигом подвергался дополнительной гидротермальной обработке. В обоих случаях на кривой ДСК в интервале 50–200°С наблюдается размытый эндоэффект, по-видимому, связанный с основной десорбцией CO2 с поверхности образца. В этом температурном интервале потеря массы составляет 0.1 мас. % для первого образца и 0.23 мас. % для образца после гидротермальной обработки. Дальнейшее повышение температуры до 800°С сопровождается монотонным уменьшением массы образцов, суммарно на 0.38 и 0.82 мас. %. Вероятно, это связано с характерной для CeO2–δ кислородной нестехиометрией [44], которая возможна у композита CeO2–δ/Mg1–xNixO и особенно у дефектного твердого раствора Ce0.9Mg0.05Ni0.05О2–δ. Таким образом, сравнительный анализ ДСК-ТГ показал, что в результате гидротермальной обработки образца Ce0.9Mg0.05Ni0.05О2 его адсорбционная способность по углекислому газу возросла в два раза.
Рис. 8.
Результаты ДСК-ТГА образца Ce0.9(Mg0.05Ni0.05)O2, синтезированного методом сжигания геля с ПВС с последующей обработкой: отжиг при 1100°С, охлаждение в атмосфере влажного CO2 (а); отжиг при 600°С, гидротермальная обработка с Н2О при 400°С; отжиг при 1100°С, охлаждение в атмосфере влажного CO2 (б). 1 – дифференциальная кривая, 2 – кривая потери массы.
ЗАКЛЮЧЕНИЕ
Полученные результаты показывают, что при получении однофазных твердых растворов Ce0.9(Mg1–xNix)O2, способных поглощать CO2, более эффективным интенсивным параметром оказывается давление водяного пара, который, кроме того, катализирует поглощение углекислого газа кубическим Ce0.9(Mg1–xNix)O2. В фундаментальном плане представлена возможность получения ограниченных твердых растворов при вариации температуры и давления.
Список литературы
Shcherbakov A.B., Zholobak N.M., Ivanov V.K. // Cerium Oxide (CeO2): Synthesis, Properties and Applications. 2020. P. 279. https://doi.org/10.1016/b978-0-12-815661-2.00008-6
Slostowski C., Marre S., Dagault P. et al. // J. CO2 Util. 2017. V. 20. P. 52. https://doi.org/10.1016/j.jcou.2017.03.023
Kanahara K., Matsushima Y. // J. Electrochem. Soc. 2019. V. 166. № 12. B978. https://doi.org/10.1149/2.0691912jes
Izu N., Matsubara I., Itoh T. et al. // J. As. Ceram. Soc. 2016. V. 4. № 2. P. 205. https://doi.org/10.1016/j.jascer.2016.04.001
Li M., Tumuluri U., Wu Z., Dai S. // Chem. Sus. Chem. 2015. V. 8. 3651. https://doi.org/10.1002/cssc.201500899
Jin S., Bang G., Liu L. et al. // Microporous and Mesoporous Mater. 2019. V. 288. P. 109587. https://doi.org/10.1016/j.micromeso.2019.109587
Martra G., Marchese L., Arena F. et al. // Top. Catal. 1994. V. 1. № 1–2. P. 63. https://doi.org/10.1007/BF01379576
Jang W.-J., Kim H.-M., Shiem J.-O. et al. // Green Chem. 2018. V. 20. № 7. P. 1621. https://doi.org/10.1039/C7GC03605A
Nguyen T.H., Kim H.B., Park E.D. // Catalysts. 2022. V. 12. № 2. P. 212. https://doi.org/10.3390/catal12020212
Preda M., Dinescu R. // Rev. Roum. Chim. 1976. V. 21. № 7. P. 1023.
Longo V., Meriani S., Ricciardiello F. et al. // Am. Ceram. Soc. 1981. V. 64. № 2. P. 38. https://doi.org/10.1111/j.1151-2916.1981.tb09574.x
Ivanova A.S., Moroz B.L., Moroz E.M. et al. // J. Solid. State Chem. 2005. V. 178. № 11. P. 3265. https://doi.org/10.1016/j.jssc.2005.08.001
Manríquez-Ramirez M.E., Elizalde I. et al. // React. Kinet. Mech. Catal. 2020. V. 131. № 2. P. 769. https://doi.org/10.1007/s11144-020-01868-8
Shafighi S., Mohammad Shafiee R.M., Ghashang M. et al. // J. Sulfur Chem. 2018. V. 39. № 4. P. 402. https://doi.org/10.1080/17415993.2018.1436710
Saito M., Itoh M., IwamotoJ. et al. // Catal. Lett. 2006. V. 106. № 3–4. P. 107. https://doi.org/10.1007/s10562-005-9615-3
Abimanyu H., Ahn B.S., Kim C.S. et al. // Ind. Eng. Chem. Res. 2007. V. 46. № 24. P. 7936. https://doi.org/10.1021/ie070528d
Chen M., Fang W.-M., Zheng X.-M. // Acta Chim. Sinica. 2004. V. 62. № 20. P. 2051.
