

Журнал неорганической химии, 2023, T. 68, № 9, стр. 1226-1234
Основность и гидридодонорная способность гидридного комплекса палладия(II) с диариламидо-бис-фосфиновым пинцетным лигандом
В. А. Куликова a, В. А. Киркина a, Е. И. Гуцул a, З. Н. Гафуров b, А. А. Кагилев b, c, И. Ф. Сахапов b, Д. Г. Яхваров b, c, О. А. Филиппов a, Е. С. Шубина a, Н. В. Белкова a, *
a Институт элементоорганических соединений им. А.Н. Несмеянова РАН
119991 Москва, ул. Вавилова, 28, Россия
b Институт органической и физической химии им. А.Е. Арбузова ФИЦ
“Казанский научный центр РАН”
420088 Казань, ул. Арбузова, 8, Россия
c Химический институт им. А.М. Бутлерова, Казанский (Приволжский)
федеральный университет
420008 Казань, ул. Кремлевская, 18, Россия
* E-mail: nataliabelk@ineos.ac.ru
Поступила в редакцию 18.04.2023
После доработки 13.06.2023
Принята к публикации 13.06.2023
- EDN: WPORKM
- DOI: 10.31857/S0044457X23600858
Аннотация
Ключевыми стадиями реакций (де)гидрирования, дегидросочетания, получения H2, восстановления CO2 с участием гидридов переходных металлов являются перенос гидрид-иона и перенос протона, а катализаторами данных превращений часто выступают комплексы с бифункциональными лигандами. Целью настоящей работы было исследование гидридодонорных свойств пинцетного гидрида палладия(II) (PNP)PdH (1; PNP = бис(2-диизопропилфосфино-4-метилфенил)амид). Для этого методами ИК- и ЯМР-спектроскопии исследовано его взаимодействие с кислотами Льюиса (BF3 · Et2O, B(C6F5)3) с привлечением квантово-химических расчетов (DFT/M06/def2-TZVP), а также использованы предложенные в литературе корреляции потенциалов электрохимического восстановления соответствующих катионов с термодинамической гидридностью. [(PNP)Pd(MeCN)][BF4] претерпевает необратимое двухэлектронное восстановление в ацетонитриле ($E_{p}^{{{\text{red}}}}$ = –1.82 В). Для полученного потенциала корреляции дают завышенное значение гидридодонорной способности $\Delta G_{{{{{\text{H}}}^{--}}}}^{^\circ }.$ Установлено, что реакция 1 с борсодержащими кислотами Льюиса неожиданно приводит к протонированию атома азота пинцетного лиганда примесью воды, а не к взаимодействию с гидридным лигандом. По данным DFT-расчетов, сродство к протону атома азота значительно выше, чем PdH, что обусловливает его более высокую активность в процессах протонирования.
ВВЕДЕНИЕ
Гидридные комплексы переходных металлов являются важным классом металлоорганических соединений как для лабораторного, так и для промышленного применения в различных каталитических и стехиометрических превращениях, включая реакции (де)гидрирования, дегидросочетания, гидросилилирования, изомеризации олефинов и гидроформилирования [1–3]. С точки зрения современных концепций водородной энергетики и возобновляемых источников энергии особый интерес представляет использование гидридов переходных металлов для электрохимического получения H2 [4, 5] и химического или электрокаталитического восстановления CO2 [6–11]. Ключевыми стадиями этих реакций, как и реакций (де)гидрирования, является перенос гидрид-иона (H–) от гидридного металлокомплекса к субстрату, а также перенос протона (H+) к металлу, лиганду или субстрату. Такая реакционная способность гидридов переходных металлов хорошо известна (Схема 1 ). Является ли связь M–H источником протона или гидрид-иона зависит от свойств металлокомплекса и реагента-партнера. Нами показано, что полярность связи М–Н настраивается на стадии образования нековалентного комплекса, предшествующей гетеролитической диссоциации связи М–Н [12].
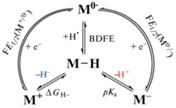
Схема 1 .
Общепризнанной характеристикой основности, в том числе металлокомплексов, является константа равновесия реакции протонирования pKb, хотя часто ее заменяют константой диссоциации соответствующей сопряженной кислоты pKa. В качестве аналогичной по смыслу характеристики реакционной способности гидрида металла как источника H– в последние годы используют термодинамическую гидридность – свободную энергию Гиббса для реакции отрыва гидрид-иона – $\Delta G_{{{{{\text{H}}}^{--}}}}^{^\circ }$ или HDA (hydride donating ability) [13, 14]. Разрыв связи металл–гидрид также может происходить посредством гомолитической диссоциации атома водорода (H•), характеристикой которой является свободная энергия диссоциации связи (BDFE). Эти три характеристики – гидридность $\Delta G_{{{{{\text{H}}}^{--}}}}^{^\circ }$, BDFE и pKa гидрида металла – взаимосвязаны через термодинамические циклы, включающие одноэлектронные потенциалы восстановления исходного комплекса металла [M]n+ и известные термодинамические параметры для гидрида, атома водорода и протона (уравнения (1)–(5)) [6, 13]:
(1)
${{\left[ {{\text{M}}--{\text{H}}} \right]}^{{(n}}}^{{--1) + }} \rightleftharpoons {{{\text{M}}}^{{(n}}}^{{--1) + }} + {{{\text{H}}}^{ \bullet }}~\,\,\,\,\,\,\,{\text{BDFE}},\,\,\,\,$(2)
${{{\text{M}}}^{{(n}}}^{{--1) + }} \rightleftharpoons {{{\text{M}}}^{n}}^{ + } + {\text{ }}{{{\text{e}}}^{--}}\,\,\,\,n{\text{F}}{{E}_{{1/2}}}({{{\text{M}}}^{n}}{{^{{ + /(n}}}^{{--1) + }}}),\,\,\,\,$(3)
${{{\text{H}}}^{ \bullet }} + {\text{ }}{{{\text{e}}}^{--}} \rightleftharpoons {{{\text{H}}}^{--}}\,\,\,\,\,\,\,\,\,\Delta G_{{{{{\text{H}}}^{{{ \bullet }}}}{\text{/}}{{{\text{H}}}^{--}}}}^{^\circ },\,\,\,\,$(4)
${{\left[ {{\text{M}}--{\text{H}}} \right]}^{{(n}}}^{{--1) + }} \rightleftharpoons {{{\text{M}}}^{n}}^{ + } + {\text{ }}{{{\text{H}}}^{--}}\,\,\,\,\Delta G_{{{{{\text{H}}}^{--}}}}^{^\circ },\,\,\,\,$(5)
$\Delta G_{{{{{\text{H}}}^{--}}}}^{^\circ }~ = {\text{ BDFE }} + n{\text{F}}{{E}_{{1/2}}}({{{\text{M}}}^{n}}{{^{{ + /(n}}}^{{--1) + }}}){\text{ }} + \Delta G_{{{{{\text{H}}}^{ \bullet }}{\text{/}}{{{\text{H}}}^{--}}}}^{^\circ }.\,\,\,\,$Связь гидридности или pKa гидрида металла и потенциала восстановления катионной формы соответствующего комплекса [M]n+ хорошо известна [13]. Недавно авторы [6] сгруппировали и обобщили данные для большого ряда гидридов переходных металлов с известными значениями $\Delta G_{{{{{\text{H}}}^{--}}}}^{^\circ }$ и pKa и продемонстрировали их линейную зависимость от потенциалов одноэлектронного восстановления катионной формы [M]n+ (E1/2(Mn+/(n– 1)+) и E1/2(M(n– 1) + /(n– 2)+) соответственно), полученных в ацетонитриле и приведенных относительно редокс-пары ферроцена. Следует отметить, что полученная сходимость справедлива только для значений потенциалов процессов восстановления, являющихся обратимыми, однако и в случае необратимых электрохимических реакций полученные значения $\Delta G_{{{{{\text{H}}}^{--}}}}^{^\circ }$ и pKa могут быть использованы для оценки реакционной способности рассматриваемого гидрида.
В качестве катализаторов описанных выше реакций (де)гидрирования и др. часто используют комплексы с так называемыми бифункциональными лигандами, в которых металлический центр и лиганд взаимодействуют в процессах образования и разрыва связи [2, 15–18]. Во многих случаях такие лиганды содержат дополнительный протоноакцепторный центр либо в составе пендантной функциональной группы, либо непосредственно координированный к атому металла. Исследуя реакционную способность одного из таких комплексов – гидрида палладия(II) с амидо-бис-фосфиновым пинцетным лигандом (PNP)PdH (1) [19], где PNP – бис(2-диизопропилфосфино-4-метилфенил)амид (схема 2), мы показали [20] существенное увеличение основности атома азота при координации с образованием комплекса 1 по сравнению с соответствующими аминами (pKa(Ph2${\text{NH}}_{2}^{ + }$) = 5.98, pKa(Ph2MeNH+) = 6.52 в MeCN [21]). В результате комплекс 1 протонируется по атому азота относительно слабыми кислотами – фторированными спиртами (CF3)3COH (pKa = 20.5 в MeCN [22]), (CF3)2CHOH (pKa = 29.9 в MeCN [23]) и даже CF3CH2OH (pKa = 35.3 в MeCN [23]11). Об образовании продукта протонирования – катионного комплекса [(PN(H)P)PdH]+ (2) – свидетельствует не только появление новых сигналов в спектрах ЯМР (δPdH = –12.26 м.д., δNH = = 7.07 м.д., δP = 55.8 м.д. в чистом (CF3)2CHOH), но и высокочастотное смещение полосы валентных колебаний Pd–H в ИК-спектрах (ΔνPdH = 68–92 см–1) [20]. Согласно данным ЯМР 1H, при низких температурах/большом избытке спирта наблюдается сильнопольное смещение гидридного резонанса (например, в толуоле-d8 при 190 K δPdH (1) = –10.18 м.д., в присутствии (CF3)2CHOH δPdHH···H = –10.46 м.д.) и уменьшение времени его спин-решеточной релаксации T1 (784.0–605.4 мс) [20], свидетельствующие об образовании диводородной связи (PNP)PdH···HOR [24]. Однако, согласно данным квантово-химических расчетов, водородно-связанные по атому азота (OH···N) комплексы характеризуются той же энергией, что и PdH···HOR [20]. Это не позволяет однозначно говорить об их отсутствии в условиях эксперимента и характеризовать протоноакцепторную способность Ej [24] гидридного лиганда комплекса 1. В настоящей работе нами предпринята попытка охарактеризовать гидридность данного комплекса, исследуя его взаимодействие с кислотами Льюиса (BF3 · Et2O, B(C6F5)3), а также используя предложенные в литературе [6] корреляции потенциалов электрохимического восстановления с ΔGH– и квантово-химические расчеты.
Схема 2 .
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Ацетонитрил очищали по стандартной методике и хранили в инертной атмосфере азота над молекулярными ситами 4 Å. Коммерчески доступные дейтеробензол (99.5 ат. % D, Carl Roth) и дейтеротолуол (99.8 ат. % D, Carl Roth) перегоняли над металлическим натрием и хранили в инертной атмосфере аргона. D2O дегазировали и также хранили в инертной атмосфере аргона. Соединения (PNP)PdCl и (PNP)PdH получали по ранее опубликованной методике [19]. Коммерческие препараты AgBF4 (98%, Aldrich), n-Bu4NBF4 (98%, Acros Organics), ферроцен (99%, Alfa Aesar) и BF3 · Et2O (Aldrich) хранили в инертной атмосфере азота и использовали без дополнительной очистки. Коммерчески доступный B(C6F5)3 (95%, Aldrich) перед проведением реакции очищали методом возгонки.
Масс-спектральные исследования с ионизацией электрораспылением (ИЭР). Масс-спектры с ИЭР регистрировали на масс-спектрометре AmazonX (Bruker Daltonik GmbH, Бремен, Германия). Детектирование положительных ионов осуществляли в интервале m/z = 100–2800. Напряжение на капилляре составляло –4500 В. В качестве газа-осушителя использовали азот с температурой 250°С и расходом 8 л/мин. Ввод образца выполняли со скоростью 4 мкл/мин при помощи шприцевого насоса. Данные ИЭР-экспериментов обрабатывали с помощью программы DataAnalysis 4.0 (Bruker Daltonik GmbH, Бремен, Германия).
Исследования методом циклической вольтамперометрии (ЦВА). В исследованиях методом ЦВА концентрация комплекса [(PNP)Pd(MeCN)][BF4] составляла 0.005 M в 3 мл ацетонитрила в качестве растворителя с n-Bu4NBF4 (0.1 M) в качестве поддерживающего электролита. Все эксперименты проводили в атмосфере инертного газа (азота) в трехканальной электрохимической ячейке, оснащенной рабочим электродом, вспомогательным и электродом сравнения. В качестве рабочего электрода использовали стеклоуглеродный электрод (рабочая поверхность 3.14 мм2). В качестве вспомогательного электрода применяли платиновую проволоку диаметром 1 мм. Ag/AgNO3 (0.01 М раствор в ацетонитриле) использовали в качестве электрода сравнения. ЦВА-кривые записывали при постоянной скорости развертки потенциала рабочего электрода 100 мВ/с при помощи потенциостата E2P фирмы BASi Epsilon (США) с программным обеспечением Epsilon-EC-USB-V200. Значения потенциалов были откалиброваны относительно потенциала редокс-пары ферроцена Fc+/Fc.
Синтез комплекса [(PNP)Pd(MeCN)][BF4]. К раствору комплекса (PNP)PdCl (8.5 мг, 0.015 ммоль) в 2 мл ацетонитрила по каплям добавляли раствор AgBF4 (2.9 мг, 0.015 ммоль) в 1 мл ацетонитрила. Полученную смесь перемешивали в течение 30 мин. В ходе реакции одновременно с изменением цвета раствора от ярко-красного до фиолетового в реакционной смеси наблюдали образование белого осадка, который далее отделяли фильтрацией. Полученный раствор анализировали методом масс-спектрометрии ИЭР и использовали в ЦВА-исследованиях.
ЯМР- и ИК-спектральные исследования. ЯМР-спектральные исследования проводили с использованием приборов Bruker Avance 300 (Bruker, Billerica, MA, USA; рабочая частота для 1Н 300.13 МГц, для 31Р{1H} 121.49 МГц) и Varian Inova 400 (Varian, Palo Alto, CA, USA; рабочая частота для 1Н 400.01 МГц, для 31Р{1H} 161.94 МГц, для 11B{1H} 128.34 МГц).
ИК-спектральные исследования проводили на приборе Nicolet iS50 FTIR (Thermo Scientific, Waltham, MA, USA) с использованием кюветы из CaF2 (l = 0.1 см), строение которой позволяет наполнять ее реакционной смесью в инертной атмосфере аргона.
Стандартная процедура проведения спектральных исследований взаимодействия 1 с кислотами Льюиса. Все операции выполняли с использованием техники Шленка в инертной атмосфере аргона.
Раствор (PNP)PdH (1, с = 0.02–0.06 М) в дейтеробензоле или дейтеротолуоле готовили в трубке Шленка, для лучшего растворения 1 использовали ультразвуковую баню. Затем к полученному раствору добавляли необходимое количество (1–1.1 экв) чистой кислоты Льюиса (B(C6F5)3, BF3 · Et2O) или предварительно приготовленного ее стокового раствора (1 М). После этого раствор помещали в ИК-кювету или ЯМР-ампулу и регистрировали спектры.
При исследовании взаимодействия с D2O к раствору 1, приготовленному описанным выше способом, добавляли 5 экв D2O, после чего полученный раствор помещали в ЯМР-ампулу, снабженную септой. После регистрации спектра в ампулу через септу добавляли 1 экв BF3 · Et2O и регистрировали новый спектр.
DFT-расчет. Для расчета гидридодонорной споcобности была применена модификация подхода, использованного нами ранее для гидридов бора [14, 25, 26]. Модификация заключается в смене базиса с 6-311++G(d, p) на def2-TZVP и вызвана наличием переходного металла в системе. Таким образом, оптимизацию геометрии исследуемых гидридов проводили в программе Gaussian09 [27] с использованием функционала M06 [28] и базисного набора def2-TZVP [29], снабженного ECP для остовных электронов атома Pd [30]. Неспецифическое влияние растворителя – ацетонитрила – учитывали с использованием SMD-модели [31]. Гидридодонорная способность, или термодинамическая гидридность, определенная как энергия Гиббса реакции отрыва гидрид-иона от исходного гидрида (MH = M+ + H–), была рассчитана для (PNP)PdH (∆G°H–(MeCN) = 62.8 ккал/моль), а также для сравнения с полученными ранее данными для серии гидридов бора: Li[BF3H] ($\Delta G_{{{{{\text{H}}}^{--}}}}^{^\circ }$(MeCN) = 55.0 ккал/моль), Li[BCl3H] ($\Delta G_{{{{{\text{H}}}^{--}}}}^{^\circ }$(MeCN) = 70.8 ккал/моль), Li[BH4] ($\Delta G_{{{{{\text{H}}}^{--}}}}^{^\circ }$(MeCN) = 65.9 ккал/моль).
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Электрохимическое исследование комплекса [(PNP)Pd(MeCN)]+
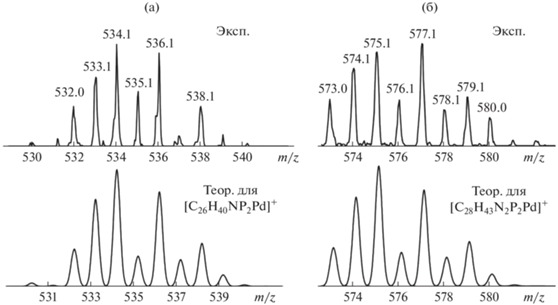
С целью оценки основности (значений $\Delta G_{{{{{\text{H}}}^{--}}}}^{^\circ }$ и pKa) гидрида палладия (PNP)PdH (1) были исследованы электрохимические параметры комплекса [(PNP)Pd(MeCN)][BF4] (3). Этот катионный комплекс получен из хлоридного аналога (PNP)PdCl путем обработки тетрафтороборатом серебра в ацетонитриле. Для подтверждения образования комплекса 3 был использован метод масс-спектрометрии ИЭР. В масс-спектрах реакционной смеси наблюдаются два пика положительно заряженных ионов, соответствующих катионным комплексам состава [(PNP)Pd]+ (m/z = 534.1) и [(PNP)Pd(MeCN)]+ (m/z = 575.1) (рис. 1).
Рис. 1.
Теоретические и экспериментальные изотопные распределения для катионных комплексов [(PNP)Pd]+ (а) и [(PNP)Pd(MeCN)]+ (б).
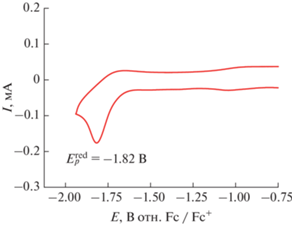
На ЦВА полученного комплекса [(PNP)Pd(MeCN)][BF4] в ацетонитриле (рис. 2) наблюдается один необратимый двухэлектронный пик восстановления ($E_{p}^{{{\text{red}}}}$ = –1.82 В). Препаративное восстановление комплекса при потенциале этого пика приводит к образованию частиц металлического палладия вследствие полного разрушения исходной структуры комплекса. Электрохимическая стадия, сопровождаемая таким химическим процессом, является необратимой, и зафиксировать соответствующий ей пик реокисления на ЦВА-кривой не удается даже в экспериментах с пониженной температурой (до –45°C) и высокой скоростью развертки потенциала (до 25000 мВ/с). Таким образом, наблюдаемый пик восстановления отнесен к MII/0-превращению катионного комплекса [(PNP)PdII(MeCN)]+.
Рис. 2.
ЦВА комплекса [(PNP)Pd(MeCN)][BF4] в ацетонитриле в присутствии n-Bu4NBF4 (0.1 M). Развертка потенциала от –0.75 до –1.90 В и далее до –0.75 В.
Для комплексов, претерпевающих двухэлектронное восстановление, в работе [6] была получена линейная корреляция гидридности с потенциалом восстановления (уравнение (6)):
(6)
$\Delta G_{{{{{\text{H}}}^{--}}}}^{^\circ }~ = {\text{ }}20.75{{E}_{{1/2}}}({{{\text{M}}}^{n}}^{ + }{\text{/}}{{{\text{M}}}^{{(n--2) + }}}){\text{ }} + 79.53.$Расчет по этому уравнению с использованием экспериментально полученных электрохимических данных дает значение гидридности для (PNP)PdH 41.8 ккал/моль, что предполагает очень высокую реакционную способность комплекса 1 в качестве донора гидрид-иона. Сопоставимые значения $\Delta G_{{{{{\text{H}}}^{--}}}}^{^\circ }$(MeCN) были получены, например, для комплексов [Pd(depe)2H]+, где depe = Et2PCH2CH2PEt2 (43.0 ккал/моль), [Pt(dmpe)2H]+, где dmpe = = Me2PCH2CH2PMe2 (41.4 ккал/моль) и [W(CO)5H]– (40.0 ккал/моль) [13]. Однако значение $\Delta G_{{{{{\text{H}}}^{--}}}}^{^\circ }$(MeCN), полученное нами по уравнению (6), значительно отличается от данных, полученных методом DFT. Расчет методом DFT M06/def2-TZVP для (PNP)PdH дает значение $\Delta G_{{{{{\text{H}}}^{--}}}}^{^\circ }$(MeCN) = 62.8 ккал/моль. Для сравнения с полученными ранее данными [14] нами также была рассчитана гидридодонорная способность для серии гидридов бора : Li[BF3H] ($\Delta G_{{{{{\text{H}}}^{--}}}}^{^\circ }$(MeCN) = = 55.0 ккал/моль), Li[BCl3H] ($\Delta G_{{{{{\text{H}}}^{--}}}}^{^\circ }$(MeCN) = = 70.8 ккал/моль), Li[BH4] ($\Delta G_{{{{{\text{H}}}^{--}}}}^{^\circ }$(MeCN) = = 65.9 ккал/моль).
Следует отметить, что уравнение (6) вытекает из уравнения (5) при условии, что в ряду комплексов переходных металлов с бидентатными лигандами, рассмотренных в работе [6], изменение BDFE незначительно по сравнению с влиянием на гидридность окислительно-восстановительного потенциала E1/2(Mn+/(n– 1)+). Однако в пинцерных системах в транс-положении к гидриду обычно находятся сильные σ-доноры, что приводит к лабилизации гидридного лиганда, тем самым увеличивая значение BDFE [32]. Потому использование корреляции, представленной в уравнении (6), становится ненадежным. Действительно, расчет BDFE для комплекса 1 по уравнению (5) с использованием значения $\Delta G_{{{{{\text{H}}}^{--}}}}^{^\circ }$, полученного методом DFT, и потенциала восстановления, полученного из ЦВА на рис. 2, дает значение 78.8 ккал/моль при том, что авторы [6] допустили среднюю величину BDFE, равную 51 ккал/моль. Таким образом, более реалистичной оценкой гидридодонорной способности комплекса (PNP)PdH является значение $\Delta G_{{{{{\text{H}}}^{--}}}}^{^\circ }$(MeCN) = 62.8 ккал/моль, полученное методом DFT. Соединения бора характеризуются близкими расчетными значениями $\Delta G_{{{{{\text{H}}}^{--}}}}^{^\circ }$(MeCN): [BF3H]– является чуть более сильным донором гидрид-иона, чем комплекс 1, а [BH4]– и аналогичный ему по величине $\Delta G_{{{{{\text{H}}}^{--}}}}^{^\circ }$(MeCN) комплекс [B(C6F5)3H]– [14] – немного более слабыми. Таким образом, BF3 и B(C6F5)3 представляются перспективными кислотами Льюиса для исследования нековалентных взаимодействий и переноса гидрид-иона с участием (PNP)PdH. Эти соединения недавно были успешно использованы нами при исследовании гидридности комплексов марганца L2Mn(CO)3H [33, 34].
Экспериментальное исследование взаимодействия (PNP)PdH с кислотами Льюиса
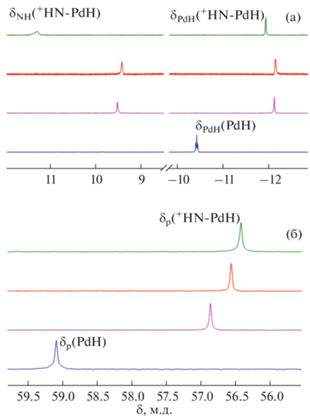
Для установления гидридных свойств (PNP)PdH нами исследовано взаимодействие гидрида палладия с кислотами Льюиса BF3 · Et2O и B(C6F5)3 в некоординирующих растворителях (бензол, толуол). Установлено, что в результате взаимодействия гидрида палладия с 1.1 экв BF3 · Et2O в дейтеротолуоле в ИК-спектрах в области валентных колебаний связи Pd–H появляется новая полоса νPdH при 1992 см–1, смещенная в более высокочастотную область по сравнению с ее положением в спектре исходного гидрида 1 (νPdH(PdH) = 1923 см–1) (табл. 1, рис. 3), и полоса νNH в области 2800 см–1. В спектрах ЯМР 1H в аналогичных условиях наблюдается смещение резонанса гидридного лиганда в сильное поле (δPdH = –12.16 по сравнению с δPdH(1) = –10.42), а также появление нового слабопольного сигнала δH = 9.41 (рис. 4). Указанные спектральные изменения аналогичны наблюдаемым при добавлении к комплексу 1 HBF4 · Et2O [20] и свидетельствуют о протонировании атома азота с образованием комплекса 2 (Схема 3 ). При взаимодействии гидрида палладия с эквимолярным количеством B(C6F5)3 спектральные изменения аналогичны (табл. 1).
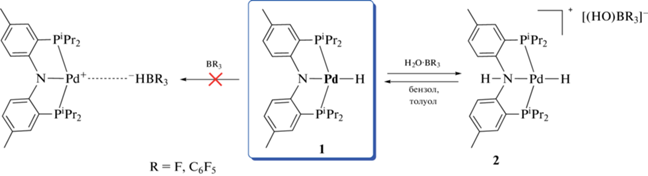
Схема 3 .
Таблица 1.
Основные ИК- и ЯМР-спектральные характеристики комплексов палладия(II) в толуоле-d8
| Комплекс | νPdH, см–1 | νOH, см–1 | δPdH, м.д. | δNH, м.д. | δP, м.д. |
|---|---|---|---|---|---|
| (PNP)PdH | 1923 | –10.42 | 59.1 | ||
| (PNP)PdH + HBF4 · Et2O | 1992 | 3140 | –12.12 | 9.53 | 56.9 |
| (PNP)PdH + BF3 · Et2O | 1992 | 3140 | –12.16 | 9.41 | 56.6 |
| (PNP)PdH + B(C6F5)3 | 1988 | 3670 | –11.95 | 11.27 | 56.4 |
Рис. 3.
ИК-спектры в области валентных колебаний νPdH (а) и νOH (б) (PNP)PdH (с = 0.02 М, синий) и при взаимодействии с 1.1 экв HBF4 · Et2O (розовый) и кислот Льюиса BF3 · Et2O (зеленый) и B(C6F5)3 (красный); 295 K, толуол-d8, l = 0.1 см.
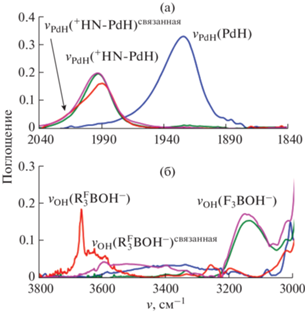
Рис. 4.
Спектры ЯМР 1Н (а) и 31Р{1Н} (б) (PNP)PdH (с = 0.02 М, синий) и при взаимодействии с 1.1 экв HBF4 · Et2O (розовый) и кислот Льюиса BF3 · Et2O (зеленый) и B(C6F5)3 (красный); 295 K, толуол-d8.
На основании полученных экспериментальных данных можно предположить, что источником протонов является остаточная вода (из растворителя и/или BF3 · Et2O/B(C6F5)3), “активированная” за счет координации к кислоте Льюиса. Известно, что в системе BF3–H2O наблюдается сложное равновесие, приводящее к образованию различных кислотных соединений, таких как HBF3OH, HBF4, HBF2(OH)2 и даже HF [35]. Вероятно, образование 2 происходит за счет депротонирования молекул воды, координированных атомом кислорода к атому бора H2O · BR3, или HBR3OH (R = F, C6F5), образующихся в результате гидролиза борных соединений при достаточно малом содержании воды. Анализ спектров ЯМР 11В{1H} позволяет предположить образование [BF3OH]– (δB = –0.13 [36]) и [B(С6F5)3OH]– (δB = = –2.9 м.д. [37]) в условиях реакции с комплексом 1.
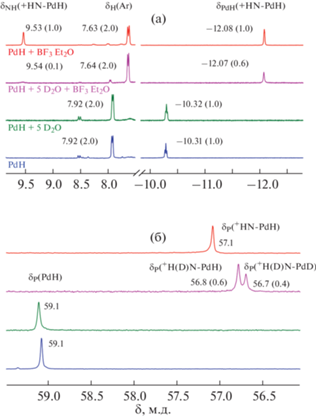
Для подтверждения выдвинутого нами предположения было исследовано взаимодействие гидрида палладия с дейтерированной водой. Добавление 5 экв D2O к раствору (PNP)PdH в C6D6 не приводит к какому-либо взаимодействию (рис. 5): по данным спектров ЯМР 1Н, ни протонирования атома азота пинцетного лиганда, ни обмена Pd–H на дейтерий не наблюдается. Добавление же к этой смеси 1 экв BF3 · Et2O приводит к количественному расходованию гидрида палладия 1, о чем свидетельствует отсутствие в спектрах ЯМР соответствующих сигналов δPdH = –10.31 м.д. и δP = = 59.1 м.д. При этом в спектре ЯМР 1Н появляются сигналы, аналогичные наблюдаемым при взаимодействии комплекса 1 с BF3 · Et2O и HBF4 · · Et2O и отнесенные к продукту протонирования по атому азота пинцетного лиганда: δPdH = –12.07 м.д. и δNH = 9.54 м.д. (рис. 5). Интегрирование этих сигналов относительно сигналов ароматического кольца лиганда (δH = 7.64 м.д.) показывает, что в данных условиях 90% комплекса протонировано именно D+, что говорит о взаимодействии с D2O. Также 40% Pd–H в процессе реакции обменивается на дейтерид, что подтверждается как интегрированием гидридного резонанса в спектре ЯМР 1Н (0.6 : 2.0 для δH = 7.64 м.д.), так и наличием в спектре ЯМР 31Р{1Н} двух близко расположенных синглетов (δP = 56.8 и 56.7 м.д.) в соотношении 0.6 : 0.4, которые относятся к комплексам гидрида и дейтерида палладия соответственно (рис. 5).
Рис. 5.
Спектры ЯМР 1Н (а) и 31Р{1Н} (б) комплекса (PNP)PdH (1; с = 0.04 М, синий), 1 в присутствии BF3 · Et2O (с = 0.04 М, красный) или D2O (с = 0.2 М, зеленый) и их трехкомпонентной смеси (розовый); C6D6, 295 K. Указаны положения сигналов (хим. сдвиги, м.д.) и значения интегралов (в скобках).
Таким образом, показано, что реакция комплекса 1 с борсодержащими кислотами Льюиса неожиданно приводит к протонированию атома азота пинцетного лиганда, а не к взаимодействию с гидридным лигандом. Вероятной причиной является большая основность/протоноакцепторная способность атома азота по сравнению с PdH. Действительно, данные DFT-расчетов показывают, что величины сродства к протону (PA, энтальпия присоединения протона –ΔH298.15 K для реакции B + H+ → BH+, где B – основание) различаются для N и PdH (табл. 2). При этом рассчитанное в газовой фазе [38] сродство к протону атома азота оказывается на уровне других гидридов переходных металлов [39], в то время как PA(PdH) – существенно ниже. Расчет сродства к протону с учетом растворителя (MeCN) дает более высокие значения PA и уменьшает разницу в характеристиках двух центров (табл. 2). В литературе описаны и различные корреляции сродства к протону органических оснований и pKa их сопряженных кислот в растворе [40–42]. Применение этих подходов для оценки кислотности [(PN(H)P)PdH]+ и [(PNP)Pd(η2-H2)]+ дает сильно различающиеся значения pKa, которые тем не менее объясняют протонирование (PNP)PdH (1) слабыми кислотами именно по атому азота. При этом [(PNP)Pd(η2-H2)]+ является более сильной кислотой и может участвовать в качестве интермедиата/кинетического продукта в процессах активации H2 или депротонирования [(PN(H)P)PdH]+. В случае реакции с кислотами Льюиса для BF3, чья рассчитанная гидридодонорная способность $\Delta G_{{{{{\text{H}}}^{--}}}}^{^\circ }$(MeCN) несколько выше, чем 1, можно было ожидать образования нековалентно связанного комплекса PdH$ \cdots $BF3. Для более сильной кислоты Льюиса B(C6F5)3 также не наблюдается ожидаемой реакции переноса гидрид-иона. В обоих случаях происходит протонирование активированной водой (R3B · OH2), которое, по-видимому, энергетически более выгодно, чем отрыв гидрида.
Таблица 2.
Сродство к протону (PA, ккал/моль)a атома азота (N–Pd) и гидридного лиганда (Pd–H) комплекса (PNP)PdH (1) и оценочные величины констант кислотности (pKa) соответствующих протонированных форм ([(PN(H)P)PdH]+ и [(PNP)Pd(η2-H2)]+)
| Параметр | N–Pd | Pd–H | N–Pd | Pd–H |
|---|---|---|---|---|
| PA | 253.6b | 236.8b | 279.1c | 269.5c |
| pKa(H2O)d | 20.8 | 11.5 | ||
| pKa(MeCN)e | 32.0 | 19.8 | ||
| pKa(MeCN)f | 18.4 | 13.8 |
a Сродство к протону (PA) рассчитано как энтальпия присоединения протона в газе –ΔH298.15 K для реакции B + H+ → BH+, где B – основание. b Данные DFT-расчета в газовой фазе из работы [38]. c DFT-расчет в MeCN в настоящей работе. d Оценка с использованием корреляции pKa(H2O)–PA(газ) из работы [40]. e Оценка с использованием корреляции pKa(MeCN)–pKa(H2O) из работы [21]. f Оценка с использованием корреляции PA(MeCN)–pKa(MeCN) из работы [41].
ЗАКЛЮЧЕНИЕ
Присоединение бис(арилфосфин)амида к атому Pd(II) с образованием пинцетного комплекса приводит к повышению основности атома азота по сравнению с диариламинами (например, Ph2NH и Ph2MeN) и обусловливает парадоксальную реакционную способность гидридного комплекса (PNP)PdH. Результаты эксперимента и квантово-химических расчетов показывают, что более высокое сродство к протону атома азота по сравнению с гидридным лигандом является движущей силой его протонирования различными кислотами, включая не только фторированные спирты, но и примеси воды в присутствии кислот Льюиса BF3 и B(C6F5)3. Трансвлияние атома азота повышает энергию связи Pd–H, для которой получены достаточно высокие значения как гомолитического (BDFE), так и гетеролитического разрыва ($\Delta G_{{{{{\text{H}}}^{--}}}}^{^\circ }$). Это объясняет невозможность применения описанных в литературе методов оценки гидридности на основе потенциалов электрохимического восстановления. Однако такая высокая основность атома азота и энергия связи Pd–H в комплексе (PNP)PdH делают его многообещающим прекурсором для получения в условиях окисления стабильного аминил-радикального гидридного комплекса, потенциально обладающего высокой реакционной способностью в радикальных реакциях. В настоящее время нашей командой продолжаются исследования, направленные на изучение электроокислительного поведения комплекса (PNP)PdH.
Список литературы
Wang D., Astruc D. // Chem. Rev. 2015. V. 115. P. 6621. https://doi.org/10.1021/acs.chemrev.5b00203
Werkmeister S., Neumann J., Junge K. et al. // Chem. Eur. J. 2015. V. 21. P. 12226. https://doi.org/10.1002/chem.201500937
Pospech J., Fleischer I., Franke R. et al. // Angew. Chem. Int. Ed. 2013. V. 52. P. 2852. https://doi.org/10.1002/anie.201208330
Dutta A., Appel A.M., Shaw W.J. // Nature Rev. Chem. 2018. V. 2. P. 244. https://doi.org/10.1038/s41570-018-0032-8
DuBois D.L. // Inorg. Chem. 2014. V. 53. P. 3935. https://doi.org/10.1021/ic4026969
Waldie K.M., Ostericher A.L., Reineke M.H. et al. // ACS Catal. 2018. V. 8. P. 1313. https://doi.org/10.1021/acscatal.7b03396
Stanbury M., Compain J.-D., Chardon-Noblat S. // Coord. Chem. Rev. 2018. V. 361. P. 120. https://doi.org/10.1016/j.ccr.2018.01.014
Sordakis K., Tang C., Vogt L.K. et al. // Chem. Rev. 2018. V. 118. P. 372. https://doi.org/10.1021/acs.chemrev.7b00182
Francke R., Schille B., Roemelt M. // Chem. Rev. 2018. V. 118. P. 4631. https://doi.org/10.1021/acs.chemrev.7b00459
Buss J.A., VanderVelde D.G., Agapie T. // J. Am. Chem. Soc. 2018. V. 140. P. 10121. https://doi.org/10.1021/jacs.8b05874
Artz J., Müller T.E., Thenert K. et al. // Chem. Rev. 2018. V. 118. P. 434. https://doi.org/10.1021/acs.chemrev.7b00435
Filippov O.A., Golub I.E., Osipova E.S. et al. // Russ. Chem. Bull. 2014. V. 63. P. 2428. https://doi.org/10.1007/s11172-014-0758-5
Wiedner E.S., Chambers M.B., Pitman C.L. et al. // Chem. Rev. 2016. V. 116. P. 8655. https://doi.org/10.1021/acs.chemrev.6b00168
Golub I.E., Filippov O.A., Belkova N.V. et al. // J. Organomet. Chem. 2018. V. 865. P. 247. https://doi.org/10.1016/j.jorganchem.2018.03.020
Khusnutdinova J.R., Milstein D. // Angew. Chem. Int. Ed. 2015. V. 54. P. 12236. https://doi.org/10.1002/anie.201503873
Gunanathan C., Milstein D. // Acc. Chem. Res. 2011. V. 44. P. 588. https://doi.org/10.1021/ar2000265
Cohen S., Bilyachenko A.N., Gelman D. // Eur. J. Inorg. Chem. 2019. V. 2019. P. 3203. https://doi.org/10.1002/ejic.201801486
Yang W., Filonenko G.A., Pidko E.A. // Chem. Commun. 2023. V. 59. P. 1757. https://doi.org/10.1039/D2CC05625A
Fan L., Foxman B.M., Ozerov O.V. // Organometallics. 2004. V. 23. P. 326. https://doi.org/10.1021/om034151x
Kirkina V.A., Kulikova V.A., Gutsul E.I. et al. // Inorganics. 2023. V. 11. P. 212. https://doi.org/10.3390/inorganics11050212
Tshepelevitsh S., Kütt A., Lõkov M. et al. // Eur. J. Org. Chem. 2019. V. 2019. P. 6735. https://doi.org/10.1002/ejoc.201900956
Raamat E., Kaupmees K., Ovsjannikov G. et al. // J. Phys. Org. Chem. 2013. V. 26. P. 162. https://doi.org/10.1002/poc.2946
Kuejtt A., Leito I., Kaljurand I. et al. // J. Org. Chem. 2006. V. 71. P. 2829. https://doi.org/10.1021/jo060031y
Belkova N.V., Epstein L.M., Filippov O.A. et al. // Chem Rev. 2016. V. 116. P. 8545. https://doi.org/10.1021/acs.chemrev.6b00091
Golub I.E., Filippov O.A., Kulikova V.A. et al. // Molecules. 2020. V. 25. P. 2920. https://doi.org/10.3390/molecules25122920
Golub I.E., Filippov O.A., Belkova N.V. et al. // Russ. J. Inorg. Chem. 2021. V. 66. P. 1639. https://doi.org/10.1134/S0036023621110073
Frisch M.J., Trucks G.W., Schlegel H.B. et al. // Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2016.
Zhao Y., Truhlar D.G. // Theor. Chem. Acc. 2008. V. 120. P. 215. https://doi.org/10.1007/s00214-007-0310-x
Weigend F., Ahlrichs R. // Phys. Chem. Chem. Phys. 2005. V. 7. P. 3297. https://doi.org/10.1039/B508541A
Andrae D., Haussermann U., Dolg M. et al. // Theor. Chim. Acta 1990. V. 77. P. 123. https://doi.org/10.1007/bf01114537
Marenich A.V., Cramer C.J., Truhlar D.G. // J. Phys. Chem. B. 2009. V. 113. P. 6378. https://doi.org/10.1021/jp810292n
Alig L., Fritz M., Schneider S. // Chem. Rev. 2019. V. 119. P. 2681. https://doi.org/10.1021/acs.chemrev.8b00555
Osipova E.S., Kovalenko S.A., Gulyaeva E.S. et al. // Molecules. 2023. V. 28. P. 3368. https://doi.org/10.3390/molecules28083368
Osipova E.S., Gulyaeva E.S., Kireev N.V. et al. // Chem. Commun. 2022. V. 58. P. 5017. https://doi.org/10.1039/D2CC00999D
Wamser C.A. // J. Am. Chem. Soc. 1951. V. 73. P. 409. https://doi.org/10.1021/ja01145a134
Zhou J., Litle E.D., Gabbaï F.P. // Chem. Commun. 2021. V. 57. P. 10154. https://doi.org/10.1039/D1CC04105C
Longobardi L.E., Mahdi T., Stephan D.W. // Dalton Trans. 2015. V. 44. P. 7114. https://doi.org/10.1039/C5DT00921A
Gregor L.C., Chen C.-H., Fafard C.M. et al. // Dalton Trans. 2010. V. 39. P. 3195. https://doi.org/10.1039/B925265G
Belkova N.V., Epstein L.M., Shubina E.S. // ARKIVOC. 2008. V. iv. P. 120. https://doi.org/10.3998/ark.5550190.0009.413
Pankratov A.N., Shchavlev A.E. // J. Analyt. Chem. 2001. V. 56. P. 123. https://doi.org/10.1023/A:1009438517429
Kovačević B., Maksić Z.B. // Org. Lett. 2001. V. 3. P. 1523. https://doi.org/10.1021/ol0158415
Glasovac Z., Kovačević B. // Int. J. Mol. Sci. 2022. V. 23. P. 10576. https://doi.org/10.3390/ijms23181057
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии