

Журнал неорганической химии, 2023, T. 68, № 9, стр. 1154-1164
Катехолатные комплексы меди(II) с полипиридильными лигандами
О. Ю. Трофимова a, *, К. И. Пашанова a, И. В. Ершова a, М. В. Арсеньев a, И. А. Якушев b, П. В. Дороватовский c, Р. Р. Айсин d, А. В. Пискунов a
a Институт металлоорганической химии им. Г.А. Разуваева РАН
603950 Нижний Новгород, ул. Тропинина, 49, Россия
b Институт общей и неорганической химии им. Н.С. Курнакова РАН
119991 Москва,
Ленинский пр-т, 31, Россия
c Национальный исследовательский центр “Курчатовский институт”
123182 Москва,
пл. Академика Курчатова, 1, Россия
d Институт элементоорганических соединений им. А.Н. Несмеянова РАН
119334 Москва,
ул. Вавилова, 28, Россия
* E-mail: olesya@iomc.ras.ru
Поступила в редакцию 27.04.2023
После доработки 11.05.2023
Принята к публикации 27.05.2023
- EDN: YIEZEI
- DOI: 10.31857/S0044457X23600846
Аннотация
Синтезированы катехолатные комплексы меди(II) на основе 3,6-ди-трет-бутил-о-бензохинона с N-донорными лигандами фенантролинового ряда: (3,6-Cat)Cu(Phen) (I), (3,6-Cat)Cu(DPQ) (II) и (3,6-Cat)Cu(DPPZ) (III), где 3,6-Cat – дианион 3,6-ди-трет-бутил-о-бензохинона, Phen – фенантролин, DPQ – дипиридо[3,2-d:2',3'-f]хиноксалин, DPPZ – дипиридо[3,2-a:2',3'-c]феназин. Синтезированные гетеролептические комплексы меди(II) демонстрируют внутримолекулярный перенос заряда лиганд–лиганд, что обусловливает их интенсивную фиолетовую окраску. Электронное строение синтезированных хромофоров исследовано методами электронной спектроскопии, циклической вольтамперометрии и квантово-химических расчетов. Молекулярное и кристаллическое строение синтезированных соединений установлено методом РСА (CIF files CCDC 2250975 (I · THF), 2 250 976 ([(II · THF)(II)] · 3THF), 2250977 (II)).
ВВЕДЕНИЕ
Одно из перспективных направлений развития современной координационной химии – синтез и изучение строения и оптофизических характеристик донорно-акцепторных хромофорных комплексов металлов, содержащих одновременно два типа редокс-активных лигандов [1, 2]. Дизайн комплексов металлов, в которых редокс-активные лиганды, координированные на металлоцентр, находятся в различных степенях окисления (нейтральной и дианионной), создает условия для эффективного фотоиндуцированного переноса заряда от донора к акцептору (схематичное представление простейшего хромофора такого типа: D–M–A, где D – донор, A – акцептор, М – металл). В хромофорах типа LL’CT (ligand-to-ligand charge transfer) разделение зарядов позволяет наблюдать фотоиндуцированный внутримолекулярный процесс, который заключается в переносе заряда с высшей занятой молекулярной орбитали (ВЗМО) электрононасыщенного донорного лиганда на низшую свободную молекулярную орбиталь (НСМО) электронодефицитного акцепторного лиганда [3–6]. Планарное взаимное расположение взаимодействующих молекулярных орбиталей двух лигандов в металлокомплексе является оптимальным для осуществления внутримолекулярного переноса заряда по типу LL’CT [7, 8]. Следовательно, оптические свойства хромофоров могут быть настроены путем подбора необходимых по электронным свойствам органических линкеров, участвующих в процессе комплексообразования. В некоторых случаях орбитали ВЗМО и НСМО имеют смешанный характер за счет вклада d-орбиталей металлоцентра [8–12]. Работы [13–15] посвящены синтезу и изучению свойств радикалсодержащих LL’CT-хромофоров. Так, на примере магнитоактивных комплексов Pd(II) и Pt(II), катехолатная часть которых содержит нитронилнитроксильный радикал, показано влияние электронного строения лигандов на время жизни высокоспинового возбужденного состояния.
Существенным преимуществом LL’CT-хромофоров над хорошо изученными производными типа MLCT (metal-to-ligand charge transfer) [16–20] является возможность отказаться от использования таких дорогостоящих и токсичных металлов, как осмий и рутений. Поэтому в последние годы все больший интерес вызывает получение хромофоров на основе распространенных в природе металлов. Так, недавно исследованы оптические свойства LL’CT-хромофоров на основе катехолатов непереходных (цинк и галлий) [21–23] и переходных (никель, кобальт, медь) [24, 25] металлов. Согласно [25], плоскоквадратные гетеролептические производные меди(II) позволяют получать весьма эффективные LL’CT-хромофоры наравне с традиционными для этой темы производными Ni, Pt и Pd [25]. При этом необходимо отметить, что особенности строения и свойства соединений меди с двумя типами лигандов (донор (катехолат)/акцептор (N-донорный лиганд)) в координационной сфере металла изучают довольно давно [26–33], однако внимание исследователей к процессам переноса заряда в таких комплексах уделяется достаточно редко [34–36].
Целью настоящего исследования стали синтез и изучение оптических свойств LL′CT-хромофоров меди(II) на основе 3,6-ди-трет-бутил-о-бензохинона и N-донорных лигандов фенантролинового типа. Получены гетеролептические комплексы меди(II) состава (3,6-Cat)Cu(Phen) · THF (I · THF), [((3,6-Cat)Cu(DPQ) · THF)((3,6-Cat)Cu(DPQ))] · 3THF ([(II · THF)(II)] · 3THF) и (3,6-Cat)Cu(DPPZ) (III), где 3,6-Cat – дианион 3,6-ди-трет-бутил-о-бензохинона, Phen – фенантролин, DPQ – дипиридо[3,2-d:2',3'-f]хиноксалин, DPPZ –дипиридо[3,2-a:2',3'-c]феназин.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
ИК-спектры регистрировали на ИК-Фурье-спектрометре ФСМ-1201 (суспензии в вазелиновом масле, кюветы KBr). Элементный анализ проводили на приборе Elementar Vario El cube. Электронные спектры поглощения (ЭСП) твердых образцов в области 220–1400 нм получали для их суспензий в вазелиновом масле на спектрофотометре Shimadzu UV-2600i, оснащенном интегральной сферой ISR 2600Plus, ЭСП растворов комплексов меди(II) – на спектрофотометре СФ-2000. Спектры ЭПР записывали на спектрометре Bruker MiniScope MS-5000 Magnettech. В работе использовали следующие коммерческие реактивы: Phen, тетрагидрофуран (THF), NN-диметилформамид (DMF). Диимины DPQ и DPPZ синтезировали по известным методикам [37]. Бис(3,6-ди-трет-бутил-о-семихинолят)меди(II) ((3,6-SQ)2Cu) синтезировали в соответствии с рекомендациями, описанными в работе [38].
Синтез комплексов (3,6-Cat)Cu(Phen) (I), (3,6-Cat)Cu(DPQ) (II) и (3,6-Cat)Cu(DPPZ) (III). Операции по синтезу гетеролептических производных меди(II) проводили в анаэробных условиях. К раствору (3,6-SQ)2Cu (0.5 ммоль, 0.252 г) в 15 мл тетрагидрофурана приливали 15 мл раствора одного из нейтральных N-донорных лигандов (0.5 ммоль, 0.09 г Phen, 0.116 г DPQ) 0.141 г DPPZ) в том же растворителе. Оба раствора предварительно нагревали до температуры 50°С. Сразу после смешивания растворов наблюдали изменение цвета реакционной смеси и образование мелкокристаллических интенсивно окрашенных продуктов синего цвета. Полученные образцы собирали на фильтре Шотта и промывали 5 мл THF. Производные I–III в кристаллическом состоянии устойчивы к действию влаги и кислорода воздуха. При кристаллизации из горячего раствора THF получены кристаллы I · THF и [(II · THF)(II)] · 3THF, пригодные для рентгеноструктурного анализа. Интересно, что медленное высушивание маточного раствора с [(II · THF)(II)] · 3THF на воздухе приводит к кристаллизации бессольватного производного II, строение которого также определено методом РСА. Элементный анализ и ЭСП проводили практически сразу после выделения комплексов меди из реакционной смеси и просушивания их на воздухе. Согласно данным элементного анализа, соединения I · THF и [(II · THF)(II)] · 3THF имеют состав, согласующийся с данными РСА, полученными для монокристаллических образцов. Производное III не содержит сольватных молекул растворителя.
Выход I · THF 82%.
ИК-спектр (ν, см–1): 1624 ср, 1605 ср, 1580 ср, 1514 ср, 1430 ср, 1402 с, 1355 ср, 1346 с, 1279 ср, 1257 с, 1246 с, 1221 ср, 1206 ср, 1199 ср, 1148 с, 1105 ср, 1081 сл, 1046 сл, 1025 сл, 975 с, 939 ср, 924 сл, 908 сл, 874 ср, 840 с, 807 ср, 777 ср, 766 ср, 739 с, 697 с, 649 с, 595 ср, 488 сл, 468 сл.
Электронный спектр поглощения (суспензия в вазелиновом масле), λ, нм: 270, 297, 360, 562.
Электронный спектр поглощения (THF), λ, нм/ε, л см–1 моль–1: 616/1441.
Электронный спектр поглощения (DMF), λ, нм/ε, л см–1 моль–1: 445/1235, 546/1218.
Выход [(II · THF)(II)] · 3THF 78%.
ИК-спектр (ν, см–1): 1615 ср, 1600 ср, 1581 ср, 1485 ср, 1408 с, 1385 ср, 1342 ср, 1314 сл, 1303 сл, 1281 сл, 1257 с, 1249 с, 1211 ср, 1195 сл, 1148 ср, 1131 ср, 1086 с, 1028 сл, 978 с, 938 ср, 920 сл, 843 сл, 815 с, 765 ср, 695 с, 644 ср, 596 ср, 568 сл, 489 сл, 466 ср.
Электронный спектр поглощения (суспензия в вазелиновом масле), λ, нм: 266, 340, 410, 620.
Электронный спектр поглощения (THF), λ, нм/ε, л см–1 моль–1: 640/1342.
Электронный спектр поглощения (DMF), λ, нм/ε, л см–1 моль–1: 445/1166, 568/1424.
Выход III 65%.
ИК-спектр (ν, см–1): 1621 ср, 1601 ср, 1585 ср, 1488 с, 1420 ср, 1404 с, 1352 с, 1341 с, 1323 сл, 1305 сл, 1284 ср, 1265 с, 1251 с, 1204 ср, 1145 ср, 1137 ср, 1121 сл, 1092 сл, 1074 с, 1045 сл, 1034 сл, 980 с, 941 с, 901 сл, 845 сл, 819 с, 809 ср, 779 ср, 755 с, 731 с, 670 с, 651 с, 639 сл, 617 ср, 600 ср, 576 ср, 557 сл, 509 сл, 492 ср.
Электронный спектр поглощения (суспензия в вазелиновом масле), λ, нм: 257, 298, 382, 580.
Электронный спектр поглощения (THF), λ, нм/ε, л см–1 моль–1: 644/1483.
РСА. Данные рентгеновской дифракции для комплексов I · THF и II получены на рентгеновском пучке станции “Белок” Курчатовского центра синхротронного излучения в Национальном исследовательском центре “Курчатовский институт” (Москва, Россия) в режиме φ-сканирования с использованием CCD-детектора Rayonix SX165 при 100 K (λ ~ 0.7527 Å) [39]. Определение и уточнение параметров элементарной ячейки, интегрирование отражений, а также поправка на поглощение интенсивности рефлексов выполнены с использованием программного пакета XDS [40]. Данные рентгеновской дифракции для комплекса [(II · THF)(II)] · 3THF получены на дифрактометре Bruker D8 Venture Photon в режиме φ- и ω-сканирования в Центре коллективного пользования ИОНХ РАН при температуре 100 K (λ(MoKα) = = 0.71073 Å, микрофокусный источник рентгеновского излучения Incoatec IµS 3.0). Первичное индицирование, уточнение параметров элементарной ячейки и интегрирование отражений выполнены с помощью пакета программ Bruker APEX3 [41], с учетом поправки на поглощение интенсивности отражений – по программе SADABS [41]. Структура расшифрована прямым методом [42] и уточнена полноматричным методом наименьших квадратов по F2 [43] в анизотропном приближении для всех неводородных атомов. В случае структуры II разупорядоченная часть атомов сольватного тетрагидрофурана была уточнена с применением ограничений на геометрические (инструкция SADI) и тепловые смещения атомов (инструкции SIMU, RIGU). Атомы водорода помещали в рассчитанные положения и уточняли в модели “наездника” с Uiso(H) = 1.5Ueq(C) для атомов водорода метильных групп и Uiso(H) = = 1.2Ueq(C) для остальных атомов водорода. Расчеты выполнены с помощью программного пакета SHELXTL [43] и программы визуализации и обработки структурных данных OLEX2 [44].
Кристаллографические данные для соединений I · THF, [(II · THF)(II)] · 3THF и II депонированы в Кембриджском банке структурных данных (№ 2250975–2250977 соответственно), deposit@ccdc.cam.ac.uk или http://www.ccdc.cam.ac.uk/ data_request/cif.
Методы квантово-химических расчетов. Расчеты проводили при помощи программы Gaussian 09 [45] методом теории функционала плотности (DFT) в приближении B3LYP/6-311++G(2d,2p) c поправками Гримме D3BJ. Локализацию стационарных точек осуществляли путем полной оптимизации геометрии молекулярных структур. Отсутствие мнимых частот свидетельствует о том, что молекулы находятся в минимуме потенциальной энергии.
Циклическая вольтамперометрия (ЦВА). Потенциалы окисления измеряли методом циклической вольтамперометрии в трехэлектродной ячейке с использованием потенциостата Smartstat PS-50 в атмосфере аргона. В качестве рабочего электрода использовали стеклоуглеродный электрод (d = 2 мм), в качестве вспомогательного электрода – платиновую проволоку, в качестве электрода сравнения – Ag/AgCl/KCl(нас.) с водонепроницаемой диафрагмой. Потенциалы пересчитаны относительно пары FcH/FcH+ (E1/2 = 0.43 В). Скорость развертки потенциала 0.2 В/с. Фоновый электролит – 0.1 М Bu4NClO4 (99%, Aldrich), дважды перекристаллизованный из водного EtOH и высушенный в вакууме при 50°C. Концентрация комплексов составляла 2 ммоль/л.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Взаимодействие (3,6-SQ)2Cu с нейтральными N-донорными лигандами фенантролинового типа в среде THF в анаэробных условиях приводит к вытеснению одной молекулы хинона (3,6-Q) из координационной сферы меди. В результате образуются синие игольчатые кристаллы комплексов меди(II) состава (3,6-Cat)Cu(Phen) · THF (I · THF), [((3,6-Cat)Cu(DPQ) · THF)((3,6-Cat)Cu(DPQ))] · · 3THF ([(II · THF)(II)] · 3THF) и (3,6-Cat)Cu(DPPZ) (III). Медленное упаривание маточного раствора [(II · THF)(II)] · 3THF на воздухе приводит к формированию кристаллов бессольватного производного (3,6-Cat)Cu(DPQ) (II). Аналитическая чистота и состав комплексов I · THF, [(II · THF)(II)] · 3THF и III, выделенных из реакционной смеси, подтверждены данными ИК-спектроскопии и элементного анализа. Полученные соединения парамагнитны и плохо растворяются в большинстве органических растворителей. Спектры ЭПР кристаллических образцов представляют собой широкий синглет с gi = = 2.064, 2.065 и 2.057 для комплексов I · THF, [(II · THF)(II)] · 3THF и III соответственно. Полученные значения хорошо согласуются с конфигурацией d9 иона двухвалентной меди в плоскоквадратном координационном окружении [46].
Молекулярное и кристаллическое строение производных I · THF, [(II · THF)(II)] · 3THF и II определено методом рентгеноструктурного анализа. Молекулярные структуры изображены на рис. 1–3. Кристаллографические данные и параметры рентгеноструктурных экспериментов I · THF, [(II · THF)(II)] · 3THF и II представлены в табл. 1, избранные длины связей – в табл. 2. Комплекс I · THF кристаллизуется в моноклинной пр. гр. P21/c. Независимая часть кристаллической ячейки I · THF содержит две молекулы комплекса и две сольватные молекулы THF (рис. 1). Атом меди(II) в I · THF имеет слегка искаженное плоско-квадратное окружение. Двугранный угол между плоскостями катехолатного O(1)C(27)C(28)O(2) и фенантролинового N(1)C(45)C(43)N(2) лигандов составляет 7.10° (во второй молекуле комплекса между плоскостями O(3)C(1)C(6)O(4) и N(3)C(19)C(23)N(4) – 7.2°).
Рис. 1.
Независимая часть кристаллической ячейки I · THF. Тепловые эллипсоиды приведены с 50%-ной вероятностью. Атомы водорода и сольватные молекулы THF не показаны.
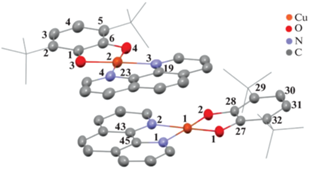
Рис. 2.
Независимая часть кристаллической ячейки [(II · THF)(II)] · 3THF. Тепловые эллипсоиды приведены с 50%-ной вероятностью. Атомы водорода и сольватные молекулы THF не показаны.
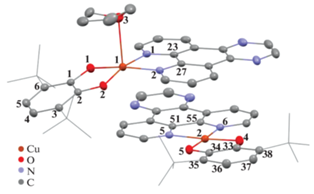
Рис. 3.
Независимая часть кристаллической ячейки II. Тепловые эллипсоиды приведены с 50%-ной вероятностью. Атомы водорода не показаны.
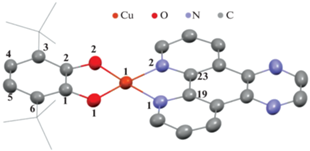
Таблица 1.
Кристаллографические данные, параметры эксперимента и уточнения структур I · THF, [(II · THF)(II)] · 3THF и II
| Параметр | Значение | ||
|---|---|---|---|
| I · THF | [(II · THF)(II)] · 3THF | II | |
| T, K | 100(2) | 100(2) | 100(2) |
| Сингония | Моноклинная | Триклинная | Моноклинная |
| Пр. гр. | P21/c | P-1 | C2/c |
| а, Å | 13.372(2) | 14.8024(6) | 29.351(13) |
| b, Å | 17.539(3) | 15.3550(6) | 13.782(4) |
| c, Å | 22.621(4) | 16.0972(6) | 13.380(6) |
| α, град | 90 | 74.8186(14) | 90 |
| β, град | 99.890(19) | 66.7282(13) | 115.594(17) |
| γ, град | 90 | 84.8508(14) | 90 |
| V, Å3 | 5226.5(15) | 3243.4(2) | 4881(3) |
| Z | 8 | 2 | 8 |
| ρвыч, г/см3 | 1.363 | 1.352 | 1.404 |
| µ, мм–1 | 1.006 | 0.719 | 1.073 |
| θmin–θmax, град | 1.637–26.581 | 1.898–30.536 | 2.247–28.388 |
| Число наблюдаемых отражений | 64332 | 54629 | 18133 |
| Число независимых отражений | 9147 | 19778 | 5134 |
| Rint | 0.1039 | 0.0384 | 0.0294 |
| S(F2) | 1.024 | 1.030 | 1.033 |
| R1/wR2 (I > 2σ(I)) | 0.0505/0.1198 | 0.0385/0.0917 | 0.0455/0.1174 |
| R1/wR2 (по всем параметрам) | 0.0869/0.1383 | 0.0609/0.0996 | 0.0555/0.1246 |
| Коэффициент поглощения | n/a | n/a | 0.0022(4) |
| Δρmax/Δρmin, e Å–3 | 0.527/–0.485 | 0.585/–0.535 | 0.556/–0.792 |
Таблица 2.
Избранные длины связей в комплексах I · THF, [(II · THF)(II)] · 3THF и II
| I · THF | [(II · THF)(II)] · 3THF | II | |||
|---|---|---|---|---|---|
| Связь | d, Å | Связь | d, Å | Связь | d, Å |
| Cu(1)–O(2) | 1.858(3) | Cu(1)–O(1) | 1.8789(11) | Cu(1)–O(1) | 1.863(2) |
| Cu(1)–O(1) | 1.862(2) | Cu(1)–O(2) | 1.8849(10) | Cu(1)–O(2) | 1.8692(18) |
| Cu(1)–N(1) | 2.001(3) | Cu(1)–N(1) | 2.0094(13) | Cu(1)–N(2) | 1.996(2) |
| Cu(1)–N(2) | 2.003(3) | Cu(1)–N(2) | 2.0200(13) | Cu(1)–N(1) | 2.000(2) |
| O(1)–C(27) | 1.351(5) | Cu(1)–O(3) | 2.5140(12) | O(1)–C(1) | 1.350(3) |
| O(2)–C(28) | 1.365(4) | O(1)–C(1) | 1.3506(18) | O(2)–C(2) | 1.354(3) |
| C(27)–C(32) | 1.411(5) | O(2)–C(2) | 1.3473(18) | C(1)–C(6) | 1.404(3) |
| C(27)–C(28) | 1.417(6) | C(1)–C(6) | 1.405(2) | C(1)–C(2) | 1.426(3) |
| C(28)–C(29) | 1.397(6) | C(1)–C(2) | 1.427(2) | C(2)–C(3) | 1.405(3) |
| C(29)–C(30) | 1.409(5) | C(2)–C(3) | 1.407(2) | C(3)–C(4) | 1.393(4) |
| C(30)–C(31) | 1.380(6) | C(3)–C(4) | 1.399(2) | C(4)–C(5) | 1.388(4) |
| C(31)–C(32) | 1.390(6) | C(4)–C(5) | 1.388(2) | C(5)–C(6) | 1.395(4) |
| Cu(2)–O(4) | 1.854(3) | C(5)–C(6) | 1.401(2) | ||
| Cu(2)–O(3) | 1.873(2) | Cu(2)–O(4) | 1.8655(11) | ||
| Cu(2)–N(4) | 1.985(3) | Cu(2)–O(5) | 1.8676(10) | ||
| Cu(2)–N(3) | 2.002(3) | Cu(2)–N(6) | 1.9943(13) | ||
| O(3)–C(1) | 1.357(5) | Cu(2)–N(5) | 2.0011(13) | ||
| O(4)–C(6) | 1.359(4) | O(4)–C(33) | 1.3536(17) | ||
| C(1)–C(2) | 1.414(5) | O(5)–C(34) | 1.3535(17) | ||
| C(1)–C(6) | 1.414(6) | C(33)–C(38) | 1.404(2) | ||
| C(2)–C(3) | 1.393(6) | C(33)–C(34) | 1.423(2) | ||
| C(3)–C(4) | 1.377(6) | C(34)–C(35) | 1.407(2) | ||
| C(4)–C(5) | 1.411(5) | C(35)–C(36) | 1.398(2) | ||
| C(5)–C(6) | 1.405(6) | C(36)–C(37) | 1.388(2) | ||
| C(37)–C(38) | 1.401(2) | ||||
Соединение [(II · THF)(II)] · 3THF кристаллизуется в триклинной группе симметрии P-1. Независимая часть кристаллической ячейки [(II · THF)(II)] · 3THF содержит две молекулы комплекса (на атом меди одной из них координирована молекула THF) и три сольватные молекулы THF (рис. 2). Координационное окружение Cu(1) образует тетрагональную пирамиду, в основании которой лежат атомы кислорода катехолатного лиганда и атомы азота DPQ. Координация молекулы THF на ион меди приводит к “вытягиванию” Cu(1) из плоскости O(1)O(2)N(1)N(2) на 0.164 Å, а сумма углов, образованных связями вокруг атома меди, составляет 358.37°, что близко по значению к идеальному плоскоквадратному основанию (360°). Диэдральный угол между плоскостями O(1)C(1)C(1)O(2) и N(5)C(51)C(55)N(6) составляет 18.53°. Координационное окружение Cu(2) – плоский квадрат, двугранный угол между плоскостями O(4)C(33)C(34)O(5) и N(5)C(51)C(55)N(6) равен 4.28°.
Согласно данным РСА, бессольватный комплекс меди II кристаллизуется в моноклинной пр. гр. C2/c. Атом меди в II имеет плоско-квадратное окружение.
Длины связей С–О в хиноновых фрагментах I · THF, ([(II · THF)(II)] · 3THF и II лежат в интервале 1.347–1.365 Å, что позволяет однозначно характеризовать структуру лиганда как катехолатную [47, 48]. Длины связей Cu–O во всех комплексах (1.854–1.884 Å) также соответствуют значениям, характерным для катехолатных комплексов меди(II) [32, 49]. Длины связей Cu–N в производных I · THF, ([(II · THF)(II)] · 3THF и II значительно превышают сумму ковалентных радиусов элементов (1.11 и 0.74 Å для Cu и N соответственно [50]), что указывает на донорно-акцепторную природу связывания фенантролиновых лигандов с катионами меди(II).
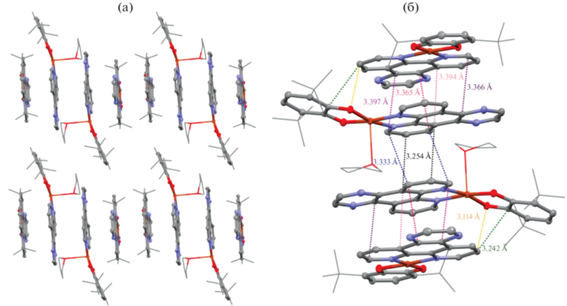
Катехолаты меди с N-донорными лигандами фенантролинового типа плохо растворимы в большинстве доступных органических растворителей. Низкая растворимость соединений обусловлена наличием большого числа коротких межмолекулярных контактов в кристаллических образцах. По данным рентгеноструктурного анализа, в производном I · THF наблюдается диполь-дипольное взаимодействие между донорной и акцепторной частями двух соседних молекул (рис. 4). В комплексе II присутствуют контакты, представляющие собой π–π-взаимодействия между лигандами фенантролинового типа соседних молекул (рис. 4).
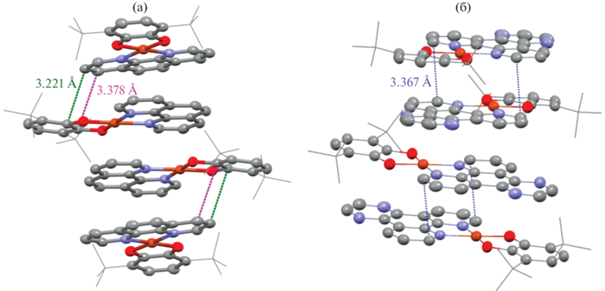
Производное [(II · THF)(II)] · 3THF характеризуется наличием межмолекулярных контактов обоих типов (рис. 5), присутствующих в соединениях I · THF и II. В кристаллической упаковке наблюдается парное чередование комплексов II · THF и II (рис. 5).
Рис. 5.
Вид упаковки [(II · THF)(II)] · 3THF вдоль вектора (001) (а); межмолекулярные контакты в [(II · THF)(II)] · 3THF (б).
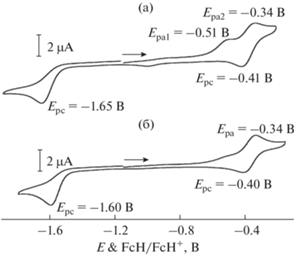
Электрохимические свойства комплексов I · THF и [(II · THF)(II)] · 3THF в растворе дихлорметана изучены методом циклической вольтамперометрии (рис. 6). Исследовать в этих условиях комплекс III не удалось из-за его недостаточной растворимости в CH2Cl2 и других органических растворителях. В исследуемых катехолатных комплексах присутствуют три редокс-центра: ион меди(II), диоксоленовый и фенантролиновый лиганды. На кривых ЦВА для обоих комплексов наблюдается обратимое окисление (E1/2 = –0.37 В), связанное с одноэлектронным окислением катехолатного лиганда Cat2–/SQ–, а также необратимое восстановление при потенциале ⁓ –1.6 В (для I · THF E1/2 = –1.61 В (Eonset = –1.54 В), для [(II · THF)(II)] · 3THF E1/2 = –1.57 В (Eonset = –1.51 В)), обусловленное восстановлением фенантролинового лиганда. Следует отметить, что в случае комплекса I · THF процесс окисления является двухступенчатым (пиковые значения потенциалов окисления равны –0.51 В (Eonset = –0.63 В) и –0.34 В), в то время как обратный процесс на циклической вольтамперограмме протекает в одну стадию (Ep = –0.41 В). Это можно объяснить сохранением димерной структуры комплекса в растворе хлористого метилена. Аналогичная ассоциация наблюдается и для комплекса [(II · THF)(II)] · 3THF (Eonset = –0.64 В), однако она менее выражена. На основании полученных значений потенциалов полуволн были рассчитаны энергии граничных орбиталей [51] – ВЗМО/НСМО, которые составили –4.43/–3.19 эВ (I · THF) и –4.43/–3.24 эВ ([(II · THF)(II)] · 3THF).
Рис. 6.
Кривые ЦВА комплексов I · THF (а) и [(II · THF)(II)] · 3THF (б) (CH2Cl2, Ar, Ag/AgCl/KCl(нас.), С = 2 ммоль/л, 0.2 М NBu4ClO4, скорость развертки 50 мВ/с).
Электронные спектры поглощения соединений были получены как для суспензий твердых образцов комплексов I · THF, [(II · THF)(II)] · 3THF и III в вазелиновом масле (рис. 7), так и для растворов производных в THF и DMF, за исключением раствора III в DMF (рис. 8). Для всех кристаллических образцов наблюдается схожий характер электронных спектров поглощения (рис. 7). Данные спектры характеризуются наличием широкой полосы переноса заряда в видимой и ближней ИК-областях спектра (450–800 нм).
Рис. 7.
Электронные спектры поглощения суспензий I · THF, [(II · THF)(II)] · 3THF и III в вазелиновом масле.
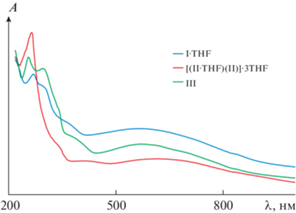
Рис. 8.
Электронные спектры поглощения растворов комплексов I · THF (С = 5.6 × 10–4 моль/л), [(II · THF)(II)] · 3THF (С = 5.6 × 10–4 моль/л в пересчете на бессольватный комплекс II) и III (С = = 6.4 × 10–4 моль/л).
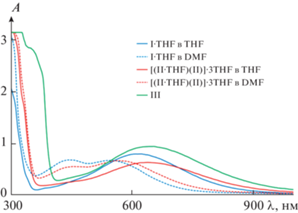
Электронные спектры поглощения растворов I · THF, [(II · THF)(II)] · 3THF и III также демонстрируют наличие широкой полосы переноса заряда, захватывающей видимую и ближнюю ИК-области спектра (рис. 8). Ключевой особенностью спектров поглощения комплексов I · THF и [(II · THF)(II)] · 3THF является наличие выраженного отрицательного сольватохромного эффекта, проявляющегося в гипсохромном смещении на ⁓70 нм длинноволновых пиков поглощения при переходе от тетрагидрофурана к более полярному [52] диметилформамиду. Схожее сольватохромное поведение наблюдалось ранее для родственного 3,6-ди-трет-бутилкатехолата меди(II), содержащего 4,4'-ди-трет-бутил-2,2'-дипиридил в качестве акцепторного лиганда (583 (THF) и 523 нм (DMF)). При этом введение фенантролина вместо дипиридила приводило к красному сдвигу полосы поглощения в электронном спектре катехолатного комплекса меди более чем на 40 нм. Необходимо отметить, что использование в качестве дииминового лиганда фенантролинов, аннелированных пиразиновым или хиноксалиновым фрагментом, сопровождается дополнительным батохромным сдвигом длинноволновой полосы поглощения в ряду комплексов I (616 нм в ТHF)–II (640 нм в ТHF)–III (644 нм в ТHF).
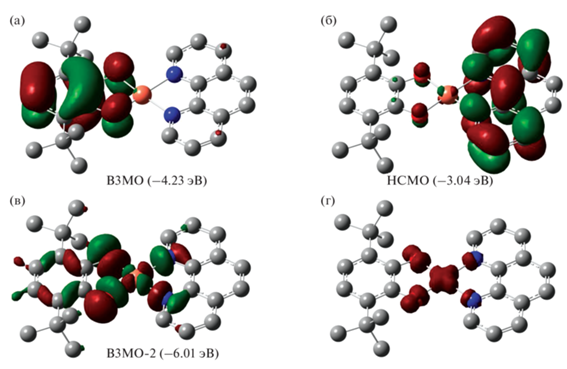
Для комплексов I–III выполнены квантово-химические расчеты методом DFT для дублетного спинового состояния на уровне B3LYP/6-311++G(2d,2p). Оптимизированная геометрия хорошо воспроизводит данные, полученные в рамках рентгеноструктурного анализа. Анализ молекулярных орбиталей показал, что граничные орбитали преимущественно располагаются на редокс-активных лигандах. В соединении I ВЗМО (–4.23 эВ) и НСМО (–3.04 эВ) располагаются на донорном катехолатном и акцепторном дииминовом фрагментах соответственно (рис. 9). Наивысшая для плоскоквадратной конфигурации иона двухвалентной меди частично заполненная dx2–y2-орбиталь в случае I расположена на ВЗМО-2 (–6.01 эВ). Эта орбиталь ответственна за дублетное спиновое состояние комплекса меди (рис. 9). Щель ВЗМО–НСМО, по данным квантово-химических расчетов для I (1.19 эВ), хорошо согласуется с аналогичной величиной, полученной на основе электрохимически измерений (1.24 эВ). Таким образом, на основании анализа состава граничных орбиталей можно утверждать, что интенсивная длинноволновая полоса поглощения в электронном спектре соединения I соответствует переносу заряда между редокс-активными лигандами, связанными с атомом металла, а сам комплекс меди следует отнести к LL’CT-хромофорам.
Рис. 9.
Виды граничных орбиталей (а–в) и распределение спиновой плотности (г) для комплекса I по данным DFT-расчетов.
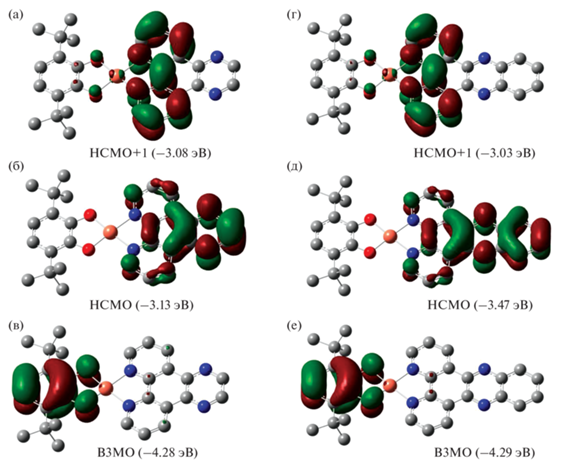
Вид граничных орбиталей комплексов II и III в целом повторяет картину, наблюдаемую для соединения I (рис. 10). ВЗМО в II и III имеют практически такую же энергию, как и в комплексе I, и располагаются на катехолатных лигандах. При этом орбиталь, принадлежащая фенантролиновому фрагменту дииминового лиганда, становится НСМО+1, а низшая свободная орбиталь занимает преимущественно аннелированный к фенантролину пиразиновый или хиноксалиновый фрагмент в II и III соответственно. НСМО в указанных соединениях располагается значительно ниже по энергии, что приводит к сужению щели ВЗМО–НСМО, это закономерно отражается на батохромном сдвиге в электронном спектре комплексов II и III по сравнению с фенантролиновым комплексом I.
Таким образом, синтезированы новые гетеролептические плоско-квадратные 3,6-ди-трет-бутилкатехолатные комплексы меди с координированными на атом металла фенантролиновыми лигандами. Установлено, что электронный спектр полученных комплексов демонстрирует поглощение, отвечающее возникновению LL’CT-переходов и обусловленное наличием двух разнозаряженных редокс-активных лигандов – донорного катехолатного и акцепторного дииминового.
Список литературы
Sobottka S., Nößler M., Ostericher A.L. et al. // Chem. Eur. J. 2020. V. 26. P. 1314. https://doi.org/10.1002/chem.201903700
Romashev N.F., Abramov P.A., Bakaev I.V. et al. // Inorg. Chem. 2022. V. 61. P. 2105. https://doi.org/10.1021/acs.inorgchem.1c03314
Shultz D.A., Stephenson R., Kirk M.L. // Dalton Trans. 2023. V. 52. P. 1970. https://doi.org/10.1039/D2DT03385B
Yang J., Kersi D.K., Giles L.J. et al. // Inorg. Chem. 2014. V. 53. P. 4791. https://doi.org/10.1021/ic500217y
Kramer W.W., Cameron L.A., Zarkesh R.A. et al. // Inorg. Chem. 2014. V. 53. P. 8825. https://doi.org/10.1021/ic5017214
Shavaleev N.M., Davies E.S., Adams H. et al. // Inorg. Chem. 2008. V. 47. P. 1532. https://doi.org/10.1021/ic701821d
Benedix R., Hennig H., Kunkely H. et al. // Chem. Phys. Lett. 1990. V. 175. P. 483. https://doi.org/10.1016/0009-2614(90)85568-W
Cameron L.A., Ziller J.W., Heyduk A.F. // Chem. Sci. 2016. V. 7. P. 1807. https://doi.org/10.1039/C5SC02703A
Ghosh P., Begum A., Herebian D. et al. // Angew. Chem. Int. Ed. 2003. V. 42. P. 563. https://doi.org/10.1002/anie.200390162
Best J., Sazanovich I.V., Adams H. et al. // Inorg. Chem. 2010. V. 49. P. 10041. https://doi.org/10.1021/ic101344t
Scattergood P.A., Jesus P., Adams H. et al. // Dalton Trans. 2015. V. 44. P. 11705. https://doi.org/10.1039/C4DT03466J
Yang J., Kersi D.K., Richers C.P. et al. // Inorg. Chem. 2018. V. 57. P. 13470. https://doi.org/10.1021/acs.inorgchem.8b02087
Kirk M.L., Shultz D.A., Marri A.R. et al. // J. Am. Chem. Soc. 2022. V. 144. P. 21005. https://doi.org/10.1021/jacs.2c09680
Kirk M.L., Shultz D.A., Hewitt P. et al. // Chem. Sci. 2021. V. 12. P. 13704. https://doi.org/10.1039/D1SC02965G
Kirk M.L., Shultz D.A., Chen J. et al. // J. Am. Chem. Soc. 2021. V. 143. P. 10519. https://doi.org/10.1021/jacs.1c04149
Hagberg D.P., Yum J.-H., Lee H. et al. // J. Am. Chem. Soc. 2008. V. 130. P. 6259. https://doi.org/10.1021/ja800066y
García-Cañadas J., Meacham A.P., Peter L.M. et al. // Angew. Chem. Int. Ed. 2003. V. 42. P. 3011. https://doi.org/10.1002/anie.200351338
Ward M.D. // J. Solid State Electrochem. 2005. V. 9. P. 778. https://doi.org/10.1007/s10008-005-0668-4
Sekar N., Gehlot V.Y. // Resonance. 2010. V. 15. P. 819. https://doi.org/10.1007/s12045-010-0091-8
Atallah H., Taliaferro C.M., Wells K.A. et al. // Dalton Trans. 2020. V. 49. P. 11565. https://doi.org/10.1039/D0DT01765E
Ершова И.В., Малеева А.В., Айсин Р.Р. и др. // Изв. Академии наук. Сер. хим. 2023. Т. 72. С. 193.
Maleeva A.V., Ershova I.V., Trofimova O.Y. et al. // Mendeleev Commun. 2022. V. 32. P. 83. https://doi.org/10.1016/j.mencom.2022.01.027
Малеева А.В., Трофимова О.Ю., Якушев И.А. и др. // Коорд. химия. 2023. Т. 49 (в печати).
Pashanova K.I., Bitkina V.O., Yakushev I.A. et al. // Molecules. 2021. V. 26. P. 4622. https://doi.org/10.3390/molecules26154622
Pashanova K.I., Ershova I.V., Trofimova O.Y. et al. // Molecules. 2022. V. 27. P. 8175. https://doi.org/10.3390/molecules27238175
Rall J., Wanner M., Albrecht M. et al. // Chem. Eur. J. 1999. V. 5. P. 2802. https://doi.org/10.1002/(SICI)1521-3765(19991001)5:10<2802::AID-CHEM2802>3.0.CO;2-5
Abakumov G.A., Krashilina A.V., Cherkasov V.K. et al. // Russ. Chem. Bull. 2001. V. 50. P. 2193. https://doi.org/10.1023/A:1015022006445
Kaizer J., Zsigmond Z., Ganszky I. et al. // Inorg. Chem. 2007. V. 46. P. 4660. https://doi.org/10.1021/ic062309a
Ovcharenko V.I., Gorelik E.V., Fokin S.V. et al. // J. Am. Chem. Soc. 2007. V. 129. P. 10512. https://doi.org/10.1021/ja072463b
Fursova E.Yu., Ovcharenko V.I., Gorelik E.V. et al. // Russ. Chem. Bull. 2009. V. 58. P. 1139. https://doi.org/10.1007/s11172-009-0148-6
Davidson R.A., Hao J., Rheingold A.L. et al. // Polyhedron. 2017. V. 133. P. 348. https://doi.org/10.1016/j.poly.2017.05.038
Cherkasova A.V., Kozhanov K.A., Zolotukhin A.A. et al. // Russ. J. Coord. Chem. 2019. V. 45. P. 489. https://doi.org/10.1134/S1070328419070029
Kuropatov V.A., Cherkasova A.V., Martyanov K.A. et al. // Eur. J. Inorg. Chem. 2021. V. 2021. P. 3292. https://doi.org/10.1002/ejic.202100517
Buchanan R.M., Wilson-Blumenberg C., Trapp C. et al. // Inorg. Chem. 1986. V. 25. P. 3070. https://doi.org/10.1021/ic00237a029
Verma P., Weir J., Mirica L. et al. // Inorg. Chem. 2011. V. 50. P. 9816. https://doi.org/10.1021/ic200958g
Lakk-Bogáth D., Csonka R., Lorencz N. et al. // Polyhedron. 2015. V. 102. P. 185. https://doi.org/10.1016/j.poly.2015.09.026
van der Tol E.B., van Ramesdonk H.J., Verhoeven J.W. et al. // Chem. Eur. J. 1998. V. 4. P. 2315. https://doi.org/10.1002/(SICI)1521-3765(19981102)4: 11<2315::AID-CHEM2315>3.0.CO;2-E
Abakumov G.A., Cherkasov V.K., Bubnov M.P. et al. // Russ. Chem. Bull. 1992. V. 41. P. 1813. https://doi.org/10.1007/BF00863815
Svetogorov R.D., Dorovatovskii P.V., Lazarenko V.A. // Cryst. Res. Technol. 2020. V. 55. P. 1900184. https://doi.org/10.1002/crat.201900184
Kabsch W. // Acta Crystallogr., Sect. D. 2010. V. 66. P. 125. https://doi.org/10.1107/S0907444909047337
Bruker. APEX3, SAINT and SADABS. Bruker AXS Inc., Madison, Wisconsin, USA. 2016.
Sheldrick G.M. // Acta Crystallogr. 2015. V. A71. P. 3. https://doi.org/10.1107/S2053273314026370
Sheldrick G.M. // Acta Crystallogr. 2015. V. C71. P. 3. https://doi.org/10.1107/S2053229614024218
Dolomanov O.V., Bourhis L.J., Gildea R.J. et al. // J. Appl. Crystallogr. 2009. V. 42. P. 339. https://doi.org/10.1107/S0021889808042726
Frisch M.J., Trucks G.W., Schlegel H.B. et al. // Revision D.01. Gaussian, Inc. Wallingford CT. 2013.
Hathaway B.J., Billing D.E. // Coord. Chem. Rev. 1970. V. 5. P. 143. https://doi.org/10.1016/S0010-8545(00)80135-6
Piskunov A.V., Maleeva A.V., Mescheryakova I.N. et al. // Eur. J. Inorg. Chem. 2012. P. 4318. https://doi.org/10.1002/ejic.201200535
Chegerev M.G., Piskunov A.V., Maleeva A.V. et al. // Eur. J. Inorg. Chem. 2016. P. 3813. https://doi.org/10.1002/ejic.201600501
Davidson R.A., Hao J., Rheingold A.L. et al. // Polyhedron. 2017. V. 136. P. 176. https://doi.org/10.1016/j.poly.2017.10.003
Batsanov S.S. // Russ. J. Inorg. Chem. 1991. V. 36. P. 1694.
Zairov R.R., Yagodin A.V., Khrizanforov M. et al. // J. Nanopart. Res. 2019. V. 21. P. 12.
Райхардт К. Растворители и эффекты среды в органической химии. М.: Мир, 1991. 764 с.
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии