

Журнал общей биологии, 2022, T. 83, № 1, стр. 3-28
Концепция современных молекулярных часов и опыт оценки времен дивергенции насекомоядных и грызунов
А. А. Банникова 1, *, В. С. Лебедев 2, **
1 Московский государственный университет им. М.В. Ломоносова, биологический факультет
119234 Москва, Ленинские горы, 1, стр. 12, Россия
2 Зоологический музей МГУ им. М.В. Ломоносова
125009 Москва, ул. Большая Никитская, 2, Россия
* E-mail: hylomys@mail.ru
** E-mail: wslebedev@gmail.com
Поступила в редакцию 19.07.2021
После доработки 07.10.2021
Принята к публикации 24.10.2021
- EDN: SBKROS
- DOI: 10.31857/S0044459622010031
Аннотация
Почти 60 лет прошло с тех пор, как было обнаружено, что степень различия ДНК двух видов является функцией времени с момента их дивергенции. В дальнейшем стало ясно, что универсальных молекулярных часов не существует, скорость молекулярной эволюции широко варьирует в зависимости от гена и филогенетической линии, коррелируя с биологическими характеристиками (продолжительность поколения, размер тела, плодовитость) и особенностями генома. Развитие концепции молекулярных часов связано с прогрессом методов, позволяющих учитывать и измерять степень непостоянства скорости молекулярной эволюции. В настоящее время большинство датировок выполняется с помощью методов нестрогих часов (Relaxed clock), которые опираются на разнообразные модели скорости молекулярной эволюции (например, с автокорреляцией скоростей в соседних ветвях или без нее). Другой актуальный фактор, позволяющий уменьшить ошибки молекулярных датировок – увеличение объема данных вплоть до геномного уровня, что повышает требования к вычислительной эффективности алгоритмов и методам разложения данных на партиции. Отдельная проблема – нелинейная зависимость оценок скорости эволюции от времени (феномен rate decay), что особенно существенно для анализа недавней истории. В отсутствие адекватной модели эволюции последовательностей нуклеотидов оценки времен могут оказаться существенно смещенными, от чего часто страдают результаты, полученные по мтДНК. Одно из важнейших направлений – развитие методов получения калибровочной информации, прежде всего – более полное использование постоянно и быстро растущего объема палеонтологических данных, анализ палеоДНК и других вариантов гетерохронных данных. Несмотря на то, что точность оценок уровней молекулярных дивергенций продолжает расти, неопределенность датировок сохраняется во многом из-за неоднозначности калибровок и недостатков существующих моделей эволюции.
Начавшись с простой предпосылки, что эволюционные изменения на молекулярном уровне происходят с относительно постоянной скоростью (Zuckerkandl, Pauling, 1962), наши представления о молекулярных часах за прошедшие 60 лет претерпели существенную эволюцию. На протяжении этого времени значительные усилия были приложены к тому, чтобы понять причины изменения скорости эволюции на филогенетических древах и применить сам принцип молекулярных часов для оценки временных масштабов эволюции. Появился широкий спектр моделей молекулярных часов, которые допускают варьирование скорости эволюции и позволяют оценивать времена даже при их сильном различии в разных филогенетических линиях (Ho, Duchêne, 2014). Как результат, на основе молекулярного подхода возникла новая шкала времени. Причем в отличие от палеонтологических часов современные молекулярные часы предоставляют возможность датировать события дивергенции, относящиеся к широкому диапазону времен не только на филогенетическом, но и на популяционном уровне (Arbogast et al., 2002). В современной геномной эпохе откалиброванные по времени филогенетические древа – это фундаментальная отправная точка для исследования эволюции организмов.
ИСТОРИЯ КОНЦЕПЦИИ МОЛЕКУЛЯРНЫХ ЧАСОВ
Открытие молекулярных часов
Гипотеза молекулярных часов заключается в том, что скорость накопления замен в нуклеотидной или аминокислотной последовательности постоянна, из чего следует линейный характер зависимости между числом мутационных шагов (которые являются функцией числа различий в последовательностях) и временем дивергенции видов. Термин “молекулярные часы” (molecular evolutionary clock) был предложен Цукеркандлем и Полингом в 1965 г. (Zuckerkandl, Pauling, 1965). Сравнивая последовательности белков (гемоглобинов) у разных видов, они получили линейную зависимость числа аминокислотных замен от возраста видов, оцененного по ископаемым остаткам, и заявили о возможности использования этого свойства для оценки времени дивергенции между видами (Zuckerkandl, Pauling, 1962).
Изучая эволюцию гемоглобинов у приматов, Цукеркандль и Полинг оценили возраст дивергенции между гориллой и человеком в 11 млн лет (Zuckerkandl, Pauling, 1962), отметив, что это время соответствует минимальной (самой поздней) палеонтологической датировке (11–35 млн лет назад). Эти данные, так же как и другие молекулярные оценки времен дивергенции гоминоидов 60-х годов (Sarich, Wilson, 1967), оказались несовместимы с представлениями палеонтологов о большом эволюционном разрыве (масштабе фенотипической дивергенции) между человеком и человекообразными обезьянами (Wilson et al., 1977). В настоящее время относительно недавняя дивергенция человека и шимпанзе считается доказанной (Easteal, Herbert, 1997; Kumar et al., 2005). Вслед за работами Цукеркандля и Полинга по гемоглобинам появились сообщения о постоянстве скорости эволюции аминокислотой последовательности цитохрома С (Margoliash, 1963) и фибринопептидов (Doolittle, Blombäck, 1964), которые показали, что разные белки и гены отличаются по скорости эволюции и поддержали гипотезу молекулярных часов.
Цукеркандль и Полинг предвидели проблемы, которые могли затруднить применение молекулярных часов (Zuckerkandl, Pauling, 1962). Они указывали на повторяющиеся замены в одном и том же аминокислотном сайте (включая обратные мутации), воздействие естественного отбора и влияние размера популяции. Исходно их концепция включала важную роль естественного отбора, но позже они предположили, что “изменения, которые происходят с довольно регулярной общей скоростью, должны быть такими, которые относительно мало изменяют функциональные свойства молекулы” (Zuckerkandl, Pauling, 1965, р. 148). Эта цитата предвосхищает тесную связь гипотезы молекулярных часов Цукеркандля и Полинга с нейтральной теорией молекулярной эволюции Кимуры (Kimura, 1968, 1969), неотъемлемой частью которой они стали.
Теория нейтральности молекулярной эволюции (neutral theory of molecular evolution)
Мотоо Кимура и Томоко Ота объяснили постоянную скорость замен аминокислот в белках их нейтральностью в том смысле, что эти замены не влияют на приспособленность организма и, следовательно, не находятся под давлением отбора (Kimura, 1968; Kimura, Ohta, 1971). Предложенная модель отличалась от более ранних тем, что в прежних моделях (Fisher, 1936; Mayr, 1963) большинство замен предполагались благоприятными (попадают под положительный отбор), либо вредными (удаляются отбором). Важное следствие теории нейтральности состоит в том, что скорость, с которой нейтральные мутации фиксируются в популяции (substitution rate), примерно равна скорости, с которой мутации спонтанно возникают (Kimura, 1968). Решающий аргумент в пользу теории нейтральности – скорость несинонимичных (аминокислотных) замен в кодирующих последовательностях ниже, чем скорость синонимичных (“молчащих”) замен, и скорость замещений в интронах и псевдогенах.
Развитием теории нейтральности молекулярной эволюции стала теория почти (эффективно) нейтральных мутаций (the nearly neutral theory of molecular evolution), разработанная Ота (Ohta, 1972a, 1973). Теория почти нейтральных мутаций предполагает, что часть нейтральных мутаций имеет небольшое влияние на приспособленность, являясь умеренно вредными или (реже) умеренно полезными. В малочисленной популяции, где отбор слаб по сравнению с дрейфом, такие мутации ведут себя практически как нейтральные (эффективно нейтральные). Таким образом, при низкой эффективной численности доля нейтральных мутаций относительно вредных оказывается выше и скорость эволюции увеличивается. Тем не менее это не противоречит постоянству скорости эволюции в единицу времени благодаря отрицательной корреляции между размером популяции и продолжительностью жизни поколения (Ohta, 1972b, 1974).
Концепция молекулярных часов вызвала резкую критику со стороны крупных биологов-эволюционистов своего времени (например, Stebbins, Lewontin, 1972; Easteal et al., 1995). Дальнейшее накопление эмпирических данных показало, что поведение молекулярных часов не соответствует в полной мере модели постоянной скорости – часы оказываются “небрежными” (“sloppy” clock), так как дисперсия числа генетических различий в конкретном временном периоде оказывается выше ожидаемой исходя из простого пуассоновского процесса (Gillespie, 1991). Теоретически на скорость молекулярной эволюции должны влиять скорость мутирования, соотношение сайтов различной селективной значимости и (в случае почти нейтральных мутаций) размер популяции (Ohta, 2002). Эти факторы различаются между видами, генами и временными периодами. Непостоянство часов есть следствие изменчивости скорости замен внутри одной линии и неравенства скоростей замен в разных линиях. Оба фактора порождают ошибки в молекулярных датировках (Bromham, Penny, 2003).
Таким образом, уже вскоре после появления концепции молекулярных часов стало ясно, что универсальных часов не существует. Все дальнейшее развитие методологии опиралось на допущение, что степень непостоянства скорости молекулярной эволюции можно измерить и учесть (Bromham, Penny, 2003). За последние два десятилетия было предпринято множество усилий по разработке методов работы с варьирующими скоростями, что привело к значительному прогрессу в методах молекулярного датирования.
СКОРОСТЬ МОЛЕКУЛЯРНОЙ ЭВОЛЮЦИИ
Варьирование скорости молекулярной эволюции в зависимости от разных факторов
Скорость появления мутаций зависит от нескольких факторов и по современным представлениям определяется балансом между ожидаемым (в основном негативным) влиянием спонтанных мутаций на приспособленность с одной стороны и затратами на исправление ошибок репликации с другой (Lynch, 2010а). В рамках этой концепции скорость молекулярной эволюции не может быть меньше порога, определяемого эффективностью отбора в условиях той или иной величины дрейфа (drift-barrier hypothesis; Lynch, 2010b). Снижение скорости спонтанных мутаций организма может быть ограничено биохимическими и физиологическими затратами, связанными с улучшением точности репликации (Drake et al., 1998). Чтобы ограничить приток вредных мутаций, очищающий отбор может снижать частоту спонтанных мутаций за счет повышения точности репликации. Однако очищающий отбор подавляется генетическим дрейфом, если повышение приспособленности от дальнейшего снижения скорости спонтанных мутаций в диплоидном организме становится меньше, чем 1/2 Ne (где Ne – эффективная численность). Это объясняет тот факт, что виды с высокой Ne популяций (т.е. более эффективным отбором) обычно демонстрируют более низкую частоту спонтанных мутаций на поколение, чем виды с низкой Ne.
Скорость эволюции нуклеотидных и аминокислотных последовательностей меняется в зависимости от локуса, временного диапазона и конкретной филогенетической линии. Хорошо известно и многократно показано, что у многоклеточных животных частота мутаций в митохондриальном геноме много выше, чем в ядерном (Brown et al., 1979; Wilson et al., 1985), например, у человека и мыши примерно в 40 раз (Fu et al., 2013; Hardouin, Tautz, 2013). Особенно велики эти различия для синонимичных замен (Pesole et al., 1992). У насекомых и паукообразных это различие между скоростью митохондриальной и ядерной эволюции значительно ниже, чем у позвоночных (Allio et al., 2017). Варьирование скорости в зависимости от локуса может быть связано с тем, в какой степени локусы находятся под давлением отбора, а также их различиями в скорости мутирования (Hodgkinson, Eyre-Walker, 2011). При анализе нуклеотидных последовательностей, как правило, принимается во внимание варьирование скоростей в трех позициях кодонов белок-кодирующих генов (Shapiro et al., 2006). Скорость мутаций может варьировать также в зависимости от конкретных нуклеотидов и их окружения, например, мутации в цитозин-гуаниновых динуклеотидах (CpG) происходят чаще, чем в других динуклеотидах, частично из-за склонности цитозина к дезаминированию (Bird, 1980). Cкорость CpG → TpG замен варьирует в широких пределах у разных видов, есть данные, что у человека эта скорость может быть в ~13−18 раз выше по сравнению с другим динуклеотидами (Nachman, Crowell, 2000; Kong et al., 2012). Известно, что частота мутаций повышена в тех частях генома, где присутствуют горячие точки рекомбинации (Duret, Arndt, 2008), что отчасти связано с увеличением доли цитозина и гуанина и количества CpG сайтов вследствие ассимметричной генной конверсии в процессе рекомбинации.
Скорость молекулярной эволюции и эффект времени генерации (longevity effect)
В литературе упоминается два варианта скорости эволюции: (1) число замен на сайт в единицу времени и (2) число замен на сайт на поколение (Bromham, 2020; Pfeifer, 2020). Как обсуждалось выше, согласно теории нейтральности, эволюция в нейтральных сайтах происходит со скоростью мутаций и, в соответствии с гипотезой молекулярных часов, скорость мутаций должна быть одинакова в разных филетических линиях. Однако на самом деле скорость эволюции сильно различается между линиями. Уже в первых иммунологических работах по гемоглобинам приматов было показано, что линии с большей продолжительностью жизни поколения имеют меньшую скорость эволюции (Goodman, 1961, 1962). Реальность этой модели, известной как “эффект времени генерации”, теперь подтверждена многочисленными исследованиями на разных животных (Allio et al., 2017), в том числе на птицах (Mooers, Harvey, 1994), беспозвоночных (Thomas et al., 2010), а также бактериях (Weller, Wu, 2015).
В настоящее время принято считать, что влияние продолжительности жизни поколения на скорость эволюции – наиболее очевидная причина различия скоростей молекулярной эволюции в разных линиях (Ho, 2020). Однако исходные утверждения Кимуры (Kimura, 1969) противоречивы: с одной стороны, скорость эволюции белков у разных видов примерно одинакова, а с другой стороны, примерно одинаковы скорости мутирования белков в пересчете на поколение (а не на единицу времени), но продолжительность жизни поколения сильно отличается у разных животных. Сам Кимура сначала считал, что синонимичные замены происходят с постоянной скоростью в пересчете на год, что могло бы служить подтверждением теории нейтральности (Kimura, 1969). Но позже он признал, что нейтральная теория должна предсказывать постоянную скорость замещения не на год, а на поколение, так как скорость зависит от числа циклов репликации, и доказательство постоянства эволюционных изменений в единицу времени представляет собой “трудную проблему” в его теории (Kimura, 1983).
Для объяснения этого факта привлекалась теория почти (эффективно) нейтральных мутаций (Ohta, 1972a). Суть объяснения состояла в том, что виды с долгоживущими поколениями, как правило, имеют более низкую эффективную численность популяции (Ne) и, следовательно, для них выше доля условно нейтральных мутаций. Однако полностью адекватным это объяснение не является, реальная причина сложней и зависит от нескольких факторов.
Во-первых, молекулярная эволюция неравномерна и у животных с большим временем генерации скорость нуклеотидных замен меньше (например, Nabholz et al., 2008, 2013; Welch et al., 2008; Galtier et al., 2009), однако зависимость между скоростью эволюции и величиной, обратной времени генерации отнюдь не линейна. Время генерации отличается у человека и мыши примерно в 40–50 раз, а скорость эволюции менее чем в 10 раз (Bromham, 2011).
Во время работы Кимуры над книгой (Kimura, 1983) предполагалось, что мутации возникают прежде всего в момент репликации ДНК половых клеток или их предшественников (Haldane, 1947; Müller, 1954) и что число циклов деления в линии предшественников половых клеток примерно одинаково у организмов с различной длиной поколения. Однако теперь ясно, что это допущение не верно; так, у человека в клетках-предшественниках сперматозоидов происходит в среднем 400 делений на поколение, а у самцов мыши − всего 62 деления (Bromham, 2011). Следовательно, строго обратной зависимости между временем генерации и скоростью эволюции, как ожидалось ранее, на самом деле быть не должно.
С другой стороны, если бы скорость репликации на единицу времени была постоянной, между временем генерации и скоростью эволюции вообще не было бы связи. На самом же деле число клеточных делений в единицу времени больше у короткоживущих видов, например, у мыши (самцы) более 100 делений в год, а у человека всего около 20 делений (Bromham, 2011). Отметим, что принимать во внимание число циклов деления и скорость мутаций нужно именно в сперматозоидах, а не в яйцеклетках. Кроме того, скорость спонтанных мутаций на одно деление у короткоживущих видов выше, чем у долгоживущих, например, у мыши этот показатель примерно в 3 раза выше, чем у человека (Milholland et al., 2017). Именно эти два фактора и определяют большую скорость мутирования у короткоживущих видов.
Есть и другой, осложняющий эту картину фактор: не все мутации вызываются ошибками репликации. Например, у многих организмов цитозин может химически модифицироваться путем метилирования (Suzuki, Bird, 2008). Основные мишени метилирования ДНК в геномах − это цитозины, за которыми следует гуанин (“CpG” мишени); по химическим причинам метилированные CpG сайты очень склонны к превращению в TpG сайты (Bird, 1980). Поскольку метилирование ДНК само по себе не зависит от репликации (например, Vandiver et al., 2015), на мутации, вызванные метилированием, продолжительность жизни поколения влиять не должна. Показано, что у разных видов приматов мутации в сайтах CpG происходят с одинаковой скоростью, в то время как скорости мутаций в других сайтах варьируют в зависимости от времени генерации (Kim et al., 2006; Moorjani et al., 2016). Итак, скорости эволюции в расчете на поколение и на единицу времени связаны между собой сложно и не линейно.
Причины вариабельности скоростей молекулярной эволюции
Одним из наиболее ярких примеров зависимости скорости мутирования ДНК от частоты репликации является феномен, который часто называют “male-driven evolution” или “male-biased mutation”. Мужские гаметы обычно производятся в большем количестве, чем женские, и мужские гаметы обычно являются продуктом большего количества поколений клеток, чем женские гаметы того же вида. Таким образом, во многих таксонах последовательности ДНК, которые проводят больше времени у самцов, будут проходить больше репликаций в единицу времени, чем те, которые проводят больше времени у самок, и поэтому будут накапливать больше ошибок копирования. Это было отмечено у млекопитающих (более высокая частота мутаций в хромосоме Y, чем в X) и у птиц (более высокая частота мутаций в хромосоме Z, чем в хромосоме W) (Wilson Sayres, Makova, 2011). Таким образом, средняя частота мутаций для вида будет сильно зависеть от мутаций, происходящих у самцов во время производства гамет (Gao et al., 2016).
При использовании молекулярных часов наибольшее значение имеет варьирование скоростей в разных филогенетических линиях. Одним из возможных объяснений стойких видоспецифичных различий в скорости мутаций является то, что относительная “стоимость” мутации различается между видами. Если так, можно ожидать, что вклад в репарацию также будет различаться, и, следовательно, можно модулировать среднюю частоту мутаций (Bromham, 2002). Показано, что помимо эффективного размера популяции, со скоростью молекулярной эволюции могут быть тесно связаны размер генома, интенсивность метаболизма, размер тела, плодовитость и продолжительность жизни.
Размер генома. Ранее было высказано предположение, что число мутаций за поколение на геном – величина постоянная (эффект Дрейка; Drake et al., 1998), а частота мутирования обратно пропорциональна размеру генома. Позднее было показано, что это правило соблюдается для вирусов и бактерий, в то время как у многоклеточных зависимость противоположная, частота мутаций тем выше, чем больше размер генома (Pfeifer, 2020). Чтобы более тщательно оценить эту закономерность в филогенетических линиях необходимо включение в исследования более широкого набора видов.
Интенсивность метаболизма и размер тела. Обычно считается, что скорость эволюции непрямым образом, через время генерации и размеры тела, связана с интенсивностью метаболизма (например, Martin, Palumbi, 1993; Gillooly et al., 2005). Крупные животные и высокие растения обычно имеют относительно низкую скорость молекулярной эволюции (Martin, Palumbi, 1993; Bromham, 2002; Gillooly et al., 2005; Lanfear et al., 2013; Barrera-Redondo et al., 2018). Это может объясняться, например, тем, что более крупные животные имеют более низкий метаболизм, поэтому генерируют меньше свободных радикалов в единицу времени и потому их ДНК меньше повреждается. С другой стороны, более крупное животное имеет больше клеток и, следовательно, больше копий генома, каждая из которых подвержена мутированию. Риск опасной для жизни мутации должен увеличиваться с увеличением количества поколений клеток, необходимых для создания тела, и количества клеток, необходимых в течение репродуктивной жизни. Таким образом, вид с большим средним размером тела может потребовать больших вложений в контроль повреждений ДНК или точность репликации (Nunney, 1999).
Однако убедительных доказательств того, что скорость молекулярной эволюции объясняется, например, именно интенсивностью метаболизма, пока мало, если учесть ее связь и с другими чертами биологии организмов (Bromham et al., 1996; Lanfear et al., 2007; Galtier et al., 2009). Более того, влияние продолжительности жизни на молекулярную эволюцию было отмечено у таксонов, которые не проявляют признаков старения с возрастом (Hua et al., 2015). Возможно, уровень репарации ДНК регулируется до уровня риска, возникающего из-за клеточных метаболитов, и уравновешивается средней стоимостью мутации.
Продолжительность жизни. Показано, что скорость мутирования мтДНК у млекопитающих, птиц и рыб определенно связана с продолжительностью жизни, но этого нельзя с уверенностью утверждать про скорость эволюции ядерных генов (Nabholz et al., 2008; Welch et al., 2008; Galtier et al., 2009; Hua et al., 2015). Некоторые исследователи объясняют это наблюдение метаболическим повреждением ДНК. В результате аэробного метаболизма в митохондриях образуются свободные радикалы кислорода, которые могут повредить молекулы ДНК. Если удлинение жизни приводит к увеличению продолжительности и репродуктивной жизни, то риск мутации увеличивается (Bromham, 2020).
Плодовитость. Для млекопитающих известны исследования, указывающие на связь плодовитости как со скоростью синонимичных, так и несинонимичных замен (Welch et al., 2008), что может быть связано с большей эффективностью отбора при высокой численности потомства. В целом, определение конкретных особенностей биологии вида, влияющих на скорость молекулярной эволюции, – весьма сложная задача, так как все биологические факторы тесно скоррелированы.
Молекулярная эволюция и скорость диверсификации
С точки зрения Хо (Ho, 2020), серьезные проблемы для использования методов молекулярного датирования могут создавать три формы изменения скорости нуклеотидных замен: взрыв генетических изменений при событиях видообразования (прерывистая эволюция); разная скорость эволюции в течение разных временных периодов (эффект эпох); снижение наблюдаемой скорости с течением времени (rate decay).
Теория прерывистого равновесия (punctuated equilibrium theory) была выдвинута в попытке объяснить пробелы в палеонтологической летописи, которые, по-видимому, характеризуются длительными периодами стагнации, сменяющимися быстрыми всплесками морфологических изменений (Eldredge, Gould, 1972). Вдохновленные этой теорией, молекулярные эволюционисты искали доказательства прерывистой молекулярной эволюции (вспышек молекулярной изменчивости) в связи с эффектом основателя в событиях видообразования (Webster et al., 2003; Pagel et al., 2006). Теоретически доля генетических изменений должна коррелировать с числом событий видообразования в каждой ветви эволюционного древа. Однако простоте таких тестов препятствует проблема, известная как эффект плотности узлов (node-density effect), которая порождает паттерн, подобный ожидаемому при прерывистой молекулярной эволюции (Fitch, Beintema, 1990).
Возможные механизмы, связывающие видообразование со скоростью молекулярной эволюции, должны проявляться только в скорости несинонимичных замен (Bromham, 2020). К этим механизмам относятся отбор, влияющий на полезные замены, и ускорение эффективно нейтральных процессов при прохождении демографического кризиса. Тем не менее связь между скоростью диверсификации и скоростью молекулярной эволюции продемонстрирована и для синонимичных замещений (Lanfear et al., 2010; Duchêne, Bromham, 2013; Bromham et al., 2015). Одно из возможных объяснений связи скорости видообразования и скорости мутаций (в том числе и синонимичных) состоит в том, что уменьшение размера популяции может приводить к снижению эффективности репарации за счет частично вредных мутаций в генах, кодирующих ферменты, ответственные за репарацию и исправление ошибок (proofreading) в процессе репликации ДНК (Lynch et al., 2016). Однако корреляция между скоростью диверсификации и скоростью молекулярной эволюции может быть объяснена и без использования гипотезы прерывистой генетической эволюции если допустить, что повышенная скорость мутирования сама по себе, вне зависимости от ее причин, может быть предпосылкой для ускоренного видообразования (например, через увеличение скорости появления несовместимых замен в изолированных популяциях в рамках модели Добжанского–Меллера) (Hua, Bromham, 2017). Вторая гипотеза предпочтительнее, поскольку проще объясняет связь между видовым разнообразием и скоростью синонимичных замен.
Обратная зависимость между оценкой скорости эволюции и временем (rate decay)
Среди перечисленных выше видимых форм изменения скорости эволюции наиболее распространенной является обратная зависимость между оценкой скорости эволюции и временем (rate decay). Этот феномен проявляется в отсутствии линейной связи между временем дивергенции и генетическим расстоянием, что вносит систематическую ошибку в оценку возраста дивергировавших групп. В результате скорости эволюции, измеряемые между видами (филогенетические) и в пределах вида (популяционные), оказываются различными. Эта проблема возникает, прежде всего, при работе с быстро эволюционирующей мтДНК, а сам феномен объясняется, как предполагалось, наличием горячих точек мутирования и условно нейтральных мутаций (Ho et al., 2005, 2007; Henn et al., 2008). Влияние rate decay на медленно эволюционирующие сайты и локусы менее значительно.
На недавних временах оценки темпов эволюции могут быть завышены за счет включения вредных мутаций, которые, как правило, удаляются из популяции очищающим отбором в течение более длительных периодов времени (Ho et al., 2011). Насыщение нуклеотидной последовательности (многократные замены в одном сайте), наоборот, вызывает недооценку количества генетических изменений на далеких эволюционных временах (Soubrier et al., 2012).
Поскольку скорость нуклеотидных замен для недавних событий значительно выше, чем для отдаленных, филогенетические скорости, полученные на основе древних калибровочных точек, могут оказаться непригодными для датирования внутривидовых межпопуляционных дивергенций. Следовательно, для надежной калибровки часов необходимо использовать не только отдаленные события дивергенции, но и относительно недавние, в том числе произошедшие в плейстоцене–голоцене (<1−2 млн лет). Например, скорость формообразования для оценки возраста такого дихотомического события, как разделение Sorex araneus/S. granarius равна 13.6% за миллион лет, а для разделения S. daphaenodon и S. araneus – 8.6% (Bannikova et al., 2010b); для расчета времен дивергенции между географическими популяциями S. araneus s.str. использовали популяционную скорость нуклеотидных замещений 2μ = 21–37%, в среднем 27.8% (Распопова и др., 2018; Raspopova et al., 2020).
Феномен rate decay согласуется с разницей между высокой скоростью мутирования, оцениваемой по родословным (pedigree rate – сравнение родителей и потомков) и медленной филогенетической скоростью (сравнение видов) (Santos et al., 2005). Доказательства систематических ошибок, зависящих от времени дивергенций и связанных с перенесением времени популяционных дивергенций на филогенетические дивергенции и наоборот, получены для вирусов и бактерий (Aiewsakun, Katzourakis, 2016; Duchêne et al., 2016). Для ядерных геномов Metazoa такие данные ограничены, хотя показано, что частота спонтанных мутаций выше, чем скорость долгосрочной молекулярной эволюции, оцененная с помощью филогенетических методов (но современный человек – исключение из этого правила) (Scally, 2016; Chintalapati, Moorjani, 2020).
Феномен rate decay получил подтверждение по результатам исследования палеоДНК (Ho et al., 2007; Shapiro et al., 2011). Так, филогенетические скорости (мутирования) цитохрома b для серых полевок, оцениваемые по времени радиации рода, составляют около 10−11% (Bannikova et al., 2010a). С другой стороны, скорости, полученные путем анализа гетерохронных сиквенсов (ископаемые голоценовые Microtus arvalis), значительно выше – 32.7% (Martínková et al., 2013). К этому близки скорости, рассчитанные на основе сценариев колонизации M. agrestis севера Европы (плейстоцен-голоценовая граница) – 45.7% (Herman et al., 2014).
Критика концепции rate decay
Концепция rate decay подвергалась активной критике (например, Emerson, Hickerson, 2015). Предполагалось, что увеличение оценок скорости отражает не реальный биологический феномен, а артефакт, связанный с недооценкой роли предкового полиморфизма (Emerson, 2007; Peterson, Masel, 2009; Tuffley et al., 2012), особенностей демографической истории и структурированности популяций (Navascues, Emerson, 2009). Было показано, что при использовании коротких последовательностей и недавних времен дивергенций (что часто бывает при использовании гетерохронных сиквенсов палеоДНК) оценки скорости могут быть завышены иногда в несколько раз (Debruyne, Poinar, 2009; Ho et al., 2011), т.е. смещенная оценка возникает именно вследствие низкой информативности данных. Кроме этого, эффективно нейтральные процессы вряд ли могут объяснить наблюдаемый масштаб rate decay, в силу нереалистичности требуемых для этого демографических сценариев (Woodhams, 2006).
Тем не менее вся вышеперечисленная критика скорее указывает на то, что феномен rate decay обусловлен совокупностью разных причин, относительный вклад которых требует дополнительного изучения. Это не меняет того факта, что датировки, основанные на использовании прямолинейных подходов к анализу быстро мутирующих последовательностей ограниченной длины, часто оказываются существенно смещенными (Ho et al., 2015a). До сих пор недостаточно изучено значение ошибок в определении модели эволюции и распределения скоростей изменчивости сайтов (Soubrier et al., 2012). Так, не ясно в какой степени важен эффект присутствия в мтДНК горячих точек мутирования (Galtier et al., 2006), положение которых само по себе может достаточно быстро меняться.
Исходно предполагалось, что эффект rate decay проявляется на относительно недавних временах. Остается неясным, в какой степени это явление связано со сходным фактом зависимости оценок скоростей эволюции от времени дивергенции на временных шкалах эволюционного масштаба (Molak, Ho, 2015). Очевидно, что существующие модели эволюции не могут адекватно описать характер эволюции некодирующих последовательностей при высоком уровне дивергенции: по мере приближения к насыщению третьи позиции кодонов мтДНК будут все в большей степени недооценивать реальные уровни дивергенции. Отсутствие адекватных моделей для описания эволюции сайтов, находящихся под сильным очищающим отбором (1-е и 2-е позиции кодонов), вероятно, приводит к отсутствию линейной зависимости между степенью дивергенции по синонимичным и несинонимичным заменам на разных уровнях (рис. 1).
Рис. 1.
Соотношение уровней дивергенции митохондриального генома Soricinae по разным типам замен. а – по оси абсцисс отложены глубины узлов филогенетического дерева ML, модель CF + Г (Г = 0.43; PAUP v. 4.0b10); по оси ординат – все замены 3-го положения, модель GTR + Г. б – по оси абсцисс – то же, что на графике а, по оси ординат все замены 1-го и 2‑го положения, модель GTR + I + Г. В обоих случаях регрессия достоверно не линейная (Statistica v. 8.0, Nonlinear estimation).

Возникает вопрос, каким способом использовать мтДНК для датировок? Вполне реалистично предположение, что уровень дивергенции по трансверсиям третьих положений кодона (tv3) мало подвержен влиянию rate decay (из-за медленного насыщения) и учитывая тот факт, что характер эволюции tv3 лучше соответствует строгим часам, чем в случае других типов замен (Irwin et al., 1991). Если в анализе используются данные по всем белок-кодирующим митохондриальным генам, рационально использовать трансверсии в четырехкратно вырожденных позициях, которые, в отличие от tv3, в целом демонстрируют большую выровненность скоростей в разных сайтах. Сравнение динамики дивергенции по разным группам замен может быть проведено с использованием нелинейной регрессии (например, Cosson et al., 2005; Bannikova et al., 2010a, b).
МОЛЕКУЛЯРНЫЕ ЧАСЫ КАК ИНСТРУМЕНТ ДЛЯ ПОЛУЧЕНИЯ ДАТИРОВОК
Методы молекулярного датирования – современный этап
Датирование событий дивергенции методом молекулярных часов, или просто “молекулярное датирование” подразумевает оценку времени расхождения филетических линий на основе анализа молекулярно-генетических данных. В настоящее время использование молекулярных часов для датирования эволюционных событий довольно быстро и значительно прогрессирует в связи с ростом вычислительной мощности филогенетических методов (Bromham, Penny, 2003; Kumar, 2005). Примечательно, что прогресс в использовании методов молекулярного датирования во многом связан с усложнением моделей эволюции, что отличает современные исследования от их более раннего этапа и является магистральным направлением современной филогенетики. Исследования в этой области привели к разработке новых методов расчета времен дивергенции таксонов (Heath, Moore, 2014; Ho, Duchêne, 2014; Kumar, Hedges, 2016). Прежде всего, к ним относятся методы, позволяющие справиться с вариациями скорости эволюции не только между генами, но и между линиями, т.е. методы нестрогих часов (Hasegawa et al., 1989; Sanderson, 1997, 2002; Thorne et al., 1998).
Другой фактор – увеличение объема доступных для анализа данных, что в сочетании с адекватными моделями эволюции нуклеотидных последовательностей приводит к уменьшению доверительных интервалов и, следовательно, к более точным оценкам возраста узлов ветвления и скоростей эволюции (Tajima, 1993; Bromham et al., 2000; Kumar, 2005; Reis et al., 2015). Наконец, важную роль играет развитие разнообразных подходов к использованию калибровочной информации – более полное использование постоянно и быстро растущего объема палеонтологических данных (Kumar, 2005; Reis et al., 2015).
Основные принципы получения молекулярных датировок
Основные этапы процедуры получения молекулярных датировок представлены на схеме (рис. 2) и состоят в следующем: (1) разбиение данных на партиции и получение филогенетического древа по выбранным генам, (2) проверка часов: тест на равенство скоростей, (3) подбор модели часов для каждой партиции (гена или группы генов), (4) в случае обращения к свободным часам (Relaxed clock), выбор скоррелированных или нескоррелированных часов, (5) калибровка часов, (6) получение относительных и абсолютных времен, построение хронограммы. Рассмотрим более подробно наиболее сложные из этих этапов и обсудим возможные ошибки.
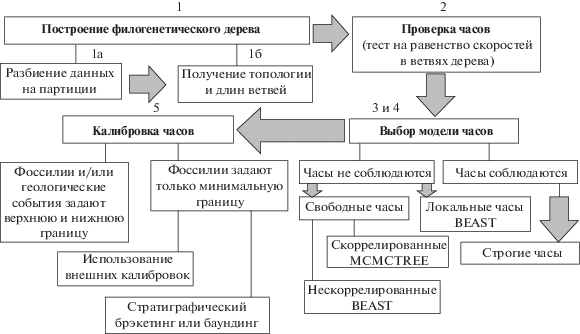
Процедуре датирования предшествует выбор и секвенирование генов, на основе которых будет проводиться датирование. Эти локусы должны быть достаточно информативны, но не насыщены. Например, мтДНК на высоких уровнях дивергенции часто дает смещенные оценки времен (Phillips et al., 2009; Brandley et al., 2011; Bannikova et al., 2014; Dornburg et al., 2014). Поэтому при уровне сходства митохондриальных последовательностей менее 85% мы не рекомендуем использовать мтДНК из-за очевидного сильного насыщения, которое не компенсируется существующими моделями эволюции.
После подбора моделей эволюции генов по имеющемуся набору данных строится филогенетическое древо. Хотя ряд методов (см. ниже) позволяет одновременно получить топологию и оценить времена дивергенций, но лучше сначала получить оценку топологии, независящую от допущений о характере изменения скорости эволюции. Важно выбрать правильную модель эволюции, а после этого правильно разбить набор данных на партиции (т.е. выделить группы последовательностей со сходным характером эволюции).
Далее проводят проверку молекулярных часов. Исходная гипотеза предполагает, что скорости молекулярной эволюции постоянны для всех филетических линий и во времени (модель строгих часов). Если это так, то все ветви филогенетического древа можно охарактеризовать с помощью единой оценки. Для проверки соблюдения часов проводится тест относительных скоростей (Relative Rate Test, RRT; Wu, Li, 1985; Tajima, 1993) или тест отношения правдоподобия (Likelihood Relative Test, LRT; Felsenstein, 1981). Тест относительных скоростей Таджимы применим для сравнения двух конкретных групп и в настоящее время в значительной степени вытеснен методами, позволяющими проверять гетерогенность скоростей между линиями по всему филогенетическому древу, такими как LRT.
Перечисленные выше тесты равенства скоростей на древе ориентированы только на очень длинные последовательности, а в случае недостаточной длины выравнивания и/или генов с низкой изменчивостью, их использование может привести к недооценке варьирования скоростей и ошибке в определении датировок (Bromham et al., 2000). В рамках байесовского подхода проверку соответствия строгим часам можно проводить путем вычисления Байес-факторов, однако этот метод требует существенных временных затрат.
В простейшем случае, если постоянство скорости не отвергается, то не составляет труда вычислить время дивергенции таксонов по формуле Tij = dij/2r, где d − ожидаемое число замещений (генетические дистанции). Для этого надо по палеонтологическим (или биогеографическим) данным определить время дивергенции хотя бы для одной пары таксонов (T0) и на основании этого получить оценку скорости r = d0/2T0. После этого можно вычислить оценки времен для остальных дивергенций. Используя эти простейшие формулы или равнозначную Tij = T0dij/d0, следует помнить, что получаемое отношение – это смещенная (завышенная) оценка, причем размер смещения тем больше, чем больше относительная ошибка (коэффициент вариации) для d0 (Nei et al., 2001; Rodríguez-Trelles et al., 2002). Поэтому, калибруя часы на основании последовательности с малым числом замен, мы рискуем получить завышенные оценки времен.
Если используются отдельные гены (или конкатенированные последовательности), то оценивается время дивергенции аллелей, а не таксонов, поэтому необходимо сделать поправку на предковый полиморфизм. Если датирование производится по конкатенированной последовательности, то временам узлов на древе соответствуют средние времена расхождения аллелей, а не собственно таксонов. В отсутствие гибридизации эти времена дивергенции аллелей старше, чем времена дивергенции таксонов. Этот фактор важно учитывать при анализе дивергенции близких видов.
Модели строгих часов. Эти модели обычно подходят для неглубоких филогений близкородственных видов и популяций в составе одного вида, где варьирование скорости молекулярной эволюции небольшое (Brown, Yang, 2011), например, если разница между последовательностями составляет менее 5% (Zhu, 2020). Но это предположение мало реалистично при сравнении далеких видов (Langley, Fitch, 1974; Yoder, Yang, 2000; Hasegawa et al., 2003). Тем не менее, если строгие часы выполняются, лучше использовать их, чем методы, допускающие варьирование скоростей, так как ошибка датировки в этом случае будет меньше (Brown, Yang, 2011).
Что делать, если модель строгих часов отвергнута? Если какой-либо таксон сильно отклоняется от остальных по скорости эволюции, простейший выход из положения – это исключить его из анализа (Takezaki et al., 1995; Hedges, Kumar, 2003). Наконец, для учета неоднородности скоростей (молекулярной эволюции) на филогенетическом древе предлагаются различные модели молекулярных часов. Эти модели примерно соответствуют двум типам: те, которые допускают всего несколько изменений скорости эволюции последовательностей, но различия между ними существенные, и те, которые предполагают множество разных скоростей, относительно мало отличающихся друг от друга (Welch, Bromham, 2005). В первом случае молекулярных скоростей обычно намного меньше, чем ветвей на древе.
Локальные часы. Методика, заключающаяся в использовании небольшого числа разных скоростей эволюции для разных участков древа, получила название “локальных часов” (Local Clock) и включает разнообразные алгоритмы многоскоростных часов (“multi-rate” clocks) (обзоры: Kumar, 2005; Welch, Bromham, 2005; Ho, Duchêne, 2014; Bromham et al., 2018). В таких моделях важной задачей является определение количества скоростей эволюции и участков на древе, к которым относятся эти скорости. Для этого в рамках метода максимального правдоподобия используются множественные тесты RRT и LRT. Эта процедура не всегда приводит к однозначным результатам (Bromham et al., 1998). Если участков дерева с разными “локальными часами” оказывается слишком много, метод становится неприменимым (Rannala, 2002; Felsenstein, 2004).
Тем не менее оказалось возможным успешно реализовать метод локальных часов в рамках байесовской статистики, где используются методы случайных локальных часов (random local clock, RLC; Drummond, Suchard, 2010, пакет BEAST) и процесс Дирихле (Dirichlet process prior, DPP-R; Heath et al., 2012, программа DPPDiv). Здесь результат соответствует не единственному оптимальному набору скоростей для ветвей, а апостериорному распределению числа локальных часов, их скоростей и положения событий смен одних часов другими на древе.
Свободные часы. Строгие часы для всего древа и независимые скорости для каждой ветви древа – это две крайности, которые в реальном эволюционном процессе маловероятны. Более вероятно, что варьирование скорости молекулярной эволюции ограничено какой-либо закономерностью, например, скорости в линиях предков–потомков более сходны, чем в отдаленных ветвях (Gillespie, 1991). Поэтому более эффективный по сравнению с локальными часами подход состоит в том, чтобы моделировать изменение скорости на филогенетическом древе (Sanderson, 1997; Huelsenbeck et al., 2000; Kishino et al., 2001).
В настоящее время широко используется модель “свободных” часов (Relaxed clock; Drummond et al., 2006). Этот метод датировок допускает варьирование скорости эволюции для каждого гена общей последовательности и каждой ветви филогенетического древа. Применение этого метода особенно эффективно при использовании нескольких калибровок, так как при этом лучше оценивается изменение скорости между ветвями древа (Thorne et al., 1998; Sanderson, 2003). Выбор модели скорости – очень важный шаг перед тем, как приступить к оценке времен дивергенций. Две основные модели свободных часов – это часы с автокорреляцией скоростей (autocorrelated branch-rate, ABR) и часы без корреляции, т.е. с независимыми скоростями (independent branch-rate, IBR) (Thorne et al., 1998; Kishino et al., 2001; Drummond et al., 2006; Lepage et al., 2006; Rannala, Yang, 2007; Ho, 2009; Ho, Duchêne, 2014).
В первом случае ожидается, что скорости в соседних ветвях древа относительно сходны; во втором – скорости всех ветвей древа распределены единообразно (экспоненциально, логнормально и т.д.), но корреляции скоростей в соседних ветвях нет. В обоих случаях моделируются либо средние скорости на ветвь, либо скорости в узлах древа. Наиболее принятый вариант модели с автокорреляцией (Thorne et al., 1998) эквивалентен модели геометрического броуновского движения, при этом логарифм скорости в узле-потомке R1 распределен нормально с математическим ожиданием log (R0) и дисперсией σ2 × t, где R0 – скорость в предковом узле, t – протяженность временного интервала между узлами 0 и 1, а σ2 – константа, отражающая относительную скорость изменения скорости.
Первой модели соответствует относительно медленная, но не ограниченная никакими пределами, эволюция скорости изменений в нуклеотидной последовательности. Этот принцип подходит для изучения близкородственных видов (Ho, 2009). Нереалистичность этой модели – в отсутствии стационарного распределения, что приводит к тому, что скорость может достигать очень больших значений.
В модели нескоррелированных часов частоты выбираются из статистического распределения и не зависят от значений на соседних ветвях (Drummond et al., 2006; Rannala, Yang, 2007). Эта модель осмысленна, когда скорости нуклеотидных замен меняются относительно быстро, но имеют ограничения и/или варьируют вокруг некого оптимума. Нереалистичность этой модели заключается в том, что даже в соседних коротких ветвях скорость может быть столь же разной, что и в дальних. Отмечается также зависимость результата от числа таксонов в анализе (Guindon, 2020).
В качестве более реалистичных моделей предлагалось использовать процессы Орнштейна–Уленбека (Aris-Brosou, Yang, 2002; Reis et al., 2016) и Кокса–Ингерсолла–Росса (Lepage et al., 2006), которые с одной стороны предполагают существование значительной стохастической компоненты в эволюции скорости нуклеотидных замен, а с другой – существование некой “оптимальной” скорости, которая является математическим ожиданием стационарного распределения. Очевидно, что при небольших временах процесс будет сходен с автокорреляционной моделью, а при больших – с моделью независимых скоростей. Недавно была предложена еще одна, пока мало исследованная, модель нестрогих часов, согласно которой вариации скорости связаны исключительно с резкими ускорениями в момент диверсификации, что представляет собой эволюцию по типу прерывистого равновесия (Manceau et al., 2020).
Выбор модели и представляет собой сложную задачу, и заметно влияет на результат. Выбор можно осуществлять с помощью Байес-факторов (Bayesi factor, BF), но чувствительность этого метода невысока (Ho et al., 2015a). Тем не менее результаты тестов чаще согласуются с представлением, что доминирующим паттерном в молекулярной филогенетике различных групп видов является автокорреляция скоростей ветвлений (например, Tao et al., 2019).
Определение партиций для генов со сходными моделями часов. Предполагается, что существуют группы локусов, в пределах которых скорости эволюции изменяются сходным образом, и поэтому к ним применимы одни и те же модели нестрогих часов. Показано, что схема разбиения данных на партиции может сильно влиять на оценки времен (Angelis et al., 2018). Если один и тот же набор данных будет адекватно разделен на большее количество партиций, оценки времени станут более точными (Zhu, 2020). Чтобы избежать больших затрат на вычисления (через BF) можно использовать, например, алгоритмы для разделения на партиции, применяемые в ML (Maximum Likelihood) анализе: Partitionfinder (Lanfear et al., 2017), Modelfinder (Kalyaanamoorthy et al., 2017). Другой (более быстрый и менее строгий) вариант – алгоритмы на основе кластеризации длин ветвей генных деревьев – ClockstaR и сходные методы (Duchêne et al., 2014, 2016).
Использование байесовских подходов для получения молекулярных датировок
Методы байесовской статистики впервые были введены в обиход филогенетики в 1990-х годах и быстро набрали популярность. Они используются в филогенетике и филогеографии для определения видовых границ, измерения популяционных параметров (потока генов между видами) и оценки времени дивергенции.
Основная цель байесовской филогенетики – оценить распределение тех эволюционных параметров, при которых с высокой вероятностью генерируется исследуемый набор данных. В число представляющих интерес параметров могут входить топология древа, а также глубины узлов, которым соответствуют времена дивергенции. Основное достоинство байесовских методов – возможность эффективной работы со сложными моделями с большим количеством параметров – возможность гибко учитывать исходную неопределенность в значении параметров, например, калибровочную информацию в форме априорного распределения. Как результат, на выходе мы получаем апостериорную оценку параметров с учетом его разброса (в форме интервала области высокой плотности апостериорного распределения вероятности – high posterior density, HPD) (Drummond et al., 2006; Yang, Rannala, 2006; Ronquist et al., 2012).
Две основные модели свободных часов реализованы в пакетах, использующих алгоритмы: MCMCTree (Brown, Yang, 2011) и BEAST (Drummond et al., 2012). Априорные распределения задаются для калибровочных данных (см. ниже), параметров моделей эволюции последовательностей (возможно для нескольких партиций), параметров модели эволюции скорости (могут быть разными для разных генов), а также для формы древа (например, модели Юла, birth–death, либо модели коалесценции в стабильной или растущей популяции). Необходимость введения априорного распределения для формы древа – сложный момент в байесовском анализе, так как это может влиять на результат (Ritchie et al., 2017), а объективное обоснование выбора с помощью Байес-факторов требует значительного компьютерного времени. В более простом случае подразумевается, что топология древа едина для всех генов.
Роль анцестрального полиморфизма и метод видового древа. Получать молекулярные оценки времени дивергенции часто удобно на основе не комбинированной последовательности генов, а так называемого видового древа (species tree). Каждый ген может иметь свою собственную историю, а время дивергенции аллелей, как правило, старше времени дивергенции таксонов. Метод видового древа позволяет решить эти две проблемы путем включения в модель оценок полиморфизма для всех внутренних узлов (multispecies coalescent model), что дает более реалистичные оценки времен, но за счет возрастания временных затрат на вычисления (Species tree dating; Heled, Drummond, 2010). Однако анализировать этим методом большие мультигенные наборы данных, в том числе и геномные, невозможно (Ogilvie et al., 2016).
Датирование филогеномных данных байесовскими методами
В последние два десятилетия вследствие развития высокопроизводительного секвенирования наблюдается стремительное накопление геномных данных. Это способствует углублению понимания эволюции на геномном уровне, но одновременно создает новые проблемы для получения молекулярных датировок из-за серьезных вычислительных проблем (Ho, 2014; Tong et al., 2016).
Тем не менее, несмотря на временные затраты, ведущим методом в филогеномном датировании остается байесовский подход (Reis et al., 2012, 2018; Springer et al., 2012; Jarvis et al., 2014; Zheng, Wiens, 2016). Значительные затраты компьютерного времени при использовании этого статистического подхода связаны с тем, что апостериорное распределение большого числа параметров генерируется с помощью метода Монте-Карло с марковскими цепями (MCMC) (Bromham et al., 2018). Повышение эффективности алгоритмов MCMC таким образом, чтобы они могли обрабатывать наборы данных в масштабе геномов, в настоящее время становится основной обсуждаемой методической темой в филогенетических исследованиях.
Например, теоретически получить топологию и времена дивергенций одновременно возможно (Drummond et al., 2006), но непрактично для очень больших филогеномных данных, потому что вычисления требуют слишком много времени и огромных вычислительных мощностей. Поэтому принято прибегать к упрощению модели, фиксируя топологию, или вводить ограничения на монофилию группировок (Drummond et al., 2006; Yang, Rannala, 2006). Наиболее важная стратегия ускорения вычислений в процессе молекулярного датирования по геномным данным – это, вероятно, использование аппроксимации правдоподобия (approximate-likelihood), что обеспечивает ускорение в 1000 раз и позволяет получить приближенную байесовскую оценку времен дивергенций по филогеномным данным (Álvarez-Carretero, Reis, 2020).
Датирование не байесовскими методами
Многие методы не байесовской статистики, использующие нестрогие часы, более приемлемы в отношении вычислительного времени и могут применяться к огромным массивам данных. Одним из широко используемых методов является метод на основе “оштрафованного правдоподобия” (penalized likelihood; Sanderson, 2002), реализованный в программах r8s (Sanderson, 2003) и treePL (Smith, O’Meara, 2012). На вход подается древо с длинами ветвей, полученными каким-либо методом, не предполагающим существование строгих часов. Алгоритм вычисляет набор скоростей нуклеотидных замен для ветвей и возрастов узлов таким образом, чтобы минимизировать величину penalized likelihood, которую образуют два слагаемых: первое из них соответствует правдоподобию длин ветвей (отклонение наблюдаемых длин ветвей от ожидаемых), второе минимизирует различия между эволюционными скоростями в ветвях предков и потомков (Sanderson, 1997, 2002), что соответствует модели автокорреляции скоростей (Thorne et al., 1998; Kishino et al., 2001). Например, с помощью программы r8s (Sanderson, 2003) при использовании 21 калибровочной точки из статьи Мередита с соавт. (Meredith et al., 2011), которые были заданы как максимальные и минимальные величины дивергенций, мы провели оценку времен дивергенции четырех ветвей Eulipotyphla по мультилокусным данным (433 гена, 235 тыс. п.о.) (Банникова, 2019).
Алгоритм RelTime, реализованный в программе MEGA-X, тоже сводит к минимуму различия между скоростью эволюции предков и потомков (Tamura et al., 2012, 2018). Скорость работы алгоритма велика благодаря дополнительным упрощениям – ошибки длин ветвей в расчет не принимаются. Существуют и другие совместимые с нестрогими часами алгоритмы, которые выигрывают в скорости за счет упрощения модели, что позволяет им работать с большими данными: датирование методом наименьших квадратов – least-squares dating, LSD (To et al., 2016), treedater (Volz, Frost, 2017), TreeTime (Sagulenko et al., 2018), wLogDate (Mai, Mirarab, 2021).
КАЛИБРОВКА МОЛЕКУЛЯРНЫХ ЧАСОВ
Итак, идея молекулярных часов родилась из наблюдения того, что степень различия ДНК двух видов является функцией времени с момента их дивергенции. Это позволяет датировать эволюционные события и сильно упрощает процедуру восстановления генеалогических связей (Bromham, Penny, 2003). Однако молекулярные данные сами по себе способны предоставить информацию только об относительных временах. Для получения абсолютной шкалы времени молекулярные часы должны быть откалиброваны (Donoghue, Benton, 2007). Калибровка часов – это использование внешней информации для ограничения возраста узлов на филогенетическом древе. Эта необходимая, помимо топологии древа, информация включает данные о скоростях нуклеотидных замен по всему древу или оценку возраста хотя бы одного внутреннего узла. По существу, калибровка молекулярных часов – это определение их скорости и масштабирование филогенетического древа в соответствии с реальным временем.
Калибровка может применяться не только к конкретным узлам филогенетического древа, но и к демографическому событию. К важным популяционным событиям относятся экспансия или сокращение эффективной численности. Изменения эффективной численности могут быть определены с помощью стандартных методов исследования демографической истории: построение скайлайнов – графического отображения динамики эффективной численности (skyline analysis; Hope et al., 2014); распределение числа замен нуклеотидов при попарном сравнении сиквенсов (mismatch distribution; Rogers, Harpending, 1992); сравнение различных демографических сценариев с использованием байесовского подхода (Bertorelle et al., 2010). В любом случае, так же, как и при датировании филогенетических событий, здесь необходимо учитывать неопределенность в сроках возникновения популяционного события.
Включение калибровок в анализ
Ранее в большинстве случаев калибровочная информация сводилась к фиксации возраста какого-либо узла на некотором определенном значении (точечная калибровка), что в настоящее время считается неоправданным упрощением (Nguyen, Ho, 2020). В современном филогенетическом анализе, как правило, используются интервальные оценки датировок. В простейшем случае задается минимальная и максимальная граница интервала равномерного распределения, например, в таких методах, как penalized likelihood (Sanderson, 2002) и RelTime (Tamura et al., 2012). Более гибкий подход, реализованный в байесовских методах, позволяет использовать калибровочную информацию как априорное распределение возраста узлов, при этом функция распределения может быть любой (Ho, Phillips, 2009), главное, чтобы она отражала исходную неопределенность возраста дивергенции клад. Обычно используются логнормальное, экспоненциальное или гамма распределение. Распределение может быть жестко ограничено (жесткая граница снизу или и снизу, и сверху), или иметь мягкие границы (Yang, Rannala, 2006).
Палеонтологические калибровки
Задача калибровки молекулярных часов не решается в рамках одной молекулярной генетики. Калибровочные данные могут включать разную информацию (биогеографическую и палеогеографическую, молекулярные датировки, полученные из прежних исследований), но основной источник информации – это палеонтологические датировки ископаемых находок изучаемой таксономической группы (Hipsley, Müller, 2014).
Неправильная калибровка часов во многом связана с неопределенностью, происходящей со стороны палеонтологии. Источники серьезных ошибок включают неверное определение палеонтологического возраста и филогенетического положения фоссилий (Benton, Donoghue, 2007; Gandolfo et al., 2008). В некоторых группах использование палеонтологических датировок малоэффективно из-за бедности летописи или невозможности морфологической диагностики (Smith, Peterson, 2002). Нередко бывает сложно однозначно определить, какой именно узел на древе должен быть откалиброван с использованием данного ископаемого материала, потому что это зависит от того, относится ли материал к стволовой группе или к ветви потомков (Benton, Donoghue, 2007). Включение в анализ большого количества калибровочных точек повышает точность оценок времени (Lee, 1999; Lukoschek et al., 2012).
Если палеонтологический возраст и клада, к которой принадлежит фоссилия, известны, логично предположить, что возраст фоссилии ограничивает снизу время дивергенции содержащей ее клады от ближайшей сестринской группы (= минимальная нижняя граница). Остается еще один существенный источник неопределенности: не известно, сколько времени прошло между моментом обособления клады и возрастом самой ранней фоссилии, заведомо к ней принадлежащей (= время существования “линии-призрака”, ghost lineage; Phillips, 2016; Springer et al., 2017). В свою очередь, время призрака включает два компонента – время от точки дивергенции до появления диагностических синапоморфий группы и интервал до первой обнаруженной фоссилии (рис. 3). Последняя величина зависит от вероятности фоссилизации с одной стороны и активности палеонтологов с другой (Gandolfo et al., 2008). Если возраст фоссилии известен с точностью до геологического диапазона, то приходится использовать его верхнюю (позднюю) границу в качестве минимального возраста узла, тогда разница между верхней границей диапазона и неизвестным истинным возрастом фоссилии оказывается третьим слагаемым оценки времени существования призрака.
Рис. 3.
Использование палеонтологической калибровочной информации в качестве минимальной границы возраста узла. Красным кружком обозначен изучаемый узел дивергенции филогенетических линий А и В. Пунктирные линии разделяют геологические периоды. Вертикальная красная стрелка указывает на самые ранние находки, уверенно относимые к линии А. Синий крестик показывает время появления морфологических синапоморфий в линии А. Другие значки обозначают: † – ископаемые остатки, безусловно относящиеся к линии А; ‡ – ископаемые остатки, безусловно относящиеся к линии В; + – ископаемые остатки, имеющие плезиоморфную морфологию, соответствующие предку таксонов А и В.
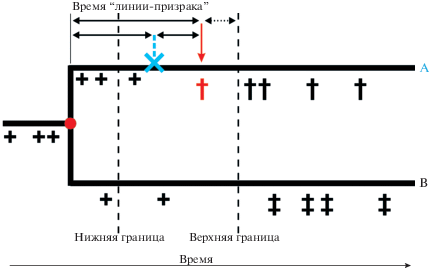
Очевидно, что минимальных ограничений для датировки недостаточно, необходимо включить как минимум хотя бы одно максимальное ограничение. Однако максимальные возрастные ограничения установить трудно, потому что они эквивалентны постулированию отсутствия определенной клады в какой-то момент прошлого. Выбор максимальных возрастных ограничений оказывает значительное влияние на получаемые молекулярные датировки (Hug, Roger, 2007; Warnock et al., 2015). В качестве примера можно привести недавнюю дискуссию вокруг временной шкалы эволюции современных птиц (Jarvis et al., 2014; Cracraft et al., 2015; Mitchell et al., 2015). Часто максимальные границы выбираются наиболее консервативным образом (т.е. избыточно древними) (Ho, Phillips, 2009), что может приводить к смещенным и неточным оценкам времен. Очевидно, что максимальные ограничения правильней задавать в виде мягких границ (Yang, Rannala, 2006), например, границы 95%-го доверительного интервала.
Для того чтобы объективно оценить доверительный интервал возраста узла исходя из времен находок фоссилий, был предложен метод стратиграфического брэкетинга (Marshall, 2008), использовавший распределение относительных возрастов самых ранних фоссилий для нескольких (многих) клад древа. Метод исходно опирался на не всегда реалистичные допущения о распределении находок на временной шкале, однако в дальнейшем были предложены его модификации, в которых применялись более сложные модели (Marshall, 2010).
Также были предложены менее формализованные методы определения максимальных границ, такие как филогенетический брэкетинг и стратиграфический баундинг (Benton, Donoghue, 2007; Meredith et al., 2011). В последнем методе в качестве верхней границы возраста узла используется нижняя (ранняя) граница геологического интервала (или двух интервалов), предшествующего тому, в котором был найден самый древний ископаемый материал. В филогенетическом брэкетинге возрастной диапазон узла ограничен возрастом его соседних узлов (т.е. таких, которые являются непосредственными предками и потомками), что может основываться на палеонтологической летописи или на независимой молекулярной оценке. Например, получение молекулярных датировок для ежовых содержит определенные трудности из-за нередкой двусмысленной интерпретации ископаемых остатков этой группы (Rich, 1981), что объясняется генерализованным строением зубной системы − основного материала палеонтологических исследований. По этой причине при подборе калибровочных точек для датирования дивергенций в сем. Erinaceidae (Bannikova et al., 2014) самая древняя калибровочная точка, связанная с олигоцен-эоценовым периодом (28–49 млн лет) радиации базальных линий современных ежовых была взята из работы Мередита с соавт. (Meredith et al., 2011). Максимальная граница для этой калибровочной точки определяется через стратиграфический ограничитель и филогенетический брэкетинг и должна приниматься с осторожностью. Согласно Лопатину (Lopatin, 2006), раннеэоценовые ископаемые ежовые относятся к примитивным гимнурам; по этой причине предпочтение отдавалось накладыванию меньших ограничений на время разделения Erinaceinae/Hylomyinae и отнесению верхнего предела этого события к границе палеоцена/эоцена.
В качестве внутренних калибровочных точек использовали: 1) время разделения Hylomys s.str. и Neotetracus (16 млн лет – минимальная граница) на основании ископаемых остатков из раннего миоцена Таиланда, относимых к этим таксонам (Mein, Ginsburg, 1997); 2) ожидаемое время базальной радиации кроновых Erinaceinae в MN13 (5.5 млн лет как минимальная граница) на основании ископаемых остатков, определенных как Erinaceus (Storch, Qiu, 1991; Qiu, Storch, 2005). Однако следует признать, что эти калибровочные точки являются вероятными, но не очевидными. Отнесение события разделения Neotetracus и Hylomys ко времени не позднее 16 млн лет назад вызывает вопросы, учитывая трудности определения этих гимнур не только по ископаемым остаткам, но и по рецентному морфологическому материалу (Frost et al., 1991; Gould, 1995). Также нет уверенности в существовании хороших признаков, позволяющих отличать поздне-миоценовых Erinaceus от других родов ежей. Некоторые авторы вообще избегают обсуждать принадлежность ископаемых остатков ежовых к тому или иному роду. Известно, что, когда такие попытки предпринимались, возникала глубокая и трудно разрешимая путаница, как, например, в случае миоценового р. Mioechinus (Depéret, 1887; Butler, 1948; Rich, 1981; Gould, 1995; Mein, Ginsburg, 2002).
Что касается стратиграфического баундинга, этот прием был применен нами при датировании дивергенций землероек: в р. Sorex, где молекулярные часы были откалиброваны по пяти палеонтологическим датировкам (Bannikova et al., 2018), а также в р. Crocidura (Bannikova et al., 2021).
Вторичная калибровочная информация
В настоящее время во многих исследованиях для калибровки часов используются уже опубликованные ранее молекулярные датировки. Их называют вторичными калибровками, поскольку они основаны не на прямых палеонтологических или биогеографических данных, а на полученных ранее временах дивергенций. Очевидно, что в некоторых ситуациях с помощью вторичной калибровки из надежного источника могут быть получены более точные оценки времени, чем с помощью первичной калибровочной информации из ненадежных палеонтологических источников (Kumar, Hedges, 1998). Эта методика уязвима для критики (Hipsley, Müller, 2014), однако минимизировать ошибку можно путем конструирования априорных распределений, которые учитывают все источники ошибок оценки, основанной на вторичной калибровке.
Вторичные калибровки обычно используют для таксонов, по которым ограничена палеонтологическая информация, например, бактерии или грибы, а также для увеличения числа калибровочных точек при датировании многовидовых филогений (например, Reis et al., 2012, 2018). В наших исследованиях использовались вторичные калибровки в случаях, когда доступные данные задавали только минимальную границу, например, при определении времен дивергенций кротовых (Bannikova et al., 2015), кутор (Igea et al., 2015) и тушканчиков (Shenbrot et al., 2017).
Калибровки, основанные на несовременных нуклеотидных последовательностях
Калибровка молекулярных часов с использованием гетерохронных данных полезна в исследованиях древней ДНК, а также для оценки темпов и временных масштабов эволюции видов с быстрой сменой поколений. Обретение опыта работы с ДНК ископаемых образцов открыло возможность использовать пробы от экземпляров, для которых имеются радиоуглеродные датировки. Однако ошибки радиоуглеродного датирования, возникающие в том числе из-за колебаний содержания 14C в атмосфере (Reimer et al., 2013), представляют собой отдельный источник неопределенности возраста (Guilderson et al., 2005). Наконец, при работе с палеоДНК существует необходимость делать поправки на химическую модификацию последовательностей (Hofreiter et al., 2001). При небольшой длине палеосиквенсов оценки времен и скоростей могут быть смещены (см. раздел о занижении оценок скорости эволюции с течением времени).
Другая область, где продуктивно использование гетерохронных последовательностей для молекулярных датировок, – это филогения быстро эволюционирующих организмов, например, вирусов (в частности, у РНК вирусов скорость накопления замен может составлять 10–3 на сайт в год (Duffy et al., 2008)). Важная практическая составляющая молекулярного датирования вирусных филогений – исследование историй эпидемий (например, Pekar et al., 2021).
Простейший способ оценить скорость и время, имея филогению для гетерохронных последовательностей, – использовать регрессию расстояния от корня древа до концов ветвей (root to tip distance) на время образцов, как это реализовано в программе TempEst (Rambaut et al., 2016). Предварительно необходимо проверить, достаточно ли информативны данные для этого анализа, что делается с помощью рандомизационного теста (Ramsden et al., 2009). Кроме этого, для гетерохронных данных активно используются методы ML (treedater; Volz, Frost, 2017) и байесовской статистики (BEAST; Heled, Drummond, 2010). Ситуация осложняется тем, что для описания эволюции вирусов часто требуется использование модели rate decay (Duchêne et al., 2014).
Модель FBD и датирование на объединенных данных
Классический метод использования калибровок сводится к двухстадийному анализу: сначала, исходя из филогенетического положения и возраста фоссилий, оценивается возраст одного или нескольких узлов дерева (node-dating), затем, используя эту информацию, определяется возраст остальных узлов древа. Принципиально иной подход – датирование на объединенных данных (total evidence dating) – заключается в построении хронограммы путем одновременного анализа молекулярных и морфологических данных, причем последние включают информацию как для современных видов, так и для ископаемых таксонов с известным возрастом (Stadler, 2010; Heath et al., 2014; Gavryushkina, Zhu, 2020). Эта процедура реализована в нескольких программах, использующих байесовский подход (MrBayes, BEAST 2). Анализ основан на модели фоссилизации – рождения–смерти (Fossilized birth–death, FBD), описывающей одновременно структуру древа, вероятность захоронения и обнаружения фоссилий. Если филогенетическое положение вымерших видов известно хотя бы приблизительно, то возможно использовать вариант анализа без привлечения морфологических данных (Heath et al., 2014). Исходный вариант FBD модели предполагал, что процессы фоссилизации и диверсификации описываются одним и тем же набором параметров во всех частях древа. Более реалистична модель фоссилизации кладообразования (fossilized birth–death process skyline model, FBD; Stadler et al., 2013; Gavryushkina et al., 2014), которая постулирует существование нескольких эпох, каждая со своей динамикой этих процессов.
Сильная сторона датирования на объединенных данных – возможность использовать максимум палеонтологической информации, большая часть которой при классическом подходе оказалась бы вне анализа, в частности, из-за неопределенности филогенетического положения ископаемого материала. Модель FBD в разных ее вариантах требует дальнейшего изучения. Пока же нельзя исключить, что результаты будут чрезмерно чувствительны к исходным допущениям (Matzke, Wright, 2016; O`Reilly, Donoghue, 2020). Отдельная проблема – надежность результатов совместного анализа морфологических и молекулярных данных. Это связано с неадекватностью существующих моделей морфологической эволюции, высокой вероятностью филогенетических ошибок из-за адаптивных параллелизмов, что, например, скомпрометировало результаты О’Лири (O`Leary et al., 2013). Наконец, насыщенность модели параметрами требует больших вычислительных мощностей.
Биогеографические калибровки
На практике датирование по ископаемым материалам осложняется целым рядом теоретических и эмпирических проблем. Хорошую альтернативу палеонтологической информации составляют биогеографические данные. К первым публикациям в этой области относится работа по гавайским цветочницам, или медоносам (Hemignathus spp.), в которой для калибровки времени радиации этих птиц использовали возраст Гавайских островов (Fleischer et al., 1998). Некоторые особенности Гавайских островов делают их подходящими для калибровки молекулярных часов. Считается, что они образовались вдоль “конвейерной ленты” и расположены последовательно по возрасту от самых старых (29.8 млн лет) островов на северо-западе до самых молодых (0.5 млн лет) на юго-востоке (Fleischer et al., 1998; Price, Clague, 2002).
Возраст островов может быть использован в качестве калибровки молекулярных часов, если соблюдается правило прогрессии (Hennig, 1966), согласно которому есть соответствие между возрастом островов и топологией филогенетического древа изучаемых таксонов. Например, в случае эндемичных гавайских цветочниц, дрозофил и бабочек самые молодые острова населяют виды, образующие внутренние ветви филогенетического древа, в то время как виды, распространенные на старых островах, располагаются на древе ближе к корню (Fleischer et al., 1998; Haines et al., 2014). Калибровка филогении по времени с помощью биогеографии и палеогеографии работает по принципу, что для данного палеогеографического контекста одни биогеографические сценарии более вероятны, чем альтернативные, и эта вероятность влияет на принимаемый в результате возраст изучаемых видов (Ho et al., 2015b; Baets et al., 2016).
МОЛЕКУЛЯРНАЯ ОЦЕНКА ВРЕМЕН ДИВЕРГЕНЦИИ ОТРЯДОВ ПЛАЦЕНТАРНЫХ КАК ПРИМЕР РАЗЛИЧИЯ РЕЗУЛЬТАТОВ, ПОЛУЧЕННЫХ РАЗНЫМИ МЕТОДАМИ НА РАЗНЫХ ДАННЫХ
Использование разного числа и набора таксонов, большего или меньшего числа генных локусов, тех или иных моделей и калибровочных точек может приводить к сильному варьированию получаемых оценок времен дивергенции. Хорошим примером служат попытки определения времени появления отрядов Placentalia. Палеонтологические данные указывают на быструю радиацию плацентарных после массового вымирания на границе мела и палеогена (М/П) 65 млн лет назад (обзор: Archibald et al., 2001; Benton, Ayala, 2003). Более жесткая модель (“hard” explosive model) предполагает быструю радиацию единственной предковой ветви, перешагнувшей М/П границу (O’Leary et al., 2013). Альтернативная, более мягкая взрывная модель (“soft” explosive model) допускает позднемеловое происхождение нескольких наиболее древних ветвей плацентарных (Simpson, 1945; Archibald, Deutschman, 2001).
Молекулярные оценки этого события в целом значительно более древние и противоречат взрывной модели палеогенового происхождения плацентарных. Они подразумевают два варианта: 1) дивергенция отрядов и дальнейшая радиация внутри них относятся в подавляющем большинстве случаев к меловому периоду (модель ускоренной диверсификации, “короткий запал” (“short fuse”); Bininda-Emonds et al., 2007); 2) предки Placentalia и большинства отрядов сформировались в меловом периоде, но дальнейшая их диверсификация происходила в палеогене (модель замедленной диверсификации, “длинный запал” (“long fuse”); Reis et al., 2012).
Таким образом, хотя результаты разных молекулярных данных совпадают в вопросе о меловом происхождении плацентарных, согласованное представление о порядке возникновения отрядов относительно М/П границы отсутствует. Например, датировки Бининда-Эмондс с соавторами (Bininda-Emonds et al., 2007) показывают, что половина отрядов плацентарных произошла более 100 млн лет назад, т.е. намного раньше того времени, на которое указывают все известные ископаемые и некоторые молекулярные данные. Однако в этом исследовании должным образом не учитывались варьирование скоростей эволюции в разных линиях и неопределенности в оценке длин ветвей, и, кроме того, применялась точечная ископаемая калибровка для корня. Другие ранние молекулярные датировки (Kumar, Hedges, 1998) тоже искажены тем, что в них не принимались во внимание такие важные источники ошибок, как (1) степень близости ископаемых калибровок ко времени события дивергенции (Yang, Rannala, 2006), (2) случайный разброс (randomness) молекулярных скоростей (Thorne et al., 1998) и (3) разброс оценок длин ветвей (Rodríguez-Trelles et al., 2002). По сравнению с прежними исследованиями, в работе Мередита с соавт. (Meredith et al., 2011) с применением байесовских методов были получены относительно недавние времена диверсификации внутри отрядов млекопитающих. При этом времена для пяти отрядов плацентарных характеризовались очень широкими доверительными интервалами (от раннего мела до позднего палеоцена), что не позволяет уверенно утверждать, в меловом периоде или в палеоцене произошла основная диверсификация плацентарных. К столь широким доверительным интервалам времен дивергенций привел ограниченный объем генетических локусов (26 ядерных генов для 164 таксонов).
Рейс и соавторы (Reis et al., 2012) провели байесовский анализ 36 ядерных и 274 митохондриальных геномов (20.6 млн п.н.) (Yang, Rannala, 2006), используя датировку по палеонтологическим данным (по методу Бентона с соавт. (Benton et al., 2009)). В результате гипотеза базальных дивергенций внутри большинства отрядов плацентарных до М/П границы была отвергнута, но при этом полученные оценки предполагают возникновение стволовых групп плацентарных до М/П границы (модель “long fuse”).
Филлипс (Phillips, 2016) получил датировки, допускающие возникновение Xenarthra, Afrotheria, Euarchontoglires и Laurasiatheria в меловом периоде, но большинство внутриотрядных дивергенций имело место в период после массового вымирания на М/П границе (“мягкая взрывная модель”). Филлипс предположил, что оценка скорости молекулярной эволюции может быть искажена такими биологическими факторами, как большие размеры тела и большая продолжительность жизни (что противоречит результатам Ромигера с соавторами (Romiguier et al., 2013)), и получил приближенные к традиционной мягкой взрывной модели датировки (более точные с его точки зрения), изымая такие таксоны из анализа времен дивергенций.
Однако Спрингер с соавт. (Springer et al., 2017) показали, что допущения, принятые Филлипсом, приводят к появлению на его древе так называемых “линий зомби” (zombie lineages) – артефакты, которые возникают, если полученные оценки времен моложе минимального возраста древнейших ископаемых остатков. Результаты Спрингера в отличие от результатов Филлипса лучше соответствуют модели “long fuse”.
Итак, молекулярные данные дают разные оценки времени филогенетических событий (табл. 1), однако никакие из них не соответствуют жесткой взрывной модели. Анализ также показывает, что, помимо сигнала от собственно данных, допущения, принятые авторами, сильно влияют на оценки и выводы.
Таблица 1.
Времена дивергенции и радиации современных групп Eulipotyphla по молекулярным данным, полученным нами (Bannikova et al., 2014, 2015; Банникова, 2019) и другими авторами (Douady et al., 2002; Douady, Douzery, 2003; Springer et al., 2003, 2017; Roca et al., 2004; Meredith et al., 2011; Reis et al., 2012), а также по палеонтологическим данным (Lopatin, 2006)
| Таксон или узел дивергенции | Наши данные | Другие молекулярные данные | Палеонтологические данные |
|---|---|---|---|
| Eulipotyphla | 70–80 млн л. н. | 70–85 млн л. н. | >70 млн л. н. |
| Solenodontidae | нет данных | ~76 млн л. н. | нет данных |
| Erinaceidae | ~66 млн л. н. | 55–65 млн л. н. | 61–63 млн л. н. |
| Hylomyinae/Erinaceinae | ~52 млн л. н. | 38 млн л. н. | ~47 млн л. н. |
| Talpidae | ~72 млн л. н. | 60–70 млн л. н. | 40 млн л. н. |
| Uropsilinae | 43–52 млн л. н. | 48–57 млн л. н. | 35–37 млн л. н. |
| Soricidae | ~66 млн л. н. | 55–65 млн л. н. | 45–47 млн л. н. |
| Soricinae/Crocidurinae | ~38 млн л. н. | 38 млн л. н. | 18 млн л. н. |
ЗАКЛЮЧЕНИЕ
История молекулярной филогенетики и развития концепции молекулярных часов содержит немало примеров несовпадения молекулярных и палеонтологических времен дивергенции и ошибочных датировок, что связано, прежде всего, со следующими причинами. Во-первых, это ошибки определения длин ветвей (и глубин узлов ветвления) из-за низкой информативности данных или неточной модели. Влияние этих источников неопределенности можно уменьшить увеличением числа локусов и длины читаемых фрагментов, а также фильтрацией генов, для которых нет адекватной модели. Во-вторых, это ошибки в калибровочной информации как биологической, так и геологической природы. Роль их должна уменьшаться с ростом объема палеонтологических данных и повышением точности определения возраста ископаемых остатков. В-третьих, в случае нестрогих часов присутствует еще и трудно устранимая неопределенность, связанная с характером варьирования скоростей как между кладами, так и между генами (ее можно лишь отчасти уменьшить, увеличивая число анализируемых генов); здесь же следует упомянуть влияние rate decay (Ho et al., 2005, 2007).
Вторая и третья причины неопределенности в совокупности приводят к тому, что даже при увеличении объема данных до геномных, ошибка времен дивергенции снижается не до нуля, а только до некоторого предела (Reis, Yang, 2013; Zhu et al., 2015), делая точное определение молекулярных времен дивергенции невозможным. Кроме того, разные варианты молекулярного датирования могут давать существенно разные результаты даже когда они применяются к аналогичным и даже идентичным наборам данных (например, Reis et al., 2014; Foster et al., 2016). Такие несовпадения показывают, что выводы часто чувствительны к предположениям, исходно заложенным в выводах.
Помимо этого, большинство методов молекулярного датирования нацелены на учет изменения скорости молекулярной эволюции без моделирования механизмов или причин изменения скорости, которая может эволюционировать в зависимости от биологии вида. Известно только несколько методов, которые моделируют скорость молекулярной эволюции в соответствии с биологией вида (Lartillot, Delsuc, 2012; Nabholz et al., 2013). С другой стороны, необходимы дальнейшие усилия в создании сложных моделей эволюции нуклеотидных последовательностей, учитывающих специфику механизмов как возникновения повреждений ДНК (например, в случае CpG), так и репарации.
Полемика по поводу молекулярных часов не умаляет их практического преимущества в изучении филогенеза, а также недавних событий популяционного уровня. Стремительное развитие геномного секвенирования способствует познанию закономерностей изменчивости скорости молекулярной эволюции, что в дальнейшем, безусловно, ликвидирует брешь в нашем понимании сопряженной эволюции видов и их геномов.
Список литературы
Банникова А.А., 2019. Молекулярная эволюция и проблемы филогенетической реконструкции истинных насекомоядных (Mammalia: Eulipotyphla). Дис. … док. биол. наук. М.: МГУ. 697 с.
Распопова А.А., Банникова А.А., Лебедев В.С., 2018. Филогеография и историческая демография обыкновенной бурозубки // Генетика. Т. 54. № 12. С. 1426–1441.
Aiewsakun P., Katzourakis A., 2016. Time-dependent rate phenomenon in viruses // J. Virol. V. 90. № 16. P. 7184–7195.
Allio R., Donega S., Galtier N., Nabholz B., 2017. Large variation in the ratio of mitochondrial to nuclear mutation rate across animals: Implications for genetic diversity and the use of mitochondrial DNA as a molecular marker // Mol. Biol. Evol. V. 34. № 11. P. 2762–2772.
Álvarez-Carretero S., Reis M., dos, 2020. Bayesian Phylogenomic Dating // The Molecular Evolutionary Clock. Cham: Springer. P. 221–249.
Angelis K., Álvarez-Carretero S., Reis M., dos, Yang Z., 2018. An evaluation of different partitioning strategies for Bayesian estimation of species divergence times // Syst. Biol. V. 67. № 1. P. 61–77.
Arbogast B.S., Edwards S.V., Wakeley J., Beerli P., Slowinski J.B., 2002. Estimating divergence times from molecular data on phylogenetic and population genetic timescales // Annu. Rev. Ecol. Syst. V. 33. № 1. P. 707–740.
Archibald J.D., Deutschman D.H., 2001. Quantitative analysis of the timing of the origin and diversification of extant placental orders // J. Mamm. Evol. V. 8. № 2. P. 107–124.
Archibald J.D., Averianov A.O., Ekdale E.G., 2001. Late Cretaceous relatives of rabbits, rodents, and other extant eutherian mammals // Nature. V. 414. № 6859. P. 62–65.
Aris-Brosou S., Yang Z., 2002. Effects of models of rate evolution on estimation of divergence dates with special reference to the metazoan 18S ribosomal RNA phylogeny // Syst. Biol. V. 51. № 5. P. 703–714.
Baets K., de, Antonelli A., Donoghue P.C.J., 2016. Tectonic blocks and molecular clocks // Phil. Trans. R. Soc. Lond. B. Biol. Sci. V. 371. № 1699. https://doi.org/10.1098/rstb.2016.0098
Bannikova A.A., Lebedev V.S., Abramov A.V., Rozhnov V.V., 2014. Contrasting evolutionary history of hedgehogs and gymnures (Mammalia: Erinaceomorpha) as inferred from a multigene study // Biol. J. Linn. Soc. V. 112. P. 499–519.
Bannikova A.A., Zemlemerova E.D., Lebedev V.S., Lavrenchenko L.A., 2021. The phylogenetic relationships within the Eastern Afromontane clade of Crocidura based on mitochondrial and nuclear data // Mamm. Biol. V. 101. P. 1005–1018.https://doi.org/10.1007/s42991-021-00120-7
Bannikova A.A., Lebedev V.S., Lissovsky A.A., Matrosova V., Abramson N.I. et al., 2010a. Molecular phylogeny and evolution of the Asian lineage of vole genus Microtus (Arvicolinae, Rodentia) inferred from mitochondrial cytb sequence // Biol. J. Linn. Soc. V. 99. № 3. P. 595–613.
Bannikova A.A., Dokuchaev E.N., Yudina E.V., Bobretzov A.V., Sheftel B.I., Lebedev V.S., 2010b. Holarctic phylogeography of the tundra shrew (Sorex tundrensis) based on mitochondrial genes // Biol. J. Linn. Soc. V. 101. № 3. P. 721–746.
Bannikova A.A., Zemlemerova E.D., Colangelo P., Sözen M., Sevindik M. et al., 2015. An underground burst of diversity – a new look at the phylogeny and taxonomy of the genus Talpa Linnaeus, 1758 (Mammalia: Talpidae) as revealed by nuclear and mitochondrial genes // Zool. J. Linn. Soc. V. 175. № 4. P. 930–948.
Bannikova A.A., Chernetskaya D., Raspopova A., Alexandrov D., Fang Y. et al., 2018. Evolutionary history of the genus Sorex (Soricidae, Eulipotyphla) as inferred from multigene data // Zool. Scr. V. 47. № 5. P. 518–538.
Barrera-Redondo J., Ramírez-Barahona S., Eguiarte L.E., 2018. Rates of molecular evolution in tree ferns are associated with body size, environmental temperature, and biological productivity // Evolution. V. 72. № 5. P. 1050–1062.
Benton M.J., Ayala F.J., 2003. Dating the tree of life // Science. V. 300. № 5626. P. 1698–1700.
Benton M.J., Donoghue P.C.J., 2007. Paleontological evidence to date the tree of life // Mol. Biol. Evol. V. 24. № 1. P. 26–53.
Benton M.J., Donoghue P.C.J., Asher R.J., 2009. Calibrating and constraining molecular clocks // The Timetree of Life. N.-Y.: Oxford Univ. Press. P. 35–86.
Bertorelle G., Benazzo A., Mona S., 2010. ABC as a flexible framework to estimate demography over space and time: some cons, many pros // Mol. Ecol. V. 19. № 13. P. 2609–2625.
Bininda-Emonds O.R.P., Cardillo M., Jones K.E., MacPhee R.D.E., Beck R.M.D. et al., 2007. The delayed rise of present-day mammals // Nature. V. 446. № 7135. P. 507–512.
Bird A.P., 1980. DNA methylation and the frequency of CpG in animal DNA // Nucleic Acids Res. V. 8. № 7. P. 1499–1504.
Brandley M.C., Wang Y., Guo X., De Oca A.N.M., Fería-Ortíz M. et al., 2011. Accommodating heterogenous rates of evolution in molecular divergence dating methods: an example using intercontinental dispersal of Plestiodon (Eumeces) lizards // Syst. Biol. V. 60. № 1. P. 3–15.
Bromham L., 2002. Molecular clocks in reptiles: Life history influences rate of molecular evolution // Mol. Biol. Evol. V. 19. № 3. P. 302–309.
Bromham L., 2011. The genome as a life-history character: Why rate of molecular evolution varies between mammal species // Phil. Trans. Roy. Soc. B. Biol. Sci. V. 366. № 1577. P. 2503–2513.
Bromham L., 2020. Causes of variation in the rate of molecular evolution // The Molecular Evolutionary Clock. Cham: Springer. P. 45–64.
Bromham L., Penny D., 2003. The modern molecular clock // Nat. Rev. Genet. V. 4. № 3. P. 216–224.
Bromham L., Rambaut A., Harvey P.H., 1996. Determinants of rate variation in mammalian DNA sequence evolution // J. Mol. Evol. V. 43. № 6. P. 610–621.
Bromham L., Penny D., Rambaut A., Hendy M.D., 2000. The power of relative rates tests depends on the data // J. Mol. Evol. V. 50. № 3. P. 296–301.
Bromham L., Hua X., Lanfear R., Cowman P.F., 2015. Exploring the relationships between mutation rates, life history, genome size, environment, and species richness in flowering plant // Am. Nat. V. 185. № 4. P. 507–524.
Bromham L., Rambaut A., Fortey R., Cooper A., Penny D., 1998. Testing the Cambrian explosion hypothesis by using a molecular dating technique // Proc. Natl. Acad. Sci. USA. V. 95. № 21. P. 12386–12389.
Bromham L., Duchêne S., Hua X., Ritchie A.M., Duchêne D.A., Ho S.Y.W., 2018. Bayesian molecular dating: Opening up the black box // Biol. Rev. V. 93. № 2. P. 1165–1191.
Brown R.P., Yang Z., 2011. Rate variation and estimation of divergence times using strict and relaxed clocks // BMC Evol. Biol. V. 11. https://doi.org/10.1186/1471-2148-11-271
Brown W.M., George M., Jr., Wilson A.C., 1979. Rapid evolution of animal mitochondrial DNA // Proc. Natl. Acad. Sci. USA. V. 76. № 4. P. 1967–1971.
Butler P.M., 1948. On the evolution of the skull and teeth in the Erinaceidae, with special reference to fossil material in the British // Proc. Zool. Soc. Lond. V. 118. № 2. P. 446–500.
Chintalapati M., Moorjani P., 2020. Evolution of the mutation rate across primates // Curr. Opin. Genet. Dev. V. 62. P. 58–64.
Cosson J.-F., Hutterer R., Libois R., Sara M., Taberlet P., Vogel P., 2005. Phylogeographical footprints of the Strait of Gibraltar and Quaternary climatic fluctuations in the western Mediterranean: A case study with the greater white-toothed shrew, Crocidura russula (Mammalia: Soricidae) // Mol. Ecol. V. 14. № 4. P. 1151–1162.
Cracraft J., Houde P., Ho S.Y.W., Mindell D.P., Fjeldså J. et al., 2015. Response to comment on “Whole-genome analyses resolve early branches in the tree of life of modern birds” // Science. V. 349. № 6255. https://doi.org/10.1126/science.aab1578
Debruyne R., Poinar H.N., 2009. Time dependency of molecular rates in ancient DNA data sets, a sampling artifact? // Syst. Biol. V. 58. № 3. P. 348–360.
Depéret C., 1887. Recherches sur la succession des Faunes de Vertébrés miocénes de la vallée du Rhone // Arch. Mus. Hist. Nat. Lyon. V. 4. P. 45–313.
Donoghue P.C., Benton M.J., 2007. Rocks and clocks: Calibrating the tree of life using fossils and molecules // Trends Ecol. Evol. V. 22. № 8. P. 424–431.
Doolittle R.F., Blombäck B., 1964. Amino-acid sequence investigations of fibrinopeptides from various mammals: evolutionary implications // Nature. V. 202. № 4928. P. 147–152.
Dornburg A., Townsend J.P., Friedman M., Near T.J., 2014. Phylogenetic informativeness reconciles ray-finned fish molecular divergence times // BMC Evol. Biol. V. 14. № 1. https://doi.org/10.1186/s12862-014-0169-0
Douady C., Douzery E., 2003. Molecular estimation of eulipotyphlan divergence times and the evolution of “Insectivora” // Mol. Phylogenet. Evol. V. 28. № 2. P. 285–296.
Douady C.J., Chatelier P.I., Madsen O., Jong W.W., de, Catzeflis F. et al., 2002. Molecular phylogenetic evidence confirming the Eulipotyphla concept and in support of hedgehogs as the sister group to shrews // Mol. Phylogenet. Evol. V. 25. № 1. P. 200–209.
Drake J.W., Charlesworth B., Charlesworth D., Crow J.F., 1998. Rates of spontaneous mutation // Genetics. V. 148. № 4. P. 1667–1686.
Drummond A.J., Suchard M.A., 2010. Bayesian random local clocks, or one rate to rule them all // BMC Biol. V. 8. https://doi.org/10.1186/1741-7007-8-114
Drummond A.J., Ho S.Y.W., Phillips M.J., Rambaut A., 2006. Relaxed phylogenetics and dating with confidence // PLoS Biol. V. 4. № 5. https://doi.org/10.1371/journal.pbio.0040088
Drummond A.J., Suchard M.A., Xie D., Rambaut A., 2012. Bayesian phylogenetics with BEAUti and the BEAST 1.7 // Mol. Biol. Evol. V. 29. № 8. P. 1969–1973.
Duchêne D., Bromham L., 2013. Rates of molecular evolution and diversification in plants: Chloroplast substitution rates correlate with species richness in the Proteaceae // BMC Evol. Biol. V. 13. https://doi.org/10.1186/1471-2148-13-65
Duchêne S., Lanfear R., Ho S.Y.W., 2014. The impact of calibration and clock-model choice on molecular estimates of divergence times // Mol. Phylogenet. Evol. V. 78. P. 277–289.
Duchêne S., Holt K.E., Weill F.-X., Le Hello S., Hawkey J. et al., 2016. Genome-scale rates of evolutionary change in bacteria // Microb. Genom. V. 2. № 11. https://doi.org/10.1099/mgen.0.000094
Duffy S., Shackelton L.A., Holmes E.C., 2008. Rates of evolutionary change in viruses: patterns and determinants // Nat. Rev. Genet. V. 9. № 4. P. 267–276.
Duret L., Arndt P.F., 2008. The impact of recombination on nucleotide substitutions in the human genome // PLoS Genet. V. 4. № 5. https://doi.org/10.1371/journal.pgen.1000071
Easteal S., Herbert G., 1997. Molecular evidence from the nuclear genome for the time frame of human evolution // J. Mol. Evol. V. 44. Suppl. 1. P. S121–S132.
Easteal S., Collet C., Betty D., Ashley M.V., 1995. Mammalian Molecular Clock. Austin: R.G. Landes Co. 170 p.
Eldredge N., Gould S.J., 1972. Punctuated equilibria: An alternative to phyletic gradualism // Models in Paleobiology. San Francisco: Freeman. P. 82–115.
Emerson B.C., 2007. Alarm bells for the molecular clock? No support for Ho et al.'s model of time-dependent molecular rate estimates // Syst. Biol. V. 56. № 2. P. 337–345.
Emerson B.C., Hickerson M.J., 2015. Lack of support for the time-dependent molecular evolution hypothesis // Mol. Ecol. V. 24. № 4. P. 702–709.
Felsenstein J., 1981. Evolutionary trees from DNA sequences: A maximum likelihood approach // J. Mol. Evol. V. 17. № 6. P. 368–376.
Felsenstein J., 2004. Inferring Phylogenies. Sunderland: Sinauer associates. 681 p.
Fisher R.A., 1936. The measurement of selective intensity // Proc. Roy. Soc. B. V. 121. P. 58–62.
Fitch W.M., Beintema J.J., 1990. Correcting parsimonious trees for unseen nucleotide substitutions: The effect of dense branching as exemplified by ribonuclease // Mol. Biol. Evol. V. 7. № 5. P. 438–443.
Fleischer R.C., McIntosh C.E., Tarr C.L., 1998. Evolution on a volcanic conveyor belt: Using phylogeographic reconstructions and K-Ar-based ages of the Hawaiian Islands to estimate molecular evolutionary rates // Mol. Ecol. V. 7. № 4. P. 533–545.
Foster C.S.P., Sauquet H., Merwe M., van der, McPherson H., Rossetto M., Ho S.Y.W., 2016. Evaluating the impact of genomic data and priors on Bayesian estimates of the angiosperm evolutionary timescale // Syst. Biol. V. 66. № 3. P. 338–351.
Frost D.R., Wozencraft W.C., Hoffmann R.S., 1991. Phylogenetic Relationships of Hedgehogs and Gymnures (Mammalia: Insectivora: Erinaceidae). Washington: Smithsonian Institution Press. 69 p.
Fu Y., Foden J.A., Khayter C., Maeder M.L., Reyon D. et al., 2013. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells // Nat. Biotechnol. V. 31. № 9. P. 822–826.
Galtier N., Enard D., Radondy Y., Bazin E., Belkhir K., 2006. Mutation hot spots in mammalian mitochondrial DNA // Genome Res. V. 16. № 2. P. 215–222.
Galtier N., Jobson R.W., Nabholz B., Glemin S., Blier P.U., 2009. Mitochondrial whims: metabolic rate, longevity and the rate of molecular evolution // Biol. Lett. V. 5. № 3. P. 413–416.
Gandolfo M., Nixon K.C., Crepet W.L., 2008. Selection of fossils for calibration of molecular dating models // Ann. Missouri Bot. Gard. V. 95. № 1. P. 34–42.
Gao Z., Wyman M.J., Sella G., Przeworski M., 2016. Interpreting the dependence of mutation rates on age and time // PLoS Biol. V. 14. № 1. https://doi.org/10.1371/journal.pbio.1002355
Gavryushkina A., Zhu C., 2020. Total-evidence dating and the fossilized birth–death model // The Molecular Evolutionary Clock. Cham: Springer. P. 175–197.
Gavryushkina A., Welch D., Stadler T., Drummond A.J., 2014. Bayesian inference of sampled ancestor trees for epidemiology and fossil calibration // PLoS Comput. Biol. V. 10. № 12. https://doi.org/10.1371/journal.pcbi.1003919
Gillespie J.H., 1991. The Causes of Molecular Evolution. Oxford: Oxford Univ. Press. 336 p.
Gillooly J.F., Allen A.P., West G.B., Brown J.H., 2005. The rate of DNA evolution: Effects of body size and temperature on the molecular clock // Proc. Natl. Acad. Sci. USA. V. 102. № 1. P. 140–145.
Goodman M., 1961. The role of immunologic differences in the phyletic development of human behavior // Hum. Biol. V. 33. № 2. P. 131–162.
Goodman M., 1962. Evolution of the immunologic species specificity of human serum proteins // Hum. Biol. V. 34. P. 104–150.
Gould G.C., 1995. Hedgehog phylogeny (Mammalia, Erinaceidae) – the reciprocal illumination of the quick and the dead / American Museum Novitates. V. 3131. N.-Y.: Americian Museum of Natural History. 45 p.
Guilderson T.P., Reimer P.J., Brown T.A., 2005. The boon and bane of radiocarbon dating // Science. V. 307. № 5708. P. 362–364.
Guindon S., 2020. Rates and rocks: Strengths and weaknesses of molecular dating methods // Front. Genet. V. 11. https://doi.org/10.3389/fgene.2020.00526
Haines W.P., Schmitz P., Rubinoff D., 2014. Ancient diversification of Hyposmocoma moths in Hawaii // Nat. Commun. V. 5. https://doi.org/10.1038/ncomms4502
Haldane J.B.S., 1947. The mutation rate of the gene for hemophilia, and its segregation ratios in males and females // Ann. Eugenics. V. 13. № 4. P. 262–271.
Hardouin E.A., Tautz D., 2013. Increased mitochondrial mutation frequency after an island colonization: positive selection or accumulation of slightly deleterious mutations? // Biol. Lett. V. 9. № 2. https://doi.org/10.1098/rsbl.2012.1123
Hasegawa M., Kishino H., Yano T., 1989. Estimation of branching dates among primates by molecular clocks of nuclear DNA which slowed down in Hominoidea // J. Hum. Evol. V. 18. № 5. P. 461–476.
Hasegawa M., Thorne J.L., Kishino H., 2003. Time scale of eutherian evolution estimated without assuming a constant rate of molecular evolution // Genes Genet. Syst. V. 78. № 4. P. 267–283.
Heath T.A., Moore B.R., 2014. Bayesian inference of species divergence times // Bayesian Phylogenetics: Methods, Algorithms and Applications. Boca Raton: CRC Press. P. 277–318.
Heath T.A., Holder M.T., Huelsenbeck J.P., 2012. A Dirichlet process prior for estimating lineage-specific substitution rates // Mol. Biol. Evol. V. 29. № 3. P. 939–955.
Heath T.A., Huelsenbeck J.P., Stadler T., 2014. The fossilized birth-death process for coherent calibration of divergence-time estimates // Proc. Natl. Acad. Sci. USA. V. 111. № 29. P. E2957–E2966.
Hedges S.B., Kumar S., 2003. Genomic clocks and evolutionary timescales // Trends Genet. V. 19. № 4. P. 200–206.
Heled J., Drummond A.J., 2010. Bayesian inference of species trees from multilocus data // Mol. Biol. Evol. V. 27. № 3. P. 570–580.
Henn B.M., Gignoux C.R., Feldman M.W., Joanna L., Mountain J.L., 2008. Characterizing the time dependency of human mitochondrial DNA mutation rate estimates // Mol. Biol. Evol. V. 26. № 1. P. 217–230.
Hennig W., 1966. Phylogenetic Systematics. Urbana: Univ. Illinois Press. 263 p.
Herman J.S., McDevitt A.D., Kawałko A., Jaarola M., Wójcik J.M., Searle J.B., 2014. Land-bridge calibration of molecular clocks and the post-glacial Colonization of Scandinavia by the Eurasian field vole Microtus agrestis // PLoS One. V. 9. № 8. https://doi.org/10.1371/journal.pone.0103949
Hipsley C.A., Müller J., 2014. Beyond fossil calibrations: Realities of molecular clock practices in evolutionary biology // Front. Genet. V. 5. https://doi.org/10.3389/fgene.2014.00138
Ho S.Y.W., 2009. An examination of phylogenetic models of substitution rate variation among lineages // Biol. Lett. V. 5. № 3. P. 421−424.
Ho S.Y.W., 2014. The changing face of the molecular evolutionary clock // Trends Ecol. Evol. V. 29. № 9. P. 496–503.
Ho S.Y.W., 2020. The molecular clock and evolutionary rates across the tree of life // The Molecular Evolutionary Clock. Cham: Springer. P. 3−24.
Ho S.Y.W., Duchêne S., 2014. Molecular-clock methods for estimating evolutionary rates and timescales // Mol. Ecol. V. 23. № 24. P. 5947–5965.
Ho S.Y.W., Phillips M.J., 2009. Accounting for calibration uncertainty in phylogenetic estimation of evolutionary divergence times // Syst. Biol. V. 58. № 3. P. 367–380.
Ho S.Y.W., Duchêne S., Duchêne D., 2015a. Simulating and detecting autocorrelation of molecular evolutionary rates among lineages // Mol. Ecol. V. 15. № 4. P. 688–696.
Ho S.Y.W., Phillips M.J., Cooper A., Drummond A.J., 2005. Time dependency of molecular rate estimates and systematic overestimation of recent divergence times // Mol. Biol. Evol. V. 22. № 7. P. 1561–1568.
Ho S.Y.W., Shapiro B., Phillips M.J., Cooper A., Drummond A.J., 2007. Evidence for time dependency of molecular rate estimates // Syst. Biol. V. 56. № 3. P. 515–522.
Ho S.Y.W., Lanfear R., Bromham L., Phillips M.J., Soubrier J. et al., 2011. Time-dependent rates of molecular evolution // Mol. Ecol. V. 20. № 15. P. 3087–3101.
Ho S.Y.W., Tong K.J., Foster C.S.P., Ritchie A.M., Lo N., Crisp M.D., 2015b. Biogeographic calibrations for the molecular clock // Biol. Lett. V. 11. № 8. https://doi.org/10.1098/rsbl.2015.0194
Hodgkinson A., Eyre-Walker A., 2011. Variation in the mutation rate across mammalian genomes // Nat. Rev. Genet. V. 12. № 11. P. 756–766.
Hofreiter M., Jaenicke V., Serre D., Haeseler A., von, Pääbo S., 2001. DNA sequences from multiple amplifications reveal artifacts induced by cytosine deamination in ancient DNA // Nucleic Acids Res. V. 29. № 23. P. 4793–4799.
Hope A.G., Ho S.Y.W., Malaney J.L., Cook J.A., Talbot S.L., 2014. Accounting for rate variation among lineages in comparative demographic analyses // Evolution. V. 68. № 9. P. 2689–2700.
Hua X., Bromham L., 2017. Darwinism for the genomic age: Connecting mutation to diversification // Front. Genet. V. 8. https://doi.org/10.3389/fgene.2017.00012
Hua X., Cowman P., Warren D., Bromham L., 2015. Longevity is linked to mitochondrial mutation rates in rockfish: A test using Poisson regression // Mol. Biol. Evol. V. 32. № 10. P. 2633–2645.
Huelsenbeck J.P., Larget B., Swofford D., 2000. A compound Poisson process for relaxing the molecular clock // Genetics. V. 154. № 4. P. 1879–1892.
Hug L.A., Roger A.J., 2007. The impact of fossils and taxon sampling on ancient molecular dating analyses // Mol. Biol. Evol. V. 24. № 8. P. 1889–1897.
Igea J., Aymerich P., Bannikova A.A., Gosálbez J., Castresana J., 2015. Multilocus species trees and species delimitation in a temporal context: Application to the water shrews of the genus Neomys // BMC Evol. Biol. V. 15. https://doi.org/10.1186/s12862-015-0485-z
Irwin D.M., Kocher T.D., Wilson A.C., 1991. Evolution of the cytochrome b gene of mammals // J. Mol. Evol. V. 32. № 12. P. 128–144.
Jarvis E.D., Mirarab S., Aberer A.J., Li B., Houde P. et al., 2014. Whole-genome analyses resolve early branches in the tree of life of modern birds // Science. V. 346. № 6215. P. 1320–1331.
Kalyaanamoorthy S., Minh B.Q., Wong T.K.F., Haeseler A., von, Jermiin L.S., 2017. ModelFinder: Fast model selection for accurate phylogenetic estimates // Nat. Methods. V. 14. № 6. P. 587–589.
Kim S.-H., Elango N., Warden C.W., Vigoda E., Yi S., 2006. Heterogeneous genomic molecular clocks in primates // PLoS Genet. V. 2. № 10. https://doi.org/10.1371/journal.pgen.0020163
Kimura M., 1968. Evolutionary rate at the molecular level // Nature. V. 217. № 5129. P. 624–626.
Kimura M., 1969. The rate of molecular evolution considered from the standpoint of population genetics // Proc. Natl. Acad. Sci. USA. V. 63. № 4. P. 1181–1188.
Kimura M., 1983. The Neutral Theory of Molecular Evolution. N.Y.; L.; Cambridge: Cambridge Univ. Press. 394 p.
Kimura M., Ohta T., 1971. On the rate of molecular evolution // J. Mol. Evol. V. 1. № 1. P. 1–17.
Kishino H., Thorne J.L., Bruno W.J., 2001. Performance of a divergence time estimation method under a probabilistic model of rate evolution // Mol. Biol. Evol. V. 18. № 3. P. 352–361.
Kong A., Frigge M.L., Masson G., Besenbacher S., Sulem P., et al., 2012. Rate of de novo mutations and the importance of father’s age to disease risk // Nature. V. 488. № 7412. P. 471–475.
Kumar S., 2005. Molecular clocks: Four decades of evolution // Nat. Rev. Genet. V. 6. № 8. P. 654–662.
Kumar S., Hedges S.B., 1998. A molecular timescale for vertebrate evolution // Nature. V. 392. № 6679. P. 917–920.
Kumar S., Hedges S.B., 2016. Advances in time estimation methods for molecular data // Mol. Biol. Evol. V. 33. № 4. P. 863–869.
Kumar S., Filipski A., Swarna V., Walker A., Hedges S.B., 2005. Placing confidence limits on the molecular age of the human–chimpanzee divergence // Proc. Natl. Acad. Sci. USA. V. 102. № 52. P. 18842–18847.
Lanfear R., Welch J.J., Bromham L., 2010. Watching the clock: Studying variation in rates of molecular evolution between species // Trends Ecol. Evol. V. 25. № 9. P. 495–503.
Lanfear R., Thomas J.A., Welch J.J., Bromham L., 2007. Metabolic rate does not calibrate the molecular clock // Proc. Natl. Acad. Sci. USA. V. 104. № 39. P. 15388–15393.
Lanfear R., Ho S.Y.W., Davies T.J., Moles A.T., Aarssen L., et al., 2013. Taller plants have lower rates of molecular evolution // Nat. Commun. V. 4. https://doi.org/10.1038/ncomms2836
Lanfear R., Frandsen P.B., Wright A.M., Senfeld T., Calcott B., 2017. Methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses // Mol. Biol. Evol. V. 34. № 3. P. 772–773.
Langley C.H., Fitch W.M., 1974. An estimation of the constancy of the rate of molecular evolution // J. Mol. Evol. V. 3. № 3. P. 161–177.
Lartillot N., Delsuc F., 2012. Joint reconstruction of divergence times and life-history evolution in placental mammals using a phylogenetic covariance model // Evolution. V. 66. № 6. P. 1773–1787.
Lee M.S.Y., 1999. Molecular clock calibrations and metazoan divergence dates // J. Mol. Evol. V. 49. № 3. P. 385–391.
Lepage T., Lawi S., Tupper P., Bryant D., 2006. Continuous and tractable models for the variation of evolutionary rates // Math. Biosci. V. 199. № 2. P. 216–233.
Lopatin A.V., 2006. Early Paleogene insectivore mammals of Asia and establishment of the major groups of Insectivora // Paleontol. J. V. 40. № 3. P. S205–S405.
Lukoschek V., Keogh J.S., Avise J.C., 2012. Evaluating fossil calibrations for dating phylogenies in light of rates of molecular evolution: a comparison of three approaches // Syst. Biol. V. 61. № 1. P. 22–43.
Lynch M., 2010a. Evolution of the mutation rate // Trends Genet. V. 26. № 8. P. 345–352.
Lynch M., 2010b. Rate, molecular spectrum, and consequences of human mutation // Proc. Natl. Acad. Sci. USA. V. 107. № 3. P. 961–968.
Lynch M., Ackerman M.S., Gout J.-F., Long H., Sung W. et al., 2016. Genetic drift, selection and the evolution of the mutation rate // Nat. Rev. Genet. V. 17. № 11. P. 704–714.
Mai U., Mirarab S., 2021. Log transformation improves dating of phylogenies // Mol. Biol. Evol. V. 38. № 3. P. 1151–1167.
Manceau M., Marin J., Morlon H., Lambert A., 2020. Model-based inference of punctuated molecular evolution // Mol. Biol. Evol. V. 37. № 11. P. 3308–3323.
Margoliash E., 1963. Primary structure and evolution of cytochrome c // Proc. Natl. Acad. Sci. USA. V. 50. № 4. P. 672–679.
Marshall C.R., 2008. A simple method for bracketing absolute divergence times on molecular phylogenies using multiple fossil calibration points // Am. Nat. V. 171. № 6. P. 726–742.
Marshall C.R., 2010. Using confidence intervals to quantify the uncertainty in the end-points of stratigraphic ranges // The Paleontological Society Papers. V. 16. P. 291–316.
Martin A.P., Palumbi S.R., 1993. Body size, metabolic rate, generation time and the molecular clock // Proc. Natl. Acad. Sci. USA. V. 90. № 9. P. 4087–4091.
Martínková N., Barnett R., Cucchi T., Struchen R., Pascal M. et al., 2013. Divergent evolutionary processes associated with colonization of offshore islands // Mol. Ecol. V. 22. № 20. P. 5205–5220.
Matzke N.J., Wright A., 2016. Inferring node dates from tip dates in fossil Canidae: The importance of tree priors // Biol. Lett. V. 12. № 8. https://doi.org/10.1098/rsbl.2016.0328
Mayr E., 1963. Animal Species and Evolution. Cambridge: Harvard Univ. Press. 797 p.
Mein P., Ginsburg L., 1997. Les mammifères du gisement miocène inférieur de Li Mae Long, Thaïlande: systématique, biostratigraphie et paléoenvironnement // Geodiversitas. V. 19. № 4. P. 783–844.
Mein P., Ginsburg L., 2002. Sur l'âge relatif des différents dépôts karstiques miocènes de La Grive-Saint-Alban (Isère) // Cahiers Sci. Mus. Hist. Nat. Lyon. V. 5. P. 7–47.
Meredith R.W., Janecka J.E., Gatesy J., Ryder O.A., Fisher C.A. et al., 2011. Impacts of the Cretaceous Terrestrial Revolution and KPg extinction on mammal diversification // Science. V. 334. № 6055. P. 521–524.
Milholland B., Dong X., Zhang L., Hao X., Suh Y., Vijg J., 2017. Differences between germline and somatic mutation rates in humans and mice // Nat. Commun. V. 8. https://doi.org/10.1038/ncomms15183
Mitchell K.J., Cooper A., Phillips M.J., 2015. Comment on “Whole-genome analyses resolve early branches in the tree of life of modern birds” // Science. V. 349. № 6255. P. 1460.
Molak M., Ho S.Y.W., 2015. Prolonged decay of molecular rate estimates for metazoan mitochondrial DNA // PeerJ. V. 3. https://doi.org/10.7717/peerj.821
Mooers A.O., Harvey P.H., 1994. Metabolic rate, generation time, and the rate of molecular evolution in birds // Mol. Phylogenet. Evol. V. 3. № 4. P. 344–350.
Moorjani P., Amorim C.E.G., Arndt P.F., Przeworski M., 2016. Variation in the molecular clock of primates // Proc. Natl. Acad. Sci. USA. V. 113. № 38. P. 10607–10612.
Müller H.J., 1954. The nature of the genetic effects produced by radiation // Radiation Biology. N.-Y.: McGraw‑Hill. P. 351–473.
Nabholz B., Glemin S., Galtier N., 2008. Strong variations of mitochondrial mutation rate across mammals–the longevity hypothesis // Mol. Biol. Evol. V. 25. № 1. P. 120–130.
Nabholz B., Uwimana N., Lartillot N., 2013. Reconstructing the phylogenetic history of long-term effective population size and life-history traits using patterns of amino acid replacement in mitochondrial genomes of mammals and birds // Genome Biol. Evol. V. 5. № 7. P. 1273–1290.
Nachman M.W., Crowell S.L., 2000. Estimate of the mutation rate per nucleotide in humans // Genetics. V. 156. № 1. P. 297–304.
Navascues M., Emerson B.C., 2009. Elevated substitution rate estimates from ancient DNA: Model violation and bias of Bayesian methods // Mol. Ecol. V. 18. № 21. P. 4390–4397.
Nei M., Xu P., Glazko G., 2001. Estimation of divergence times from multiprotein sequences for a few mammalian species and several distantly related organisms // Proc. Natl. Acad. Sci. USA. V. 98. № 5. P. 2497–2502.
Nguyen J.M.T., Ho S.Y.W., 2020. Calibrations from the fossil record // The Molecular Evolutionary Clock. Cham: Springer. P. 117–134.
Nunney L., 1999. Lineage selection and the evolution of multistage carcinogenesis // Proc. Roy. Soc. B. V. 266. № 1418. P. 493–498.
Ogilvie H.A., Heled J., Xie D., Drummond A.J., 2016. Computational performance and statistical accuracy of *BEAST and comparisons with other methods // Syst. Biol. V. 65. № 3. P. 381–396.
Ohta T., 1972a. Evolutionary rate of cistrons and DNA divergence // J. Mol. Evol. V. 1. № 2. P. 150–157.
Ohta T., 1972b. Population size and rate of evolution // J. Mol. Evol. V. 1. № 4. P. 305–314.
Ohta T., 1973. Slightly deleterious mutant substitutions in evolution // Nature. V. 246. № 5428. P. 96–98.
Ohta T., 1974. Mutational pressure as the main cause of molecular evolution and polymorphism // Nature. V. 252. № 5482. P. 351–354.
Ohta T., 2002. Near-neutrality in evolution of genes and gene regulation // Proc. Natl. Acad. Sci. USA. V. 99. № 25. P. 16134–16137.
O’Leary M.A., Bloch J.I., Flynn J.J., Gaudin T.J., Giallombardo A. et al., 2013. The placental mammal ancestor and the post–K-Pg radiation of placentals // Science. V. 339. № 6120. P. 662–667.
O’Reilly J.E., Donoghue P.C.J., 2020. The effect of fossil sampling on the estimation of divergence times with the fossilised birth death process // Syst. Biol. V. 69. № 1. P. 124–138.
Pagel M., Venditti C., Meade A., 2006. Large punctuational contribution of speciation to evolutionary divergence at the molecular level // Science. V. 314. № 5796. P. 119–121.
Pekar J., Worobey M., Moshiri N., Scheffler K., Wertheim J.O., 2021. Timing the SARS-CoV-2 index case in Hubei province // Science. V. 372. № 6540. P. 412–417.
Pesole G., Sbisá E., Preparata G., Saccone C., 1992. The evolution of the mitochondrial D-loop region and the origin of modern man // Mol. Biol. Evol. V. 9. № 4. P. 587–598.
Peterson G.I., Masel J., 2009. Quantitative prediction of molecular clock and Ka/Ks at short timescales // Mol. Biol. Evol. V. 26. № 11. P. 2595–2603.
Pfeifer S.P., 2020. Spontaneous mutation rates // The Molecular Evolutionary Clock. Cham: Springer. P. 35–44.
Phillips M.J., 2016. Geomolecular dating and the origin of placental mammals // Syst. Biol. V. 65. № 3. P. 546–557.
Phillips S.J., Dudík M., Elith J., Graham C.H., Lehmann A. et al., 2009. Sample selection bias and presence-only distribution models: implications for background and pseudo-absence data // Ecol. Appl. V. 19. № 1. P. 181–197.
Price J.P., Clague D.A., 2002. How old is the Hawaiian biota? Geology and phylogeny suggest recent divergence // Proc. Roy. Soc. B. Biol. Sci. V. 269. № 1508. P. 2429–2435.
Qiu Z.D., Storch G., 2005. The fossil record of the Eurasian Neogene Insectivores (Erinaceomorpha, Soricomorpha, Mammalia): Part I // Scripta Geol. Spec. Issue. Leiden. V. 5. P. 37–50.
Rambaut A., Lam T.T., Carvalho L.M., Pybus O.G., 2016. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen) // Virus Evol. V. 2. № 1. https://doi.org/10.1093/ve/vew007
Ramsden C., Holmes E.C., Charleston M.A., 2009. Hantavirus evolution in relation to its rodent and insectivore hosts: no evidence for codivergence // Mol. Biol. Evol. V. 26. № 1. P. 143–153.
Rannala B., 2002. Identifiability of parameters in MCMC Bayesian inference of phylogeny // Syst. Biol. V. 51. № 5. P. 754–760.
Rannala B., Yang Z., 2007. Inferring speciation times under an episodic molecular clock // Syst. Biol. V. 56. № 3. P. 453–466.
Raspopova A.A., Bannikova A.A., Sheftel B.I., Kryštufek B., Kouptsov A.V. et al., 2020. A never-ending story of the common shrew: searching for the origin // Mammal Res. V. 65. № 4. P. 729–742.
Reimer P.J., Bard E., Bayliss A., Beck J.W., Blackwell P.G. et al., 2013. IntCal13 and Marine13 radiocarbon age calibration curves 0–50,000 years cal BP // Radiocarbon. V. 55. № 4. P. 1869–1887.
Reis M., dos, Yang Z., 2013. The unbearable uncertainty of Bayesian divergence time estimation // J. Syst. Evol. V. 51. № 1. P. 30–43.
Reis M., dos, Zhu T., Yang Z., 2014. The impact of the rate prior on Bayesian estimation of divergence times with multiple loci // Syst. Biol. V. 63. № 4. P. 555–565.
Reis M., dos, Donoghue P.C.J., Yang Z., 2016. Bayesian molecular clock dating of species divergences in the genomics era // Nat. Rev. Genet. V. 17. № 2. P. 71–80.
Reis M., dos, Inoue J., Hasegawa M., Asher R.J., Donoghue P.C., Yang Z., 2012. Phylogenomic datasets provide both precision and accuracy in estimating the timescale of placental mammal phylogeny // Proc. Roy. Soc. B. V. 279. № 1742. P. 3491–3500.
Reis M., dos, Thawornwattana Y., Angelis K., Telford M.J., Donoghue P.C.J., Yang Z., 2015. Uncertainty in the timing of origin of animals and the limits of precision in molecular timescales // Curr. Biol. V. 25. № 22. P. 2939–2950.
Reis M.D., dos, Gunnell G.F., Barba-Montoya J., Wilkins A., Yang Z., Yoder A.D., 2018. Using phylogenomic data to explore the effects of relaxed clocks and calibration strategies on divergence time estimation: Primates as a test case // Syst. Biol. V. 67. № 4. P. 594–615.
Rich T.H.V., 1981. Origin and history of the Erinaceidae and Brachyericinae (Mammalia, Insectivora) in North America // Bull. Amer. Mus. Nat. Hist. V. 171. P. 1–116.
Ritchie A.M., Lo N., Ho S.Y., 2017. The impact of the tree prior on molecular dating of data sets containing a mixture of inter-and intraspecies sampling // Syst. Biol. V. 66. № 3. P. 413–425.
Roca A.L., Bar-Gal G.K., Eizirik E., Helgen K.M., Maria R. et al., 2004. Mesozoic origin for West Indian insectivores // Nature. V. 429. № 6992. P. 649–651.
Rodríguez-Trelles F., Tarrío R., Ayala F.J., 2002. A methodological bias toward overestimation of molecular evolutionary time scales // Proc. Natl. Acad. Sci. USA. V. 99. № 12. P. 8112–8115.
Rogers A.R., Harpending H., 1992. Population growth makes waves in the distribution of pairwise genetic differences // Mol. Biol. Evol. V. 9. № 3. P. 552–569.
Romiguier J., Ranwez V., Douzery E.J.P., Galtier N., 2013. Genomic evidence for large, long-lived ancestors to placental mammals // Mol. Biol. Evol. V. 30. № 1. P. 5–13.
Ronquist F., Klopfstein S., Vilhelmsen L., Schulmeister S., Murray D.L., Rasnitsyn A.P., 2012. A total-evidence approach to dating with fossils, applied to the early radiation of the Hymenoptera // Syst. Biol. V. 61. № 6. P. 973–999.
Sagulenko P., Puller V., Neher R.A., 2018. TreeTime: Maximum-likelihood phylodynamic analysis // Virus Evol. V. 4. № 1. https://doi.org/10.1093/ve/vex042
Sanderson M.J., 1997. A nonparametric approach to estimating divergence times in the absence of rate constancy // Mol. Biol. Evol. V. 14. № 12. P. 1218–1231.
Sanderson M.J., 2002. Estimating absolute rates of molecular evolution and divergence times: A penalized likelihood approach // Mol. Biol. Evol. V. 19. № 1. P. 101–109.
Sanderson M.J., 2003. r8s: Inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock // Bioinformatics. V. 19. № 2. P. 301–302.
Santos C., Montiel R., Sierra B., Bettencourt C., Fernandez E. et al., 2005. Understanding differences between phylogenetic and pedigree-derived mtDNA mutation rate: A model using families from the Azores Islands (Portugal) // Mol. Biol. Evol. V. 22. № 6. P. 1490–1505.
Sarich V.M., Wilson A.C., 1967. Immunological time scale for hominid evolution // Science. V. 158. № 3805. P. 1200–1203.
Scally A., 2016. The mutation rate in human evolution and demographic inference // Curr. Opin. Genet. Dev. V. 41. P. 36–43.
Shapiro B., Rambaut A., Drummond A.J., 2006. Choosing appropriate substitution models for the phylogenetic analysis of protein-coding sequences // Mol. Biol. Evol. V. 23. № 1. P. 7–9.
Shapiro B., Ho S.Y.W., Drummond A.J., Suchard M.A., Pybus O.G., Rambaut A., 2011. A Bayesian phylogenetic method to estimate unknown sequence ages // Mol. Biol. Evol. V. 28. № 2. P. 879–887.
Shenbrot G., Bannikova A., Giraudoux P., Quéré J.-P., Raoul F., Lebedev V., 2017. A new recent genus and species of three-toed jerboas (Rodentia: Dipodinae) from China: A living fossil? // J. Zool. Syst. Evol. Res. V. 55. № 4. P. 356–368.
Simpson G.G., 1945. The principles of classification and a classification of mammals // Bull. Amer. Mus. Nat. Hist. V. 85. P. 1–350.
Smith A.B., Peterson K.J., 2002. Dating the time of origin of major clades: molecular clocks and the fossil record // Annu. Rev. Earth Planet. Sci. V. 30. № 1. P. 65–88.
Smith S.A., O’Meara B.C., 2012. treePL: Divergence time estimation using penalized likelihood for large phylogenies // Bioinformatics. V. 28. № 20. P. 2689–2690.
Soubrier J., Steel M., Lee M.S.Y., Der Sarkissian C., Guindon S. et al., 2012. The influence of rate heterogeneity among sites on the time dependence of molecular rates // Mol. Biol. Evol. V. 29. № 11. P. 3345–3358.
Springer M.S., Murphy W.J., Eizirik E., O’Brien S.J., 2003. Placental mammal diversification and the Cretaceous-Tertiary boundary // Proc. Natl. Acad. Sci. USA. V. 100. № 3. P. 1056–1062.
Springer M.S., Meredith R.W., Gatesy J., Emerling C.A., Park J. et al., 2012. Macroevolutionary dynamics and historical biogeography of primate diversification inferred from a species supermatrix // PLoS One. V. 7. № 11. https://doi.org/10.1371/journal.pone.0049521
Springer M.S., Emerling C.A., Meredith R.W., Janečka J.E., Eizirik E., Murphy W.J., 2017. Waking the undead: Implications of a soft explosive model for the timing of placental mammal diversification // Mol. Phylogenet. Evol. V. 106. P. 86–102.
Stadler T., 2010. Sampling-through-time in birth-death trees // J. Theor. Biol. V. 267. № 3. P. 396–404.
Stadler T., Kühnert D., Bonhoeffer S., Drummond A.J., 2013. Birth–death skyline plot reveals temporal changes of epidemic spread in HIV and hepatitis C virus (HCV) // Proc. Natl. Acad. Sci. USA. V. 110. № 1. P. 228–233.
Stebbins G.L., Lewontin R.C., 1972. Comparative evolution at the levels of molecules, organisms, and populations // Darwinian, Neo-Darwinian, and Non-Darwinian Evolution. Berkeley: Univ. California Press. P. 23–42.
Storch G., Qiu S., 1991. Insectivores (Mammalia: Erinaceidae, Soricidae, Talpidae) from the Lufeng hominoid locality, Late Miocene of China // Geobios. V. 24. № 5. P. 601–621.
Suzuki M., Bird A., 2008. DNA methylation landscapes: Provocative insights from epigenomics // Nat. Rev. Genet. V. 9. № 6. P. 465–476.
Tajima F., 1993. Simple methods for testing the molecular evolutionary clock hypothesis // Genetics. V. 135. № 2. P. 599–607.
Takezaki N., Rzhetsky A., Nei M., 1995. Phylogenetic test of the molecular clock and linearized trees // Mol. Biol. Evol. V. 12. № 5. P. 823–833.
Tamura K., Tao Q., Kumar S., 2018. Theoretical foundation of the RelTime method for estimating divergence times from variable evolutionary rates // Mol. Biol. Evol. V. 35. № 7. P. 1170–1182.
Tamura K., Battistuzzi F.U., Billing-Ross P., Murillo O., Filipski A., Kumar S., 2012. Estimating divergence times in large molecular phylogenies // Proc. Natl. Acad. Sci. USA. V. 109. № 47. P. 19333–19338.
Tao Q., Tamura K., Battistuzzi F.U., Kumar S., 2019. A machine learning method for detecting autocorrelation of evolutionary rates in large phylogenies // Mol. Biol. Evol. V. 36. № 4. P. 811–824.
Thomas J.A., Welch J.J., Lanfear R., Bromham L., 2010. A generation time effect on the rate of molecular evolution in invertebrates // Mol. Biol. Evol. V. 27. № 5. P. 1173–1180.
Thorne J.L., Kishino H., Painter I.S., 1998. Estimating the rate of evolution of the rate of molecular evolution // Mol. Biol. Evol. V. 15. № 12. P. 1647–1657.
To T.-H., Jung M., Lycett S., Gascuel O., 2016. Fast dating using least-squares criteria and algorithms // Syst. Biol. V. 65. № 1. P. 82–97.
Tong K.J., Lo N., Ho S.Y.W., 2016. Reconstructing evolutionary timescales using phylogenomics // Zool. Syst. V. 41. № 4. P. 343–351.
Tuffley C., Timothy W., White J., Hendy M.D., Penny D., 2012. Correcting the apparent mutation rate acceleration at shorter time scales under a Jukes–Cantor model // Mol. Biol. Evol. V. 29. № 12. P. 3703–3709.
Vandiver A.R., Idrizi A., Rizzardi L., Feinberg A.P., Hansen K.D., 2015. DNA methylation is stable during replication and cell cycle arrest // Sci. Rep. V. 5. https://doi.org/10.1038/srep17911
Volz E.M., Frost S.D.W., 2017. Scalable relaxed clock phylogenetic dating // Virus Evol. V. 3. № 2. https://doi.org/10.1093/ve/vex025
Warnock R.C.M., Parham J.F., Joyce W.G., Lyson T.R., Donoghue P.C.J., 2015. Calibration uncertainty in molecular dating analyses: There is no substitute for the prior evaluation of time priors // Proc. Roy. Soc. B. Biol. Sci. V. 282. № 1798. https://doi.org/10.1098/rspb.2014.1013
Webster A.J., Payne R.J.H., Pagel M., 2003. Molecular phylogenies link rates of evolution and speciation // Science. V. 301. № 5632. P. 478.
Welch J.J., Bromham L., 2005. Molecular dating when rates vary // Trends Ecol. Evol. V. 20. № 6. P. 320–327.
Welch J.J., Bininda-Emonds O.R., Bromham L., 2008. Correlates of substitution rate variation in mammalian protein-coding sequences // BMC Evol. Biol. V. 8. https://doi.org/10.1186/1471-2148-8-53
Weller C., Wu M., 2015. A generation-time effect on the rate of molecular evolution in bacteria // Evolution. V. 69. № 3. P. 643–652.
Wilson A.C., Carlson S.S., White T.J., 1977. Biochemical evolution // Annu. Rev. Biochem. V. 46. № 1. P. 573–639.
Wilson A.C., Cann R.L., Carr S.M., George M., Gyllensten U.B. et al., 1985. Mitochondrial DNA and two perspectives on evolutionary genetics // Biol. J. Linn. Soc. V. 26. № 4. P. 375–400.
Wilson Sayres M.A., Makova K.D., 2011. Genome analyses substantiate male mutation bias in many species // BioEssays. V. 33. № 12. P. 938–945.
Woodhams M., 2006. Can deleterious mutations explain the time dependency of molecular rate estimates? // Mol. Biol. Evol. V. 23. № 12. P. 2271–2273.
Wu C.I., Li W.H., 1985. Evidence for higher rates of nucleotide substitution in rodents than in man // Proc. Natl. Acad. Sci. USA. V. 82. № 6. P. 1741–1745.
Yang Z., Rannala B., 2006. Bayesian estimation of species divergence times under a molecular clock using multiple fossil calibrations with soft bounds // Mol. Biol. Evol. V. 23. № 1. P. 212–226.
Yoder A.D., Yang Z., 2000. Estimation of primate speciation dates using local molecular clocks // Mol. Biol. Evol. V. 17. № 7. P. 1081–1090.
Zheng Y., Wiens J.J., 2016. Combining phylogenomic and supermatrix approaches, and a time-calibrated phylogeny for squamate reptiles (lizards and snakes) based on 52 genes and 4162 species // Mol. Phylogenet. Evol. V. 94. P. 537–547.
Zhu T., 2020. Bayesian molecular dating // The Molecular Evolutionary Clock. Cham: Springer. P. 83–100.
Zhu T., Reis M., dos, Yang Z., 2015. Characterization of the uncertainty of divergence time estimation under relaxed molecular clock models using multiple loci // Syst. Biol. V. 64. № 2. P. 267–280.
Zuckerkandl E., Pauling L., 1962. Molecular disease, evolution, and genic heterogeneity // Horizons in Biochemistry / Eds Kasha M., Pullman B. N.-Y.: Academic Press. P. 189–225.
Zuckerkandl E., Pauling L., 1965. Evolutionary divergence and convergence in proteins // Evolving Genes and Proteins. N.-Y.: Academic Press. P. 97–166.
Дополнительные материалы отсутствуют.
Инструменты
Журнал общей биологии