Chen M., Zheng H., Shi C. et al. // J. Mol. Catal. A. 2005. V. 237. № 1–2. P. 132. https://doi.org/10.1016/j.molcata.2005.04.038
Hrovat M., Hole J., Bernic S. et al. // Mater. Res. Bull. 1998. V. 33. № 8. P. 1175. https://doi.org/10.1016/S0025-5408(98)00103-2
Wang C.-C., Li J.-H., Sun Y.-F. et al. // Acta Phys.-Chim. Sin. 2011. V. 27. № 10. P. 2421. http://www.whxb.pku.edu.cn/EN/Y2011/V27/I10/2421
Pound B.G. // Solid State Ionics. 1992. V. 52. № 1–3. P. 183. https://doi.org/10.1016/0167-2738(92)90104-W
Ranlov J., Poulsen F.W., Mogensen M. // Solid State Ionics. 1993. V. 61. № 4. P. 277. https://doi.org/10.1016/0167-2738(93)90392-G
Pound B.G. // Solid State Ionics. 1993. V. 61. № 4. P. 281. https://doi.org/10.1016/0167-2738(93)90393-H
Lu B., Kawamoto K. // Mater. Res. Bull. 2014. V. 53. P. 70. https://doi.org/10.1016/j.materresbull.2014.01.043
Hilaire S., Luo L., Rechberger F. et al. // Z. Anorg. Allg. Chem. 2014. V. 640. № 5. P. 733. https://doi.org/10.1002/zaac.201300567
Huang Z., Zhao Z., Qi H. et al. // J. Energy Chem. 2020. V. 40. P. 46. https://doi.org/10.1016/j.jechem.2019.02.007
Keneko H., Tamaura Y. // J. Phys. Chem. Solids. 2009. V. 70. № 6. P. 1008. https://doi.org/10.1016/j.jpcs.2009.05.015
Thurber A., Reddy K.M., Shutthanandan V. et al. // Phys. Rev. B. 2007. V. 76. P. 165206. https://doi.org/10.1103/PhysRevB.76.165206
Zinkevich M., Geupel S., Aldinger F. // J. Alloys. Compd. 2005. V. 293. P. 154. https://doi.org/10.1016/j.jallcom.2004.09.069
Prostakova V., Chen J., Jak E. et al. // Calphad. 2012. V. 37. P. 1. https://doi.org/10.1016/j.calphad.2011.12.009
Smirnova M.N., Kop’ev M.A., Nipan G.D. et al. // Russ. J. Inorg. Chem. 2022. V. 67. P. 978. https://doi.org/10.1134/S0036023622070221
Smirnova M.N., Kop’ev M.A., Nipan G.D. et al. // Russ. J. Inorg. Chem. 2022. V. 67. P. 1823. https://doi.org/10.1134/S0036023622600824
Arkhipenko A.A., Koshel E.S., Baranovskaya V.B. // Industrial laboratory. Diagnostics of materials. 2021. V. 87. № 11. P. 19. https://doi.org/10.26896/1028-6861-2021-87-11-19-25
Miri A., Sarani M. // Ceram. Int. 2018. V. 44. № 11. P. 12642. https://doi.org/10.1016/j.ceramint.2018.04.063
Binet C., Daturi M., Lavalley J.-K. // Catal. Today. 1999. V. 50. № 2. P. 207. https://doi.org/10.1016/S0920-5861(98)00504-5
Ding Y.D., Song G., Liao Q. et al. // Energy. 2016. V.112. P. 101. https://doi.org/10.1016/j.energy.2016.06.064
Sandhya K.L., Prabhakar R.P., Lakshmipathy R.M. et al. // J. Alloys Compd. 2008. V. 461. № 1–2. P. 509. https://doi.org/10.1016/j.jallcom.2007.07.055
Brito P.C.A., Santos D.A.A., Duque J.G.S. et al. // Phys. B. Condens. Mater. 2010. V. 405. № 7. P. 1821. https://doi.org/10.1016/j.physb.2010.01.054
Zhang G., Li L., Li G. et al. // Solid State Sci. 2009. V. 11. P. 671. https://doi.org/10.1016/j.solidstatesciences.2008.10.01
Polezhaeva O.S., Yaroshinskaya N.V., Ivanov V.K. // J. Inorg. Chem. 2007. V. 52. P. 1184. https://doi.org/10.1134/S0036023607080049
Köck E.-M., Bernard J., Podewit M. et al. // Chem. Eur. J. 2020. V. 26 P. 285. https://doi.org/10.1002/chem.201904142
Kolle J.M., Fayaz M., Sayari A. // Chem. Rev. 2021. V. 121. № 13. P. 7280. https://doi.org/10.1021/acs.chemrev.0c00762
Baltrusaitis J., Schuttlefield J., Zeitler E. et al. // Chem. Eng. J. 2011. V. 170. P. 471. https://doi.org/10.1016/j.cej.2010.12.041
Knoblauch N., Simon H., Schmücker M. // Solid State Ionics. 2017. V. 301. P. 43. https://doi.org/10.1016/j.ssi.2017.01.003
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии