

Журнал неорганической химии, 2023, T. 68, № 9, стр. 1324-1336
Изучение свойств новых пятиядерных карбоксилатных комплексов меди(II) и РЗЭ(III)
О. С. Пушихина a, *, Е. В. Карпова a, **, Д. А. Царёв b, В. А. Тафеенко a, Т. Б. Шаталова a
a Московский государственный университет имени М.В. Ломоносова
119991 Москва,
Ленинские горы, 1, Россия
b Научно-исследовательский институт Митоинженерии МГУ
имени М.В. Ломоносова
119991 Москва, Ленинские горы, 1, Россия
* E-mail: pushikhina_chem@mail.ru
** E-mail: karpova@inorg.chem.msu.ru
Поступила в редакцию 28.04.2023
После доработки 13.06.2023
Принята к публикации 14.06.2023
- EDN: WLVYXQ
- DOI: 10.31857/S0044457X23601189
Аннотация
Взаимодействием индивидуальных галогенацетатов меди и РЗЭ в водном растворе получены новые пятиядерные комплексы [Cu3M2(CHF2COO)12(H2O)8]·2H2O, где M = Er (I) и Nd (II). Молекулярное строение комплекса I установлено методом РСА монокристалла (CIF file CCDC № 2159724). Особенности строения комплексов и природа карбоксилатных мостиков между металлоцентрами влияют на свойства этих комплексов, поэтому для сравнения были получены два аналогичных соединения с монохлорацетат-лигандом – [Cu3M2(СH2ClCOO)12(H2O)8]·2H2O, где M = Er (III) и Nd (IV). Соединения III и IV изоструктурны ранее изученным комплексам данного типа с другими РЗЭ. Соединения I–IV охарактеризованы методами рентгеновской дифракции и ИК-спектроскопии, изучена их термическая стабильность. Для подтверждения формирования предшественников молекулярной формы кристаллического соединения I определены формы комплексов в растворе методом ESI-MS.
ВВЕДЕНИЕ
Полиядерные гетерометаллические комплексы вызывают особый интерес с точки зрения изучения природы обменных взаимодействий между ионами переходных и редкоземельных элементов (РЗЭ), связанных между собой мостиковыми лигандами [1–3]. Обменные взаимодействия могут осуществляться не только по кратчайшим каналам металл–элемент(Э)–металл, но и через сопряженные связи М–О–Э–О–М: через нитрат-, карбонат-, фосфат-, ванадат-ионы, а также трифлаты и карбоксилаты металлов [4]. Характер обмена зависит от типа орбиталей атома металла, количества электронов на них, от расположения атомов-посредников обмена [5]. На данный момент получено большое количество карбоксилатов металлов, проявляющих магнитные свойства, среди которых можно выделить два основных типа кристаллических структур – молекулярные и обладающие протяженной структурой [6–8]. Разработка синтетических подходов для получения гетерометаллических комплексов с пониженной размерностью спиновой системы на основе 3d–4f-металлов и карбоксилатных лигандов имеет важное научное значение.
В настоящей работе получены и исследованы новые пятиядерные гетерометаллические комплексы состава [Cu3M2(CHF2COO)12(H2O)8]·2H2O, где M = Er (I) и Nd (II). Карбоксилаты с аналогичным металлическим остовом Cu3M2 ранее изучались в ряде работ, где монохлорацетатные комплексы с Gd [9], Pr, Nd, Sm, Dy, Ho [10], Yb [11] и Y [12] были получены реакцией нитратов меди и РЗЭ с монохлоруксусной кислотой. Для сравнения свойств были получены соединения с эрбием (III) и неодимом (IV). Несмотря на то, что структура соединения IV была установлена в [10], спектральные данные и сведения о термической устойчивости данного вещества отсутствуют.
Галогензамещенные ацетаты как лиганды могут участвовать в образовании различных типов полиядерных гетерометаллических структур. Из литературы [13] известно, что возможно образование соединения CuNd2(СCl3COO)8(H2O)6, структура которого построена из цепочечных фрагментов, мономерное звено которых содержит два катиона неодима и один катион меди. Нами обнаружено, что в аналогичных [13] условиях синтеза в случае с монохлоруксусной и дифторуксусной кислотами из растворов с исходным соотношением Cu : РЗЭ = 1 : 2 образуются, по-видимому, более устойчивые в этих условиях пятиядерные комплексы I–IV с соотношением металлов 3 : 2.
Понимание процессов образования предшественников кристаллической формы соединения в растворе – ключевой аспект возможности осуществления направленного дизайна координационных соединений с заданными составом и строением. С помощью метода ESI-MS возможна идентификация состава всех форм комплексов, образованных в растворе. Кроме того, данный метод позволяет исследовать не только отдельные комплексы, но и динамику образования и изменения форм этих комплексов в различных условиях.
В настоящей работе обсуждаются свойства четырех гетерометаллических галогенацетатных соединений меди и РЗЭ и проводится сравнение этих данных с литературными.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Соединения I–IV получены методами растворной химии. Для синтеза гетерометаллических галогенацетатов на первом этапе получали ряд индивидуальных (гомометаллических) соединений меди, неодима и эрбия. Для этой цели использовали коммерческие реактивы без предварительной подготовки: основный карбонат меди (CuOH)2CO3, оксиды неодима Nd2O3 и эрбия Er2O3, кислоты СH2ClCOOH и CHF2COOH квалификации “х. ч.”. Индивидуальные соли металлов, Cu(CH2ClCOO)2(H2O)1.5, Cu(CHF2COO)2(H2O)0.5, M(CH2ClCOO)3(H2O)5/3 и M(CHF2COO)3(H2O)3, где M = Er и Nd, синтезировали, опираясь на известные методики [14–16].
Синтез [Cu3M2(CHF2COO)12(H2O)8]·2H2O, где M = Er (I) и Nd (II). Навески солей 1.5 ммоль Cu(CHF2COO)2(H2O)0.5 и 1 ммоль Er(CHF2COO)3(H2O)3 растворяли в 5 мл воды. При дальнейшем концентрировании в приоткрытой виале при комнатной температуре в растворе образовались кристаллы интенсивно-синего цвета, устойчивые на воздухе. Состав и строение соединения I установлены методом рентгеноструктурного анализа монокристалла. По аналогичной методике из солей меди и неодима синтезировано соединение II. Полученные соединения изоструктурны.
ИК-спектр (ν, см–1) соединения I: 3403 сл. ν(OH); 1707 ср., 1657 с., 1612 с. νas(СОО); 1471 ср. νs(СОО); 1331 с. δ(CH); 1112 с., 1060 с. ν(CF2); 954 ср. δ(CCF); 813 ср. ν(CC).
ИК-спектр (ν, см–1) соединения II: 3402 сл. ν(OH); 1703 ср., 1656 с., 1610 с. νas(СОО); 1468 ср. νs(СОО); 1330 с. δ(CH); 1117 с., 1059 с. ν(CF2); 954 ср. δ(CCF); 813 ср. ν(CC).
Синтез [Cu3M2(СH2ClCOO)12(H2O)8]·2H2O, где M = Er (III) и Nd (IV). Соединения получены аналогично синтезу I реакцией между соответствующими индивидуальными монохлорацетатами металлов с тем лишь отличием, что здесь кристаллизация кинетически затруднена, поскольку насыщенный раствор обладает более высокой вязкостью. Новое соединение III изоструктурно ранее изученным комплексам Gd, Pr, Nd, Sm, Dy, Ho, Yb, Y [9–12]. Данная методика позволяет получить известное соединение IV, структура которого установлена авторами [10]. Необходимо отметить, что растворы, содержащие монохлорацетат-ионы, нежелательно концентрировать при нагревании, поскольку происходит их разложение с выделением хлорид-ионов, в то время как дифтор-ацетаты более устойчивы к нагреванию.
ИК-спектр (ν, см–1) соединения III: 3602 сл., 3342 ср. ν(OH); 3013 сл., 2958 сл. ν(CH2); 1670 ср., 1602 с., 1563 с. νas(СОО); 1396 с. νs(СОО); 1253 ср., 970 сл. δ(CH2); 931 ср. ν(CC); 711 ср. ν(CCl); 790 ср., 696 ср. δ(СОО).
ИК-спектр (ν, см–1) соединения IV: 3590 сл., 3353 ср. ν(OH); 3013 сл., 2958 сл. ν(CH2); 1665 ср., 1607 с., 1561 с. νas(СОО); 1399 с. νs(СОО); 1255 ср., 967 сл. δ(CH2); 933 ср. ν(CC); 710 ср. ν(CCl); 793 ср., 675 ср. δ(СОО).
Рентгеноструктурный анализ монокристалла (РСА). Структурные данные депонированы в Кембриджском центре кристаллографических данных (№ 2159724 (I); deposit@ccdc.cam.ac.uk; www: http://www.ccdc.cam.ac.uk). Основные кристаллографические характеристики и параметры съемки приведены в табл. 1, характерные межатомные расстояния и валентные углы – в табл. 2. Данные рентгеноструктурного анализа монокристаллического образца получены на дифрактометре StadiVari фирмы STOE с двухкоординатным детектором Pilatus100K (CuKα-излучениие, λ = 1.54186 Å). Структура решена прямыми методами и уточнена методом наименьших квадратов по F2 с помощью программного комплекса SHELX [17–19]. Уточнение позиционных и тепловых параметров неводородных атомов проведено в полноматричном анизотропном приближении. Положения атомов водорода рассчитаны и уточнены по методу “наездника”. Изображения структуры подготовлены с помощью программы DIAMOND [20].
Таблица 1.
Основные кристаллографические характеристики и детали рентгеноструктурного эксперимента для I
| Параметр | Значение |
|---|---|
| Формула | C12H16Cu1.5ErF12O17 |
| M | 922.82 |
| Сингония | Моноклинная |
| Пр. гр. | P21/c |
| a, Å | 10.2339(5) |
| b, Å | 17.2025(8) |
| c, Å | 15.6227(8) |
| α, град | 90 |
| β, град | 97.281(4) |
| γ, град | 90 |
| V, Å3 | 2728.2(2) |
| Z | 4 |
| T, K | 295(2) |
| ρ(выч.), г/см3 | 2.247 |
| μ, мм–1 | 8.378 |
| F(000) | 1774 |
| Размер кристалла, мм | 0.12 × 0.14 × 0.23 |
| Область сканирования по θ, град | 3.839–70.685 |
| Диапазон индексов hkl | –12 ≤ h ≤ 7 |
| –21 ≤ k ≤ 20 | |
| –19 ≤ l ≤ 15 | |
| Число измеренных/независимых отражений | 19580/5125 |
| Rint | 0.0725 |
| Число отражений с I > 2σ(I) | 4295 |
| GOF | 1.058 |
| R-факторы (I > 2σ(I)) | R1 = 0.0629 |
| wR2 = 0.1776 | |
| R-факторы (по всем отражениям) | R1 = 0.0708 |
| wR2 = 0.1838 | |
| Остаточная электронная плотность (min/max), e/Å3 | –1.344/1.829 |
Таблица 2.
Сопоставление характерных межатомных расстояний и валентных углов в ряду изотипных соединений: полученного I и известных [Cu3M2(СH2ClCOO)12(H2O)8]·2H2O, где M = Nd [10], Gd [9], Dy [10], Yb [11]. Даны значения для пары атом металла–атом кислорода карбоксилат-аниона
| Соеди-нение | Плоский квадрат [Cu(2)] | Фонарик [Cu(1)] | ||||||
|---|---|---|---|---|---|---|---|---|
| Cu–O, Å | M–O, Å | Cu–M, Å | Cu–O, Å | M–O, Å | Cu–M, Å | O–Cu–O, град | O–M–O, град | |
| I | 1.957(5) 1.955(5) |
2.282(5) | 3.5588(1) | 1.943(6)–1.986(5) | 2.295(6)–2.330(5) | 4.6472(2) | 171.7(2)–172.4(2) | 116.3(2)–117.4(2) |
| M = Nd | 1.954(3) 1.964(3) |
2.385(3) | 3.601(2) | 1.965(3)–1.990(3) | 2.366(3)–2.452(4) | 4.654(2) | 173.20(13)–175.51(3) | 113.6(1)–116.7(1) |
| M = Gd | 1.959(4) 1.970(5) |
2.337(4) | 3.561(1) | 1.956(4)–1.979(4) | 2.325(3)–2.397(5) | 4.662(1) | 172.4(2)–174.4(2) | 114.4(2)–117.9(2) |
| M = Dy | 1.950(3) 1.950(3) |
2.299(3) | 3.535(1) | 1.954(3)–1.987(3) | 2.298(4)– 2.369(3) |
4.621(1) | 173.2(1)–174.5(1) | 116.4(1)–118.5(1) |
| M = Yb | 1.936(11) 1.952(10) |
2.269(9) | 3.506(2) | 1.959(11)–1.985(12) | 2.274(9)– 2.351(11) |
4.620(1) | 172.0(4)–173.4(5) | 116.8(4)–118.8(4) |
Рентгенофазовый анализ поликристаллических образцов I–IV выполняли на дифрактометре с вращающимся анодом Rigaku D/MAX 2500 (Rigaku, Япония) в режиме отражения с использованием CuKα-излучения и графитового монохроматора, а также на дифрактометре STOE STADI IP в режиме пропускания (CuKα-излучение, монохроматор Ge (111)).
Исследование термического поведения образцов I–IV проводили на синхронном термоанализаторе Netzsch STA 409PC Luxx®, совмещенном с квадрупольным масс-спектрометром Netzsch QMS 403 C Aëolos®. Образцы (~10 мг) нагревали в динамическом токе аргона (30 мл/мин) до температуры 800°С со скоростью 2 град/мин в алундовом тигле. Во всем исследуемом интервале температур записывали масс-спектры продуктов разложения. Анализ термограмм и масс-спектров проводили при помощи программного обеспечения Netzsch Proteus®.
ИК-спектры поглощения кристаллических образцов I–IV регистрировали на приборах Nicolet 8700 (Thermo Scientific) (НПВО, разрешение 4 см–1) и Perkin Elmer Spectrum 3 (НПВО, разрешение 2 см–1) в диапазоне 4000–650 см–1.
Формирование предшественников молекулярной формы соединения I в водном растворе (1 мг/мл) изучали с помощью масс-спектрометрии с ионизацией методом электрораспыления (ESI-MS) в режимах регистрации положительных (+ESI) и отрицательных ионов (–ESI) на тандемном масс-спектрометрическом детекторе Waters ACQUITY TQD в диапазоне массовых чисел m/z от 50 до 2000.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Для получения гетерометаллических соединений с протяженными структурными фрагментами проводили серию синтезов с различным соотношением солей металлов.
Соединения I–IV образуются при явном избытке одного из металлов в реакционной смеси, например при соотношении Cu : РЗЭ = 1 : 2, что говорит о высокой устойчивости данного пятиядерного молекулярного фрагмента. Кристаллизация в таких растворах кинетически затруднена, а состав твердой фазы, формирующейся при кристаллизации, многокомпонентен (рис. S1 ). При увеличении доли солей РЗЭ в синтезах наблюдалось понижение кристалличности продуктов и образование аморфных осадков.
Методику синтеза соединений удалось оптимизировать для получения количеств однофазного продукта, достаточных для его характеризации методами РФА, ТГА, ESI-MS и ИК-спектроскопии.
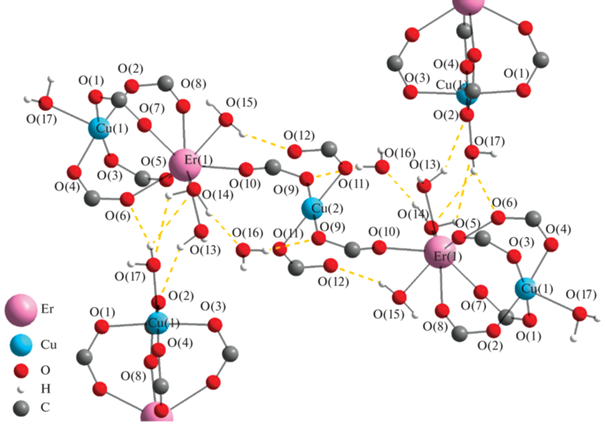
Согласно данным РСА (табл. 1, 2), комплекс I представляет собой центросимметричные цепочечные пятиядерные молекулярные фрагменты с линейной последовательностью ионов металлов Cu(1)–Er(1)–Cu(2)–Er(1)–Cu(1), связанных мостиковыми карбоксильными группами (рис. 1).
Рис. 1.
Молекулярный фрагмент структуры соединения I и его связывание водородными связями (пунктир) с соседними фрагментами. Дифторметильные группы не показаны.
Соединения I–IV обладают идентичной формулой координационного каркаса, но, несмотря на их очень близкие кристаллографические характеристики (табл. 3), не являются изоструктурными. Далее в тексте будем описывать такие соединения как изотипные. Атомные размеры и индуктивный эффект заместителя при карбоксильной группе сопоставимы и приводят к образованию веществ со схожим строением. Необходимо отметить, что подобные трифтор- и трихлорацетатные комплексы не удается синтезировать по ряду причин, таковыми являются сила кислоты, строение индивидуальных солей меди и РЗЭ, стерический фактор.
Таблица 3.
Сопоставление параметров элементарной ячейки в ряду полученных и известных из литературы изотипных соединений, пр. гр. P21/c
| Параметр | [Cu3M2СHF2COO)12(H2O)8]·2H2O | [Cu3M2(СH2ClCOO)12(H2O)8]·2H2O | ||||
|---|---|---|---|---|---|---|
| Nd* | Er** | Nd [10] | Gd [9] | Dy [10] | Yb [11] | |
| a, Å | 10.2464 (6) | 10.2339(5) | 10.4980(2) | 10.521(2) | 10.408(2) | 10.424(6) |
| b, Å | 17.5934(11) | 17.2025(8) | 17.9960(3) | 18.024(4) | 17.838(4) | 17.953(4) |
| c, Å | 15.7001(8) | 15.6227(8) | 15.0760(4) | 15.310(3) | 15.010(3) | 15.193(4) |
| β, град | 98.098(4) | 97.281(4) | 99.3500(6) | 99.85(3) | 99.65(3) | 100.18(1) |
В ряду CH3COOH, СH2ClCOOH, CHF2COOH, ССl3COOH, CF3COOH возрастает влияние заместителя, ослабевает донорная способность атома кислорода, что сказывается на характеристиках образуемых связей. Более ионный характер связи металл–кислород в случае трифторацетат-иона может приводить к невыгодности образования гетерометаллических соединений и, как итог, к образованию индивидуальных соединений. Различия в строении индивидуальных солей меди также могут являться причиной образования индивидуальных, а не гетерометаллических соединений. Так, кристаллогидраты трихлорацетата [15] и трифторацетата [16] меди имеют молекулярный мотив кристаллической структуры, образованный моноядерными частицами. Кристаллогидраты монохлорацетата [15] и ацетата [21] меди обладают структурой типа “китайский фонарик” – биядерные дискретные фрагменты, в которых ионы меди попарно соединены четырьмя мостиковыми RCOO-лигандами. Наибольшей устойчивостью такого фрагмента обладает ацетат меди, поэтому в случае уксусной кислоты образование гетерометаллических соединений не наблюдается. Дифторацетат меди известен своим слоистым 2D-полимерным строением [16]. Координационным полиэдром меди во всех перечисленных соединениях является квадратная пирамида, кроме гидрата трифторацетата меди, его полиэдром является тетрагонально искаженный октаэдр.
Кристаллическая структура трифторацетатов РЗЭ состоит из биядерных фрагментов [22, 23], что способствует первоочередной кристаллизации этих соединений. В то же время трихлорацетаты, монохлорацетаты и дифторацетаты РЗЭ обладают 1D-полимерной структурой [24–26]. Координационные возможности атомов РЗЭ определяются их размером – мостиковые анионы равномерно распределены по цепи, а молекулы воды дополняют окружение металлоцентров до максимально возможного (КЧ = 8–9).
Координационные возможности атомов меди и РЗЭ открывают огромную вариативность протяженных (полимерных) гетерометаллических структур, однако в нашем случае уместнее говорить об олигомеризации: пара “строительных” блоков CuML4 объединилась еще одним атомом меди, организовав пятиядерную устойчивую единицу, которая не может полимеризоваться дальше. Одной из возможных причин образования олигомерной структуры вместо полимерной может служить центросимметричный плоскоквадратный фрагмент CuL4 с атомом Cu(2), который в данной структуре оказался каркасообразующим. Такая форма комплекса уникальна тем, что ионы металлов связаны исключительно мостиковыми карбоксильными группами, а сама молекула не стабилизируется другими полидентатными лигандами, например, основаниями Шиффа [27–29].
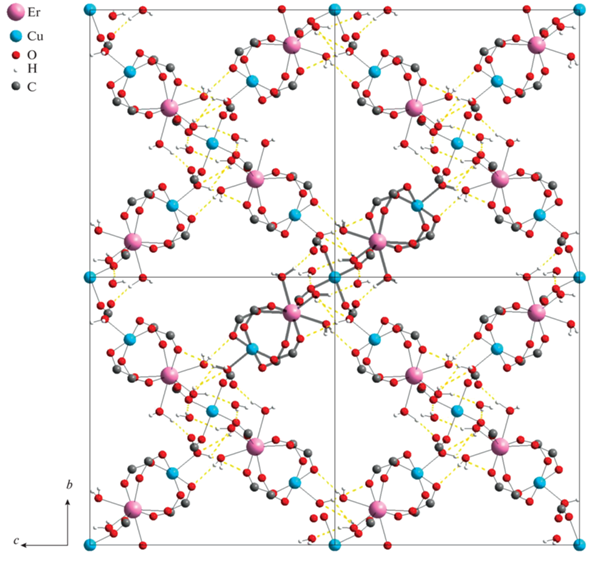
При формировании кристаллической упаковки пятиядерные молекулярные фрагменты и полостные молекулы воды связываются водородными связями, образуя трехмерную структуру (рис. 2). Большое число коротких контактов и плотное перекрестное расположение молекулярных фрагментов приводят к тому, что характерные расстояния между атомами металлов и атомами кислорода молекул воды не увеличены (Er–O 2.326–2.401(5) Å, Cu–O 2.190(6) Å), как это описано в [24]. Все десять молекул воды, входящих в структуру I, участвуют в связывании (O–H···O 2.798–3.591(9) Å), причем большинство этих атомов кислорода выступает в роли как акцепторов, так и доноров водородной связи, образуя по 2–3 подобных контакта.
Рис. 2.
Взаимное расположение фрагментов в кристаллической структуре соединения I в плоскости bc. Показаны водородные связи, дифторметильные группы скрыты для ясности.
Данные РСА согласуются с результатами рентгенофазового эксперимента (Robs = 5.18%, wRobs = 6.31%, Rall = 5.18%, wRall = 6.31%, GOF = 3.23, Rp = 9.67%, wRp = 12.70%), что подтверждает фазовую чистоту образца (рис. S3 ).
Изучение формирования предшественников молекулярной формы комплексов в растворе возможно с использованием ESI-MS. Электроспрей является очень мягким методом ионизации, поэтому масс-спектры соединений, полученные с использованием этого метода ионизации обычно не содержат пиков фрагментарных (осколочных) ионов [30]. Следует отметить, что при неудачном выборе экспериментальных условий или при анализе термолабильных соединений масс-спектры могут содержать пики, соответствующие как фрагментарным, так и аддуктным ионам.
Интенсивность аддуктных ионов и количество аддуктов сильно зависят от экспериментальных условий. Как правило, при увеличении напряжения на конусе (cone voltage) интенсивности молекулярных и псевдомолекулярных ионов растут, а суммарная интенсивность аддуктных ионов уменьшается (при этом относительные интенсивности аддуктных ионов изменяются в сторону увеличения интенсивности тех ионов, которые содержат меньшее количество присоединенных молекул растворителя). Так, в работе [31] на примере метанольного раствора CuCl2 показано, что при увеличении напряжения на конусе с 40 до 60 В интенсивность аддуктных ионов [Cu(MeOH)2]+ и [Cu(MeOH)]+ существенно снижается при одновременном увеличении интенсивности иона [Cu]+.
В результате нашего ESI-MS эксперимента в режиме –ESI и при оптимальном напряжении обнаружены ионы, приведенные в табл. 4.
Таблица 4.
Соотнесение чисел m/z с предполагаемым составом иона/аддукта. Приведены данные для иона с максимальной интенсивностью в изотопном кластере, значения округлены до целого числа
| m/z | Предполагаемый состав |
|---|---|
| 95 | [СHF2COO]– |
| 113 | [СHF2COO– + H2O]– |
| 253 | [Cu+(СHF2COO–)2] – |
| 348 | [Cu+2(СHF2COO–)3]– |
| 366 | [Cu+2(СHF2COO–)3 + H2O] – |
| 546 | [Er+3(СHF2COO–)4]– |
| 801 | [Er+3Cu+2(СHF2COO–)6]– |
| 819 | [Er+3Cu+2(СHF2COO–)6 + H2O]– |
| 1054 | [Er+3${\text{Cu}}_{{\text{2}}}^{{{\text{ + 2}}}}$(СHF2COO–)8]– |
| 1072 | [Er+3${\text{Cu}}_{{\text{2}}}^{{{\text{ + 2}}}}$(СHF2COO–)8 + H2O]– |
| 1506 | [Er2+3${\text{Cu}}_{{\text{2}}}^{{{\text{ + 2}}}}$(СHF2COO–)11] – |
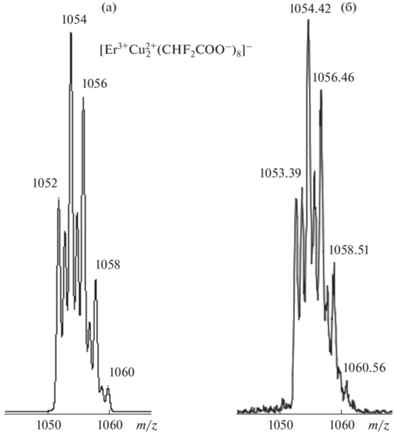
Изотопные кластеры ионов хорошо соответствуют рассчитанным (MassLynx, Isotope Model tool). В качестве примера приведены рассчитанный и экспериментальный изотопные кластеры для иона с m/z = 1054 (рис. 3).
Наличие иона с m/z = 253, содержащего в себе одновалентную медь, можно объяснить протеканием окислительно-восстановительной реакции в камере ионизации масс-спектрометра. В настоящее время возможность протекания окислительно-восстановительных реакций при ионизации методом электроспрея показана для ионов различных металлов (Cu, Hg, Fe) и является общеизвестным фактом [32].
Полный масс-спектр отрицательно заряженных ионов в диапазоне m/z от 50 до 2000 представлен на рис. 4. Исходя из сравнения экспериментальных изотопных кластеров с рассчитанными изотопными паттернами можно заключить, что ионы с m/z < 400 не содержат в своем составе эрбия, т.е. не являются целевыми.
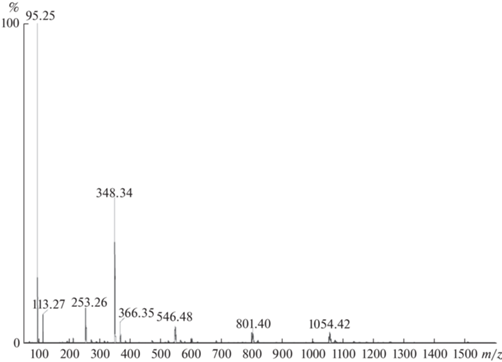
Наличие в масс-спектре образца ионов с m/z = = 801, 1054 и 1506 может служить подтверждением возможности образования в растворе относительно устойчивых ионных ассоциатов с чередующимися атомами меди и эрбия, связанными между собой карбоксильными группами анионов дифторуксусной кислоты. Эти ассоциаты являются предшественниками молекулярной формы комплекса I. Их наличие в растворе может служить подтверждением тезиса об олигомеризации.
Анализ масс-спектров, полученных в режиме +ESI, показал присутствие пиков, соответствующих медьсодержащим ионам, и отсутствие в масс-спектрах пиков, соответствующих эрбийсодержащим ионам.
ИК-спектроскопия является инструментом для определения типов координации. Карбоксилатные группы могут быть координированы ионами металлов различным образом. Известно, что если разница между частотами (∆) асимметричных и симметричных валентных колебаний O–C–O меньше, чем в свободном карбоксилат-ионе, это считается показателем мостикового бидентатного связывания или хелатирующей карбоксильной группы; если ∆ больше, то это указывает на монодентатность лиганда (псевдоэфирную координацию) [33]. Такая закономерность прослеживается при анализе спектров пропускания полученных пятиядерных комплексов, в которых карбоксильные лиганды имеют различные функции. В структуре можно выделить три группы эквивалентных лигандов: псевдоэфирные (монодентатные), мостиковые в фонариковых фрагментах и мостиковые к центральному иону меди, различающиеся длиной связей и величиной угла О–С–О. Полученные нами ИК-спектры НПВО кристаллических образцов соединений I–IV представлены на рис. 5.
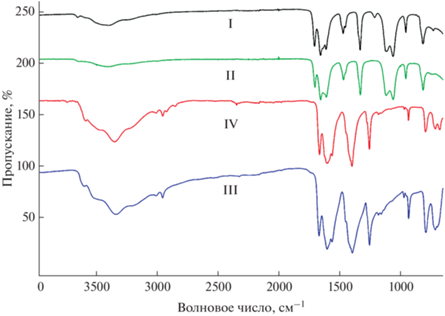
ИК-спектры дифторацетатных комплексов I и II ожидаемо повторяют контуры друг друга. Характерные интенсивные полосы валентных асимметричных (νas(COO)) и симметричных (νs(COO)) колебаний находятся при 1707–1610 и 1470 см–1 соответственно. Разрешение группы полос νas(COO) может быть объяснено с точки зрения описанной выше структурной функции лиганда. В спектре свободного дифторацетат-аниона (NaСHF2COO) величина ∆ составляет ~200 см–1 [34]. Полоса νas(COO) расщеплена на три пика: для левого, более отстраненного, ∆ = 236 см–1, что указывает на принадлежность к колебаниям νas(COO) монодентатных лигандов, а центральный и правый пики со значениями ∆ = 186 и 141 см–1 соответственно относятся к νas(COO) мостиковых лигандов.
Эффект влияния заместителей на частоты валентных колебаний карбоксильной группы возрастает в ряду СH2Cl < CHF2 ввиду большего индуктивного эффекта фторсодержащего заместителя. В спектрах монохлорацетатных комплексов III и IV полосы валентных колебаний связей O–C–O смещены в более длинноволновую область – νas(COO) при 1670–1561 см–1 и νs(COO) при 1398 см–1. Учитывая изотипность рассматриваемых соединений I–IV, можно заключить, что полосы, приписываемые колебаниям связей в лигандах с различной структурной функцией, смещены синхронно и имеют аналогичные соотнесения, и расчеты ∆ это подтверждают.
По причине того же эффекта можно наблюдать смещение полосы, которая относится к скелетным колебаниям ν(CC), при 813 см–1 в спектрах соединений I и II по сравнению с 932 см–1 в спектрах III и IV. Связь С–С нечувствительна к типу координации карбоксилатного лиганда, поэтому расщепление полосы не наблюдается.
Деформационные колебания связей С–H в монохлор- и дифторметильной группах проявляются в спектрах в виде интенсивных полос при 1254 см–1 для III и IV, приписываемых δ(CH2) в монохлорацетате, и при 1331 см–1 для I и II, отнесенных к δ(CH) в дифторацетате.
Две интенсивные полосы в спектрах I и II в интервале от 1117 до 1059 см–1 отвечают валентным колебаниям ν(CF2). Среднеинтенсивная полоса в спектрах III и IV при 711 см–1 относится к валентным колебаниям ν(CCl), а группа полос низкой интенсивности от 3020 до 2950 см–1 принадлежит валентным колебаниям ν(CH2). Поглощение в коротковолновой области всех спектров 3700–3100 см–1 соответствует валентным колебаниям ν(OH) молекул воды, участвующих в образовании водородных связей, и это согласуется с данными РСА.
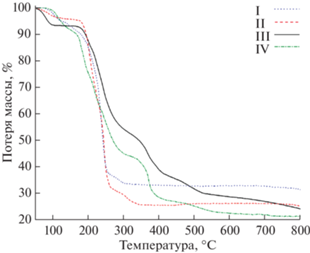
Анализ термического поведения соединений I–IV позволяет выделить две общие ступени разложения (дегидратация и декарбоксилирование), где происходит основная потеря массы, затем в зависимости от лиганда ход кривых существенно различается (рис. 6).
Рассмотрим первый этап термической деструкции соединений I и II. Дегидратация протекает в широком диапазоне температур, о чем свидетельствует увеличение интенсивностей ионных токов (рис. 7) с m/z = 18 (H2O+). Потеря массы ~10% до 180°С, судя по ДТГ-кривой и масс-спектрам ионных токов, соответствует удалению молекул воды, как полостных, связанных водородными связями, так и координированных металлоцентрами. Уход молекул воды сопровождается появлением осколочных ионов (ОН+), ввиду чего одними из промежуточных продуктов разложения могут быть гидроксокомплексы.
Рис. 7.
ТГ-кривая образца соединения I и кривые основных зарегистрированных ионных токов с m/z = 12, 16, 17, 18, 19, 44, 51.
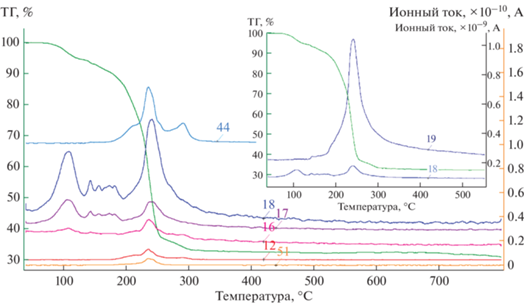
Второй этап характеризуется значительной потерей массы – 55% до 270°С. В этом диапазоне зарегистрированы ионные токи с теми же m/z, но относящиеся уже к осколочным фрагментам разложения галогенацетатных групп. Профили кривых ионных токов других осколочных частиц с m/z = 12 (С+), 19 (F+), 44 (${\text{CO}}_{2}^{ + }$) и 51 (${\text{CHF}}_{2}^{ + }$) в этом диапазоне совпадают.
Промежуточным продуктом разложения, предположительно, является фторокарбонат эрбия, о чем может свидетельствовать пик на кривой ионного тока ${\text{CO}}_{2}^{ + }$ в области 270–310°С. Иным вариантом может быть выделение углекислого газа как продукта взаимодействия фтора с материалом тигля и сажей, которая часто образуется при разложении органических частиц. Кривая ионного тока F+ показывает, что эти ионы присутствуют в камере вплоть до 500°С, кроме того, эти частицы расходуются, по всей видимости, на образование вторичных ионов ${\text{CF}}_{2}^{ + },$ которые далее через COF+ переходят в ${\text{CO}}_{2}^{ + }$ – на соответствующих кривых также имеется пик в диапазоне 270–310°С.
Твердый остаток составляет 33.5% для I и 32.4% для II, и по расчету потерь разложение происходит с образованием оксида одновалентной меди и фторида эрбия (3/2Cu2O + 2ErF3).
Для подтверждения расчета навески вещества I дополнительно разлагали в алундовом и платиновом тиглях в муфельной печи при 240 и 340°С в течение 4 ч в динамическом токе аргона. Анализ рентгенограмм твердых остатков разложения позволяет заключить, что оксид меди(I) образуется на более раннем этапе, чем фторид эрбия (рис. S4 ). Результаты РФА подтверждают расчеты по данным ТГА. Потеря массы в алундовом тигле составила 67.1%.
Отдельно стоит отметить, что продукты разложения в алундовом и платиновом тиглях различаются. После разложения в платиновом тигле в твердом остатке присутствует примесь металлической меди. Данное отличие можно объяснить отсутствием взаимодействия вещества с материалом тигля. Кроме того, на стенках кварцевого реактора, в котором проводилось разложение, в обоих случаях отмечен красноватый налет фторида эрбия. Таким образом, в системах наблюдается частичный массоперенос разлагаемого вещества с последующим разложением на более горячих стенках реактора.
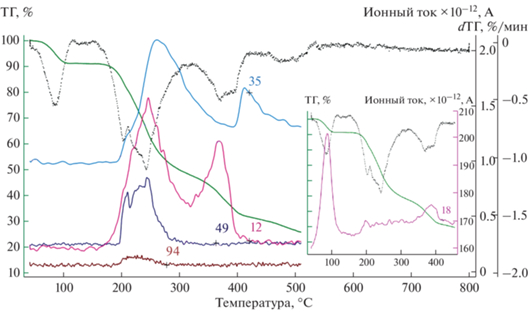
Характер термической деструкции при переходе от дифтор- к монохлорацетатам меняется в диапазоне высоких температур. Для соединений III и IV появляются третий и четвертый этапы разложения, характерные для хлорзамещенных галогенацетатов [35]. Дегидратация и декарбоксилирование протекают аналогично I и II, как было сказано выше. На втором этапе (180–310°С) можно наблюдать максимумы в аналогичном наборе кривых ионных токов с m/z = 12 (С+), 16 (О+), 35 (Cl+) и 44 (${\text{CO}}_{2}^{ + }$), а также 49 (СCl+) и 94 (СH2ClCOOH+) (рис. 8). Однако при дальнейшем повышении температуры в области 310–400°С можно наблюдать еще один не менее интенсивный пик на тех же кривых, за исключением m/z = 49 и 94. По всей видимости, на втором этапе происходит только частичное разрушение карбоксилат-аниона с разрывом связей C–Cl, и оставшиеся органические частицы представляют собой в какой-то мере видоизменившийся карбоксилат, возможно карбонат меди. Плавный ход ТГ-кривой не позволяет точно рассчитать массу промежуточных соединений, однако параллельный эксперимент по разложению образца III в муфельной печи при 400°С и РФА твердого остатка показали, что основным продуктом разложения является оксохлорид эрбия, образующийся в результате деструкции гексагидрата хлорида эрбия, который также присутствует в смеси в большом количестве (рис. S5 ). На кривых ионных токов HCl+, H2O+ наблюдаются соответствующие пики при этой температуре.
Рис. 8.
ТГ- и ДТГ-кривые образца соединения III и кривые основных зарегистрированных ионных токов с m/z = 12, 18, 35, 49, 94.
В статье [12] проведен ТГА изоструктурного соединения, [Cu3Y2(CH2ClCOO)12(H2O)8] · · 2H2O. Авторы указывают схожие температуры процессов разложения, но ввиду отсутствия регистрации ионных токов газообразных продуктов разложения и РФА твердого остатка, делают выводы исключительно на численных значениях потери массы. Так, считается, что вторым этапом (210–350°С) является полное декарбоксилирование с образованием смеси хлоридов CuCl2 + 2YCl3, а третьим (350–580°С) – разложение хлорида меди(II) до хлорида меди(I) c последующей его сублимацией и финальным остатком в виде хлорида эрбия. Мы не можем полностью согласиться со сделанными выводами. Появление широкого пика ионов хлора (m/z = 35, 37) в области 350–450°С может быть свидетельством разложения CuCl2 до CuCl, который, расплавляясь при 423°С, легче покидает среду разложения. По данным РФА, при 400°С в твердом остатке медь присутствует в основном в виде CuCl и CuO, а металлическая медь является незначительной примесью.
Потеря массы по ТГ-кривой к 530°С составляет 75%, что коррелирует с расчетом на две единицы оксохлорида эрбия с долей примеси оксида двухвалентной меди в остатке.
Более подробно данные ТГА всех четырех соединений представлены графически в дополнительных материалах (рис. S6–S9 ).
ЗАКЛЮЧЕНИЕ
Показано, что продуктом совместной кристаллизации индивидуальных галогенацетатов меди и РЗЭ являются комплексы, представляющие собой пятиядерные молекулярные единицы. Близкая природа лигандов – дифтор- и монохлорацетат-анионов – позволяет получать изотипные соединения, т.е. с идентичным металлическим остовом и параметрами элементарной ячейки при качественном атомном различии состава.
Для соединения I установлены состав и строение методом рентгеноструктурного анализа монокристалла, а также определены условия образования однофазного кристаллического образца. Для соединений I–IV установлены спектральные характеристики и изучена их термическая устойчивость. Характер термического разложения соединения I показывает восстановление до оксида одновалентной меди и металлической меди, как и в некоторых других карбоксилатах [36], и образование устойчивого фторида РЗЭ. Хлорсодержащие соединения III и IV разлагаются с образованием оксохлорида РЗЭ, оксида меди(II) и хлорида меди(I), причем CuCl сублимирует при высокой температуре. Промежуточным продуктом разложения является гексагидрат хлорида РЗЭ, устойчивый в широком диапазоне температур.
Методом ESI-MS установлено, что в водном растворе образуются многоядерные частицы-прекурсоры образования кристаллических молекулярных комплексов.
Список литературы
Can Xu, Chen S., Jia L. // Russ. J. Inorg. Chem. 2022. V. 67. P. 22. https://doi.org/10.1134/S0036023622601519
Ba Q., Qian J., Zhang C. // J. Clust. Sci. 2019. V. 30. P. 747. https://doi.org/10.1007/s10876-019-01534-7
Zhong L., Liu M., Zhang B. et al. // Chem. Res. Chin. Univ. 2019. V. 35. P. 693. https://doi.org/10.1007/s40242-019-9058-9
Васильев А., Волкова О., Зверева Е., Маркина М. Низкоразмерный магнетизм. Москва: ФИЗМАТЛИТ, 2018.
Goodenough J.B. Magnetism and the Chemical Bond. New Jersey: John Wiley & Sons, 1963.
Chen F., Lu W., Zhu Y., Wu B., Zheng X. // J. Coord. Chem. 2010. V. 63. № 20. P. 3599. https://doi.org/10.1080/00958972.2010.514904
Viola, Muhammad N., Ikram M. et al. // J. Mol. Struct. 2019. V. 1196. P. 754. https://doi.org/10.1016/j.molstruc.2019.06.095
Bovkunova A.A., Bazhina E.S., Evstifeev I.S. et al. // Dalton Trans. 2021. V. 50. № 35. P. 12275. https://doi.org/10.1039/d1dt01161h
Chen X.-M., Tong M.-L., Wu Y.-L., Luo Y.-J. // J. Chem. Soc., Dalton Trans. 1996. V. 10. P. 2181. https://doi.org/10.1039/DT9960002181
Voronkova V.K., Galeev R.T., Shova S. et al. // Appl. Magn. Reson. 2003. V. 25. P. 227. https://doi.org/10.1007/BF03166687
Cui Y., Zheng F.K., Yan D.C. et al. // Chin. J. Struct. Chem. 1998. V. 17. P. 5.
Zhang C.-G., Yan D., Ma Y., Yang F. // J. Coord. Chem. 2000. V. 51. P. 261. https://doi.org/10.1080/00958970008055132
Wojciechowski W., Legendziewicz J., Puchalska M., Ciunik Z. // J. Alloys Compd. 2004. V. 380. P. 285. https://doi.org/10.1016/j.jallcom.2004.03.056
Bateman W.G., Conrad D.B. // J. Am. Chem. Soc. 1915. V. 37. P. 2553.
Judd M.D., Plunkett B.A., Pope M. // J. Therm. Anal. 1976. V. 9. P. 83. https://doi.org/10.1007/BF01909269
Карпова Е.В., Болталин А.И., Коренев Ю.М., Троянов С.И. // Коорд. химия. 2000. Т. 26. № 5. С. 384.
Sheldrick G.M. // Acta Cryst. 2008. V. A64. P. 112. https://doi.org/10.1107/S0108767307043930
Sheldrick G.M. // Acta Cryst. 2015. V. A71. P. 3. https://doi.org/10.1107/S2053273314026370
Sheldrick G.M. // Acta Cryst. 2015. V. C71. P. 3. https://doi.org/10.1107/S2053229614024218
Brandenburg K., Berndt M. DIAMOND. Version 2.1e. Crystal Impact GbR. Bonn, 2000.
Niekerk J.N., Schoening F.R.L. // Acta Cryst. 1953. V. 6. P. 227. https://doi.org/10.1107/S0365110X53000715
Jangbo S., Rongzhi N., Xin S., Bo P. // Proceedings of SPIE. 2017. V. 10256. P. 1046357. https://doi.org/10.1117/12.2260699
Кавун В.Я., Кайдалова T.A., Костин В.И. и др. // Коорд. химия. 1984. Т. 10. № 11. С. 1502.
Анцышкина А.С., Порай-Кошиц M.A., Острикова В.Н. // Журн. неорган. химии. 1988. Т. 33. № 8. С. 1950.
Sugita Y., Ouchi A. // Bull. Chem. Soc. Jpn. 1987. V. 60. P. 171. https://doi.org/10.1246/bcsj.60.171
Oczko G., Starynowicz P. // J. Mol. Struct. 2000. V. 523. P. 79. https://doi.org/10.1016/S0022-2860(99)00391-9
Cristóvão B., Osypiuk D., Miroslaw B., Bartyzel A. // Polyhedron. 2020. V. 188. P. 114703. https://doi.org/10.1016/j.poly.2020.114703
Costes J.-P., Auchel M., Dahan F. et al. // Inorg. Chem. 2006. V. 45. № 5. P. 1924. https://doi.org/10.1021/ic050587o
Georgopoulou A.N., Pissas M., Psycharis V. et al. // Molecules. 2020. V. 25. № 10. P. 2280. https://doi.org/10.3390/molecules25102280
Herbert C.G., Johnstone R.A.W. Mass Spectrometry Basics N.Y: CRC Press, 2003. https://doi.org/10.1002/aoc.509
Schramel O., Michalke B., Kettrup A. // J. Chromatogr. A. 1998. V. 819. P. 231. https://doi.org/10.1016/S0021-9673(98)00259-3
Henderson W., McIndoe J.S. Mass Spectrometry of Inorganic, Coordination and Organometallic Compounds. New Jersey: John Wiley & Sons Ltd., 2005. https://doi.org/10.1002/0470014318
Deacon G.B., Phillips R.J. // Coord. Chem. Rev. 1980. V. 33. P. 227. https://doi.org/10.1016/S0010-8545(00)80455-5
The Matheson Company Inc. SpectraBase New Jersey: John Wiley & Sons, 1980.
Pushikhina O.S., Volkova K.R., Karpova E.V. et al. // Mendeleev Commun. 2022. V. 32. № 2. P. 208. https://doi.org/10.1016/j.mencom.2022.03.018
Judd M.D., Plunkett B.A., Pope M.I. // J. Therm. Anal. 1974. V. 6. P. 555. https://doi.org/10.1007/BF01911560
Дополнительные материалы
- скачать ESM.zip
- Приложение 1. Рис. S1. - Рис. S9.
Инструменты
Журнал неорганической химии