

Океанология, 2023, T. 63, № 3, стр. 382-391
Тиосульфат в верхней части анаэробной зоны Черного моря
А. В. Дубинин 1, *, М. Н. Римская-Корсакова 1, О. А. Очередник 2, С. В. Пахомова 1
1 Институт океанологии им. П.П. Ширшова РАН
Москва, Россия
2 Южное отделение Института океанологии им. П.П. Ширшова РАН
Геленджик, Россия
* E-mail: dubinin@ocean.ru
Поступила в редакцию 19.12.2022
После доработки 23.01.2023
Принята к публикации 16.02.2023
- EDN: SRLYNJ
- DOI: 10.31857/S0030157423030036
Аннотация
Тиосульфат – соединение серы в промежуточных степенях окисления между сульфидом и сульфатом, появляется в процессах окисления сероводорода бактериальным и химическим путем. Тиосульфат играет заметную роль в процессах окисления сульфидов и его наличие может свидетельствовать о механизме окислительно-восстановительных реакций в биогеохимическом цикле серы и углерода. В настоящей работе представлено распределение тиосульфата, полученное методом дериватизации с 2,2'-дитиобис(5-нитропиридином), в верхней части анаэробной зоны в Черном море в 2018–2021 годах. Наблюдение проводилось ежегодно на станции, расположенной мористее Геленджика в области континентального склона. Вне зависимости от времени наблюдения концентрация тиосульфата растет с глубиной вместе с ростом концентрации сероводорода. Концентрации выше предела обнаружения 0.01 мкМ тиосульфата обнаруживаются в воде с условной плотностью 16.3 и содержанием сероводорода 7–105 мкМ. Максимальных концентраций тиосульфат достигает на максимальной глубине наблюдения 600 м–0.30 мкМ. При отсутствии окислителей сероводорода в анаэробной зоне предполагается, что появление тиосульфата связано с восстановительным циклом серы в процессе сульфат редукции. Исчезновение тиосульфата происходит в верхней части анаэробной зоны раньше сероводорода.
В природных условиях сера в своих соединениях может иметь степени окисления от +6 до –2. Наиболее распространенной формой серы в морской воде является сульфат. В анаэробных условиях сульфат используется бактериальным сообществом для окисления органического вещества, образуя, в конечном счете, сероводород. Восстановление сульфата и окисление сероводорода приводит к появлению серы промежуточных степеней окисления в основном в виде элементной серы, сульфита, тиосульфата, полисульфидов и политионатов [7, 17, 22]. Тиосульфат – анион тиосерной кислоты, с двумя ионами серы, имеющими разные степени окисления (–2 внешний ион, и +6 внутренний ион). Известная концентрация тиосульфата в воде анаэробных бассейнов (Черное море, впадина Кариако) и в поровой воде осадков обычно не превышает 10 мкМ [8, 9, 11, 13, 16, 17, 20]. Низкие концентрации тиосульфата объясняются его участием в метаболизме сульфатредуцирующих бактерий в присутствии органического субстрата, при наличии окислителей он может окисляться литотрофными бактериями, в отсутствие органического субстрата и окислителей он может диспропорционировать на сульфат и сероводород [9]. По данным [10] тиосульфат – основной продукт окисления сероводорода абиогенным путем в верхней части анаэробной зоны Черного моря. Из-за дефицита кислорода и нитрат-иона в субокислительной зоне в качестве окислителей сероводорода используются оксигидроксиды марганца и железа [6]. Дальнейшее окисление тиосульфата и сульфита до сульфата происходит за счет бактериальной деятельности хемоавтотрофных бактерий [10]. Исследование бактериального потребления тиосульфата в экспериментах в воде озера Кинерет показало, что тиосульфат относительно других промежуточных форм серы (элементной серы, сульфита и полисульфидов) имеет меньшую биологическую скорость оборота [7]. В экспериментальных работах по окислению сероводорода растворенным кислородом было показано, что в воде анаэробных бассейнов тиосульфат может появляться как результат реакции сульфита с сероводородом [21], серы элементной с сульфитом [22].
До настоящего времени концентрация тиосульфата в верхней части анаэробной зоны Черного моря известна плохо. С улучшением методов пробоотбора определяемая концентрация тиосульфата в воде Черного моря [3] оказалась заметно меньше ранее полученных данных [17]. В верхней части анаэробной зоны до глубины 320 м она не превышает 0.15 мкМ [3]. Опыт исследования промежуточных форм серы показывает, что получаемые концентрации тиосульфата и сульфита уменьшаются по мере уменьшения контакта сероводородных вод с воздухом атмосферы. Однако полученные результаты методом дистилляции [3] не позволяют раздельно определить тиосульфат и сульфит. Для раздельного определения тиосульфата и сульфита мы использовали метод дериватизации с 2,2'-дитиобис(5-нитропиридином) (DTNP), предложенный [16]. Первое применение метода дериватизации для раздельного определения тиосульфата и сульфита показало, что концентрации обоих соединений постепенно растут в анаэробной зоне Черного моря, достигая в придонной области 2 мкМ для сульфита и 3 мкМ для тиосульфата [16]. Впоследствии метод дериватизации получил широкое применение при изучении концентраций тиосульфата и сульфита в воде анаэробного бассейна Кариако [8, 11, 13, 20 ] и Фрамварен фьорда [12].
Цель настоящей работы – изучение распределения тиосульфата в верхней части анаэробных вод Черного моря, оценка его долгопериодной временной изменчивости за 4 года наблюдения (2018–2021), а также за короткий период в течение одной недели в 2021 году. В задачи работы входила оценка правильности метода определений. Для этого использовали разное время дериватизации, пробы переменного объема и отбор параллельных проб из одного батометра. В 2019 году была проведена работа по сравнению данных, полученных методом дериватизации и методом дистилляции.
МАТЕРИАЛЫ И МЕТОДЫ
Отбор проб проводили с борта МНИС “Ашамба” на станции с координатами (44.489° с.ш., 37.869° в.д.), расположенной в 7 милях от берега на континентальном склоне в северо-восточной части Черного моря, глубина дна составила 1200 м. Наблюдения проводились 12 июля 2018 года (станция Ash-15), 12 июля 2019 года (Ash-19), 20 июля 2020 года (Ash-21) и 7 и 12 июля 2021 года (Ash-22 и 24). Глубина отбора проб составила 150–600 м и охватывала верхнюю часть анаэробной зоны. Измерения температуры и солености выполнялись зондом “Sea Bird 19plus”. Отбор проб проводился с помощью комплекса “Rosette”, оснащенного шестью 4-литровыми батометрами. Для вытеснения воды из батометра и минимизации контаминации кислородом воздуха использовали аргон высокой чистоты 99.998% [3]. Содержания сероводорода определяли спектрофотометрическим методом [2]. Кислород определяли методом Винклера [5]. Определение форм марганца в воде станции Ash-19 проводили спектрофотометрически с формальдоксимом по методике, описанной в [4]. Растворенное органическое вещество морской воды разлагали УФ-облучением с добавлением перекиси водорода (50 мкл 30%-ной Н2О2 на 20 мл пробы) при рН 2 в течение 1 часа. В качестве источника УФ-излучения использовалась ртутная лампа высокого давления мощностью 80 Вт. Затем фотометрически определяли общую концентрацию растворимого марганца. Разница между общим содержанием растворимого и растворенного Mn+2 является “связанным марганцем”, который представлен растворенным окисленным марганцем, Mn+3 [19]. Содержание взвешенного марганца (Mn+4) рассчитывалось, как разница концентрации общего марганца, измеренного в нефильтрованных подкисленных пробах и концентрации растворенного Mn+2. Взвешенный органический углерод получен на основании фильтрации проб морской воды (масса около 3–3.5 кг) отдельных горизонтов станции Ash-21 (20.07.2020). Определение проводилось по методике, описанной в работе [1].
Для определения сульфита и тиосульфата методом дериватизации в пробу морской воды (20 мл) с ацетатным буфером (1 мл 0.2M, pH 6) немедленно после отбора вводили 1 мл раствора реагента 2,2'-дитиобис(5-нитропиридин) (DTNP, концентрация 10 мM в ацетонитриле) для образования производных изучаемых анионов. Через 5 мин раствор с образовавшимися производными фильтровали через шприцевой фильтр 0.45 мкм и одновременно количественно экстрагировали на картриджах, заполненных октадецилкремнеземом (C18). За 2 часа до предполагаемого пробоотбора через картридж последовательно пропускали 5 мл метанола, 5 мл деионизованной воды и 5 мл ацетатного буферного раствора (20 мМ, рН 6) с тетрабутилацетатом (10 мМ). Образцы загружали на картридж со скоростью 4 мл/мин при помощи шприцевого насоса и после продували аргоном. Картриджи замораживали сразу после экстракции в атмосфере аргона и транспортировали в ИО РАН для проведения анализа.
Размораживание картриджей производили непосредственно перед определением, производные ${\text{SO}}_{3}^{{2 - }}$ и ${{{\text{S}}}_{2}}{\text{O}}_{3}^{{2 - }}$ элюировали с картриджа 1 мл метанола, разбавляли в отношении 1 : 1 деионизированной бескислородной водой и анализировали методом жидкостной хроматографии (Shimadzu LC-20) c УФ-детектированием (λ = 320 нм) на колонке заполненной обращеннофазовым сорбентом С18 (Hypersil ODS, 5 μm, 2.1 × 150 mm) c предколонкой Supelguard Discovery (20 × 4.0 mm, 5 µm). Разделение проводили при градиентном элюировании при скорости 0.2 мл/мин, температуре 25°С. Подвижная фаза А – ацетатный буферный раствор (0.05 M) в присутствии тетрабутиламмония гидросульфата (7.5 мM), подвижная фаза B – ацетонитрил. Для проведения определений была значительно модифицирована градиентная схема элюирования, применяемая в работе [16] для проб черноморской воды. Наиболее эффективное разделение достигалось в следующей схеме: 10% B 1 мин; от 10 до 28% B 6 мин; от 28 до 40% B 10.5 мин; 40% B 2 мин; от 40 до 44% B 3 мин; от 44 до 100% B 2.5 мин; 100% B 5 мин; от 100 до 10% B 7 мин; от 10 до 0% B 7 мин; от 0 до 10% B 1 мин. Время удерживания составило примерно 22.3 и 24.0 мин для ${\text{SO}}_{3}^{{2 - }}$ и ${{{\text{S}}}_{2}}{\text{O}}_{3}^{{2 - }}$ соответственно. Градуировочные графики имели строго линейный вид в пределах определяемых концентраций 0.25 мкМ – 10 мкМ (рис. 1). Предел обнаружения, рассчитанный как 3 стандартных отклонения от фонового значения в бланковом образце градуировочного графика, составил 0.03 и 0.02 мкМ для сульфита и тиосульфата соответственно. В морской воде для тиосульфата предел обнаружения составил 0.01 мкМ. Он был рассчитан аналогичным образом на основании анализа образцов субокислительной зоны моря. Полученные фоновые концентрации тиосульфата в субокислительной зоне не вычитались из данных для образцов в сероводородной зоне (табл. 1 и 2). Воспроизводимость определения сульфита и тиосульфата в морской воде с учетом всей пробоподготовки (дериватизация, фильтрование, продувка картриджей аргоном, их хранение в замороженном состоянии) оценивалась измерением пар образцов, взятых последовательно из одного батометра на разных глубинах. Для 8 пар образцов она составила для сульфита 0.05 мкМ, а для тиосульфата 0.01 мкМ. Для оценки влияния соотношения количеств реактива DTNP и пробы морской воды при определении сульфита и тиосульфата в 2019 году была отобрана серия параллельных проб переменного объема с одного горизонта при одинаковом количестве используемого реагента (рис. 2). Дополнительно в том же году рассмотрено влияние времени дериватизации на результаты определения тиосульфата и сульфита (рис. 3). Для 10 образцов станции Ash-19 проведено сравнение результатов определения суммы тиосульфата и сульфита методом дериватизации и дистиляции (рис. 4) по методике, описанной в работе [3].
Рис. 1.
Градуировочные зависимости для хроматографического определения сульфита (1) и тиосульфата (2).
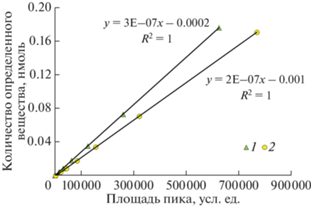
Таблица 1.
Концентрации тиосульфата, сероводорода и гидрофизические параметры водной толщи станций Ash-15, Ash-19 и Ash-21
| Глубина, м | Потенциальная температура, °C | Соленость, PSU | Условная плотность, кг/м3 | H2S, мкМ | S2O3, мкМ |
|---|---|---|---|---|---|
| Ашамба-15 (2018 г.) | |||||
| 140 | 8.67 | 20.84 | 16.09 | <0.1 | – |
| 150 | 8.68 | 20.97 | 16.20 | 0.25 | – |
| 160 | 8.70 | 21.10 | 16.30 | 6.9 | <0.01 |
| 170 | 8.70 | 21.18 | 16.36 | 11.1 | <0.01 |
| 203 | 8.73 | 21.37 | 16.50 | 25.1 | 0.01 |
| 361 | 8.83 | 21.88 | 16.89 | 105 | 0.13 |
| 480 | 8.84 | 22.02 | 16.99 | 151 | 0.24 |
| 600 | 8.85 | 22.12 | 17.07 | 196 | 0.30 |
| Ашамба-19 (2019 г.) | |||||
| 152 | 8.70 | 20.93 | 16.17 | 0.40 | <0.01 |
| 170 | 8.71 | 21.16 | 16.34 | 10.6 | – |
| 180 | 8.72 | 21.25 | 16.42 | 19.0 | 0.01 |
| 200 | 8.74 | 21.40 | 16.52 | 30.5 | 0.02 |
| 250 | 8.78 | 21.62 | 16.69 | 54.6 | 0.02 |
| 301 | 8.81 | 21.76 | 16.80 | 78.7 | 0.08 |
| 350 | 8.83 | 21.86 | 16.87 | 98.1 | 0.09 |
| 401 | 8.84 | 21.93 | 16.93 | 114 | 0.13 |
| 450 | 8.85 | 22.00 | 16.98 | 139 | 0.08 |
| 501 | 8.85 | 22.05 | 17.02 | 156 | 0.13 |
| 550 | 8.85 | 22.09 | 17.05 | 180 | 0.17 |
| 600 | 8.85 | 22.12 | 17.08 | 198 | 0.13 |
| Ашамба-21 (2020 г.) | |||||
| 160 | 8.72 | 21.05 | 16.26 | 3.4 | – |
| 170 | 8.73 | 21.12 | 16.31 | 7.1 | 0.02 |
| 179 | 8.73 | 21.22 | 16.38 | 12.7 | 0.02 |
| 199 | 8.75 | 21.39 | 16.51 | 23.9 | 0.02 |
| 250 | 8.79 | 21.61 | 16.69 | 50.4 | 0.04 |
| 301 | 8.81 | 21.74 | 16.78 | 70.7 | 0.06 |
| 350 | 8.83 | 21.83 | 16.85 | 87.3 | 0.07 |
| 400 | 8.84 | 21.91 | 16.91 | 109 | 0.10 |
| 450 | 8.85 | 21.98 | 16.96 | 127 | 0.11 |
| 499 | 8.85 | 22.04 | 17.01 | 154 | 0.19 |
| 550 | 8.85 | 22.09 | 17.05 | 167 | 0.20 |
| 599 | 8.85 | 22.12 | 17.08 | 182 | 0.22 |
Таблица 2.
Концентрации тиосульфата, сероводорода, кислорода и гидрофизические параметры водной толщи станций Ash-22 и Ash-24
| Глубина, м | Потенциальная температура, °C | Соленость, PSU | Условная плотность, кг/м3 | H2S, мкМ | O2, мкМ | S2O3, мкМ |
|---|---|---|---|---|---|---|
| Ашамба-22 (2021 г.) | ||||||
| 144 | 8.78 | 20.95 | 16.17 | 0.79 | – | – |
| 159 | 8.77 | 21.15 | 16.32 | 7.54 | – | – |
| 170 | 8.78 | 21.23 | 16.39 | 12.4 | – | – |
| 183 | 8.78 | 21.31 | 16.45 | 18.6 | – | 0.01 |
| 200 | 8.78 | 21.40 | 16.52 | 28.1 | – | 0.01 |
| 299 | 8.82 | 21.76 | 16.80 | 78.7 | – | 0.04 |
| 350 | 8.84 | 21.86 | 16.87 | 101 | – | 0.05 |
| 401 | 8.85 | 21.93 | 16.93 | 119 | – | 0.04 |
| 591 | 8.85 | 22.11 | 17.07 | 185 | – | 0.11 |
| Ашамба-24 (2021 г.) | ||||||
| 130 | 8.77 | 20.35 | 15.70 | – | 13.0 | – |
| 140 | 8.79 | 20.62 | 15.91 | – | 6.9 | 0.01 |
| 149 | 8.78 | 20.73 | 16.00 | – | 3.1 | 0.01 |
| 161 | 8.79 | 20.89 | 16.12 | <0.1 | 0.01 | |
| 165 | 8.81 | 20.97 | 16.18 | <0.1 | 0.01 | |
| 170 | 8.83 | 21.01 | 16.21 | 0.10 | 0.01 | |
| 182 | 8.77 | 21.15 | 16.33 | 7.66 | 0.01 | |
| 199 | 8.78 | 21.29 | 16.43 | 17.4 | 0.01 | |
| 300 | 8.81 | 21.70 | 16.75 | 64.7 | 0.04 | |
| 399 | 8.84 | 21.89 | 16.90 | 115 | 0.10 | |
| 499 | 8.85 | 22.01 | 16.98 | 154 | 0.18 | |
| 548 | 8.85 | 22.06 | 17.02 | 174 | 0.17 | |
| 600 | 8.85 | 22.10 | 17.06 | 206 | 0.22 | |
Рис. 2.
Влияние изменения отношения объема пробы относительно реагента на результат определения сульфита (1) и тиосульфата (2) для горизонта 600 м стации Ash-19.
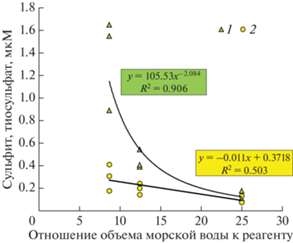
Рис. 3.
Влияние времени реакции дериватизации на результат определения сульфита (1) и тиосульфата (2) для горизонта 450 м станция Ash-19. Приведены средние для двух параллельных проб.
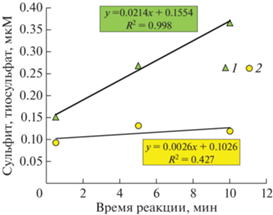
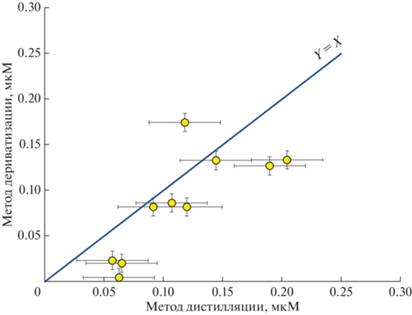
Рис. 4.
Сравнение концентрации суммы тиосульфат+сульфит, полученной методом дистилляции и пересчитанной на тиосульфат, с концентрацией тиосульфата, полученной методом дериватизации, для образцов воды станции Ash-19. Сплошная линия – функция y = x. Доверительный интервал соответствует стандартному отклонению 0.03 и 0.01 мкМ для метода дистилляции и дериватизации соответственно.
РЕЗУЛЬТАТЫ
Несмотря на высокую чувствительность метода дериватизации, данные для тиосульфата и сульфита в параллельных пробах показали различную воспроизводимость. Стандартное отклонение для сульфита в пробах морской воды составило 0.05 мкМ при полученном пределе обнаружения при построении калибровочных графиков 0.03 мкМ. Для тиосульфата эти показатели заметно лучше (0.02 и 0.01 мкМ). Причина такого различия, вероятно, связана с мешающим влиянием дополнительного источника сульфита при подготовке проб морской воды. Мы провели дериватизацию трех параллельных проб с одного горизонта с разным временем реакции. Две параллельные пробы после добавления реактива через 30 с начали загружать на картридж, вторые и третьи две пробы выдерживали 5 и 10 мин соответственно (рис. 3). Осреднение параллельных проб показывает, что концентрация деривата тиосульфата практически не меняется, в то время как концентрация сульфита направленно возрастает и по прошествии 10 мин превышает исходную более чем в два раза. Это могло бы свидетельствовать о реакции сероводорода в пробе со следами кислорода и формировании сульфита при окислении сероводорода.
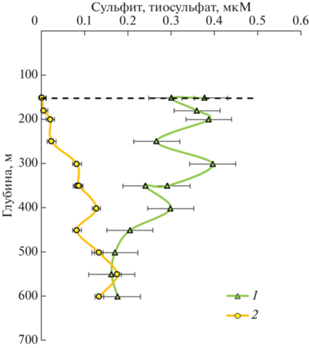
Анализ параллельных проб переменного объема с горизонта 600 м этой же станции Ash-19 (концентрация H2S 198 мкМ) показал, что определяемая концентрация сульфита уменьшалась с увеличением отношения объема пробы относительно количества реагента (рис. 2). Это может свидетельствовать о деградации реагента с образованием определяемого производного деривата сульфита. Предположение о появлении дополнительного сульфита за счет окисления сероводорода выглядит в данном случае маловероятно, так как с уменьшением объема морской воды абсолютное количество сероводорода пропорционально уменьшается, а реагента DTNP увеличивается. Практически на всех станциях максимум сульфита был обнаружен в области интерфейса, с увеличением концентрации сероводорода с глубиной концентрации сульфита уменьшались, что противоречит предположению появления сульфита в связи с контаминацией проб кислородом воздуха (рис. 5). Для тиосульфата определяемые концентрации практически не зависели от объема пробы. Таким образом, мы полагаем, что при взаимодействии DTNP с компонентами морской воды происходит распад реагента, который вносит в результаты анализа дополнительный сигнал сульфита. В этой связи, данные по этой форме серы мы не рассматриваем.
Рис. 5.
Распределение сульфита (1) и тиосульфата (2) на станции Ash-19. Доверительный интервал равен стандартному отклонению. Пунктирная линия соответствует глубине верхней границы анаэробной зоны.
Сравнение результатов анализа методов дистилляции (S2O3 + SO3) и дериватизации для тиосульфата показало, что данные близки, и в методе дериватизации концентрация тиосульфата ожидаемо меньше, чем сумма тиосульфата и сульфита (рис. 4). Следует отметить, что воспроизводимость и предел обнаружения метода дистилляции [3] в 3 раза хуже метода дериватизации.
На рис. 6 показана общая закономерность распределения тиосульфата в водной толще Черного моря за 4 года наблюдений. Виден плавный рост его концентраций с глубиной и ростом концентрации H2S. В 2018 году тиосульфат выше предела обнаружения появлялся на глубине 361 м (σθ 16.89; здесь и далее условная плотность приводится в кг/м3), там его содержание составляло 0.13 мкМ. В 2019 году его появление (0.02 мкМ) было зафиксировано на глубине 200 м (σθ ~ 16.52). В 2020 году на самом верхнем из отобранных горизонтов – на глубине 170 м (σθ 16.30) содержание тиосульфата уже составляло 0.02 мкМ. На двух станциях в 2021 году тиосульфат появлялся на глубине 299 м (σθ 16.30). Его начальная концентрация составила 0.04 мкМ. На станции Ash-24 (12.07.2021) концентрации тиосульфата анализировали, начиная с субокислительной зоны при содержании кислорода 7 мкМ. Вплоть до глубины 300 м (σθ 16.75), где концентрация сероводорода была 64.7 мкМ, концентрация тиосульфата практически не менялась, находясь на уровне фоновых содержаний (0.01 мкМ) (табл. 2).
Рис. 6.
Изменение концентрации тиосульфата от глубины водной толщи в 2018-2021 годах на станциях Ash-15 (12.07.2018) (1), Ash-19 (12.07.2019) (2), Ash-21 (20.07.2020) (3), Ash-22 (07.07.2021) (4) и Ash-24 (12.07.2021) (5).

Максимальные концентрации тиосульфата 0.30 мкМ достигаются на максимальной глубине наблюдения 600 м (рис. 6). Результаты определения тиосульфата всех станций показывают, что тиосульфат всегда растет с глубиной вместе с концентрациями сероводорода. Однако, его максимальные концентрации заметно отличаются между годами и даже в течение одной недели. Такие вариации концентрации тиосульфата не вполне понятны, так как концентрации сероводорода практически не меняются год от года.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Методом дериватизации с DTNP были получены первые результаты по раздельному распределению тиосульфата и сульфита в Черном море [16]. Концентрации обеих форм серы росли с глубиной вместе с концентрацией сероводорода. К сожалению, авторы не привели свои данные в табличном виде, так как их целью было описание нового метода определения. Их выводы относительно устойчивости дериватов тиосульфата и сульфита несколько противоречат нашим полученным результатам. Они указывали, что дериват сульфита более устойчив, чем тиосульфата. Однако в наших экспериментах с различным временем дериватизации и соотношением реагента и морской воды, дериват тиосульфата показал хорошую воспроизводимость результатов, а сульфита плохую. Авторы работы указывали, что увеличение количества реагента не оказывает влияния на воспроизводимость результатов. Однако мы вполне согласны с их выводами только относительно тиосульфата.
Трудности применения метода дериватизации для определения тиосульфата и сульфита встречаются в работах, посвященных исследованию этих форм серы в анаэробных водах впадины Кариако [8, 11, 13] и фиорда Фрамварен [12]. В работе [8] полевые бланки для тиосульфата и сульфита были слишком высоки (0.8 ± 0.3 и 2.2 ± 0.2 мкМ соотвественно) и авторы предпочли их не вычитать из полученных результатов. Они считали, что из-за проблем с контаминацией кислородом при пробоотборе образцов с содержанием H2S выше 10 мкМ данные по тиосульфату и сульфиту для глубинных горизонтов приводить нельзя. Достаточно высокие бланковые образцы были получены в работе [13]. Для сульфита они составили 0.8 ± 1.0 мкМ, для тиосульфата в лучшем случае 0.4 ± 0.4 мкМ, что также свидетельствует о трудностях пробоподготовки. Полагая, что причиной не очень хороших результатов является растворенный кислород воздуха в пробах, авторы работы [11] попытались вводить поправки на продукцию сульфита 0.017 мкМ/мин в пробе, исходя из данных по экспериментам с окислением сероводорода кислородом вод впадины Кариако [21]. И здесь следует отметить, что в экспериментах со временем дериватизации мы получили близкую скорость появления “дополнительного” сульфита, которая составила 0.021 мкМ/мин (рис. 3). Видимо, подобные трудности с определением сульфита данным методом встречены во Фрамварен фиорде [12]; автор указывает на мешающее влияние высоких концентраций сероводорода (в придонной области концентрация сероводорода достигает 6000 мкМ). Несмотря на предварительное удаление сероводорода в виде ZnS, автору упомянутой работы не удалось получить данные по тиосульфату и сульфиту с хорошей воспроизводимостью.
В Черном море максимальная концентрация сульфита (0.90 мкМ) нами отмечена в слое с условной плотностью порядка 16.3; с увеличением условной плотности (глубины и концентрации сероводорода) концентрация сульфита уменьшалась, обычно достигая минимума (0.11 мкМ) на глубине 600 м (рис. 5). У нас нет достаточных оснований считать, что на определение сульфита влияет случайное окисление сероводорода, концентрация которого на глубине 600 м в исследуемом районе достигает 200 мкМ (табл. 1 и 2). Если бы контаминация кислородом воздуха была постоянным фактором, влияющим на результат анализа, то мы имели бы систематическую ошибку, которая бы не могла ухудшить воспроизводимость анализа. Мы предполагаем, что на определение сульфита влияет деградация реагента под влиянием реакций с морской водой, что приводит к появлению дериватов сульфита. На получение результатов может влиять скорость загрузки пробы на картридж, во время которой возможен распад реагента.
Как показали наши исследования, тиосульфат в определяемых количествах выше 0.01 мкМ появляется в верхней части анаэробной зоны при концентрациях сероводорода 7–105 мкМ. По данным [10] тиосульфат является основным продуктом окисления сероводорода (68–82% в верхней части анаэробной зоны). Однако ни в одном случае в верхней части анаэробной зоны повышенных концентраций тиосульфата мы не нашли. Появление тиосульфата вследствие окисления сероводорода кислородом в субокислительных условиях невозможно ввиду отсутствия кислорода в необходимом количестве:
Другим потенциальным окислителем сероводорода может быть Mn+3 и Mn+4. Окисление сероводорода за счет MnO2 может протекать по реакции
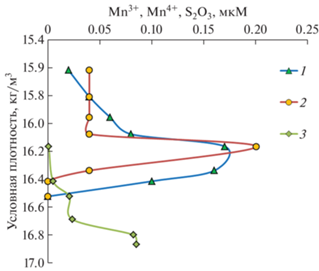
В этом случае в верхней части анаэробной зоны необходимо наличие окисленных форм марганца. На станции Ash-19 мы определили концентрацию окисленных форм марганца, их содержание достигало 0.17 и 0.20 мкМ для Mn+3 и Mn+4 соответственно на горизонте, где концентрация сероводорода составляла 0.4 мкМ (рис. 7 и табл. 2). Но уже при концентрации сероводорода 19 мкМ, концентрации форм марганца уменьшились до 0.1 мкМ Mn+3 и ниже предела обнаружения для Mn+4, при этом концентрация тиосульфата достигла всего 0.01 мкМ. При 30 мкМ сероводорода концентрация тиосульфата выросла до 0.02 мкМ (табл. 2), а сколь-нибудь значимые количества окисленных форм марганца обнаружены не были. Из этого можно сделать вывод, что тиосульфат не является продуктом окисления сероводорода окисленными формами марганца.
Рис. 7.
Изменение концентраций Mn+3 (1), Mn+4 (2) и ${{{\text{S}}}_{2}}{\text{O}}_{3}^{{2 - }}$ (3) на станции Ash-19 (12.07.2019) в зависимости от условной плотности (кг/м3).
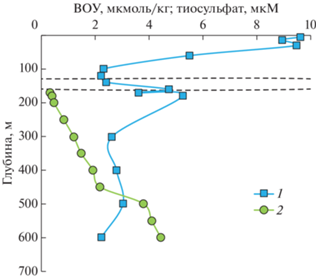
Рост концентрации тиосульфата с глубиной коррелирует с ростом концентраций сероводорода. Отсутствие окислителей в этой части водной толщи, способных окислять сероводород предполагает, что тиосульфат мог появиться в восстановительной части биогеохимического цикла серы. Данные о появлении промежуточных форм серы при восстановлении сульфата до сероводорода немногочисленны. Экспериментальным путем показано, что тиосульфат может быть продуктом неполного восстановления сульфата при условии лимитирования количества органического субстрата [15]. Исчезновение тиосульфата приурочено к верхней части анаэробной зоны, которая характеризуется повышенным количеством взвешенного органического углерода (ВОУ) (рис. 8). Этот промежуточный максимум ВОУ в верхней части анаэробной зоны хорошо известен и, как было показано ранее, связан с повышенным содержанием массы бактериопланктона, что приводит к высоким скоростям окисления сульфида, восстановления сульфата и темновой ассимиляции CO2 [10, 14, 18].
Рис. 8.
Изменение концентраций взвешенного органического вещества (ВОУ) (1) и концентраций тиосульфата (2) в воде станции Ash-21 (20.07.2020). Данные для тиосульфата увеличены в 20 раз. Пунктиром показана субокислительная зона (129–159 м), ограниченная сверху концентрацией кислорода менее 10 мкМ и снизу появлением сероводорода в количестве более 3 мкМ.
ЗАКЛЮЧЕНИЕ
На основании применения метода дериватизации с DTNP получено распределение тиосульфата и сульфита в верхней части анаэробной зоны Черного моря. Эксперименты с переменным временем дериватизации и объемом проб показали, что на данные по концентрации сульфита влияют продукты деградации реактива DTNP в результате его реакции с морской водой. В связи с этим распределение сульфита в работе не рассматривалось.
Распределение тиосульфата рассмотрено в верхней части анаэробной зоны в течение 4 лет наблюдений на 5 станциях. Вне зависимости от времени наблюдения изменение тиосульфата характеризуется ростом концентраций с глубиной. Концентрации выше предела обнаружения 0.01 мкМ обнаруживаются в воде с условной плотностью 16.3 и содержанием сероводорода 7–105 мкМ. Максимальных концентраций тиосульфат достигает на максимальной глубине наблюдения 600 м– 0.30 мкМ. Отсутствие вероятных окислителей сероводорода в анаэробной зоне предполагает, что появление тиосульфата в воде и рост его концентраций с увеличением содержаний сероводорода обусловлены неполным восстановлением сульфата до сероводорода бактериальным путем. Исчезновение тиосульфата происходит в зоне повышенных концентраций взвешенного органического вещества, которое связано с увеличением массы бактериопланктона. К этой зоне приурочена высокая скорость окисления сероводорода, восстановления сульфатов и темновой ассимиляции CO2.
Благодарности. Авторы приносят свою благодарность за помощь в получении результатов по взвешенному органическому углероду Н.А. Беляеву и В.Ю. Федулову.
Источники финансирования. Работа выполнена в рамках госзадания ИО РАН проект FMWE-2021-0004.
Список литературы
Беляев Н.А., Пересыпкин В.И., Поняев М.С. Органический углерод воды, взвеси и верхнего слоя донных осадков западной части Карского моря // Океанология. 2010. Т. 50. № 5. С. 748–757.
Дубинин А.В., Демидова Т.П., Кременецкий В.В. и др. Определение восстановленных форм серы в анаэробной зоне Черного моря: сравнение методов спектрофотометрии и иодометрии // Океанология. 2012. Т. 52. № 2. С. 200–209.
Дубинин А.В., Демидова Т.П., Римская-Корсакова М.Н. и др. Определение восстановленных форм серы в воде анаэробных бассейнов // Морской гидрофизический журнал. 2019. Т. 35. № 1 37–51. https://doi.org/10.22449/0233-7584-2019-1-37-51
Пахомова С.В., Розанов А.Г., Якушев Е.В. Растворенные и взвешенные формы железа и марганца в редокс-зоне Черного моря // Океанология. 2009. Т. 49. № 6. С. 835–850.
Современные методы гидрохимических исследований океана. М.: ИОАН СССР, 1992. 200 с.
Dellwig O., Leipe T., März et al. A new particulate Mn-Fe-P-shuttle at the redoxcline of anoxic basins // Geochim. Cosmochim. Acta. 2010. V. 74. P. 7100–7115.
Findlay A.J., Kamyshny A. Turnover Rates of Intermediate Sulfur Species (${\text{S}}_{x}^{{2 - }}$, S0, ${{{\text{S}}}_{2}}{\text{O}}_{3}^{{2 - }}$, ${{{\text{S}}}_{4}}{\text{O}}_{6}^{{2 - }}$, ${\text{SO}}_{3}^{{2 - }}$) in Anoxic Freshwater and Sediments // Front. Microbiol. 2017. V. 8. P: 2551. https://doi.org/10.3389/fmicb.2017.0255110.3389/fmicb.2017.02551
Hayes M.K., Taylor G.T., Astor Y. et al. Vertical distributions of thiosulfate and sulfite in the Cariaco Basin // Limnol. Oceanogr. 2006. V. 51. № 1. P. 280–287.
Jørgensen B.B. A thiosulfate shunt in the sulfur cycle of marine sediments // Science. 1990. V. 249. P. 152–153.
Jørgensen B.B., Fossing H., Wirsen C.O. et al. Sulfide oxidation in the anoxic Black Sea chemocline // Deep-Sea Res. 1991. V. 38. № 2. S1083–S1103.
Li X., Taylor G.T., Astor Y. et al. Relationship of sulfur speciation to hydrographic conditions and chemoautotrophic production in the Cariaco Basin // Marine Chem. 2008. V. 112. P. 53–64.
Millero F.J. The oxidation of H2S in Framvaren Fjord // Limnol. Oceanogr. 1991. V. 36. № 5. P. 1007–1014.
Percy D., Li X., Taylor G.T. et al. Controls on iron, manganese and intermediate oxidation cate sulfur compounds in the Cariaco Basin // Marine Chem. 2008. V. 111. P. 47–62.
Pimenov N.V., Neretin L.N. Composition and activities of microbial communities involved in carbon, sulfur, nitrogen and manganese cycling in the oxic/anoxic interface of the Black Sea // In: Neretin L.N. (Ed.) Past and present water column anoxia. Elsevier, 2006. P. 501–521.
Vainshtein M.B., Matrosov A.G., Baskunov V.P. et al. Thiosulfate as an intermediate product of bacterial sulfate reduction // Microbiology. 1980. P. 672–675.
Vairavamurthy A., Mopper K. Determination of sulfite and thiosulfate in aqueous samples including anoxic seawater by liquid chromatography afterderivatization with 2,2'-dithiobis(5-nitropyridine) // Environment Sci. Technol. 1990. V. 24. P. 333–337.
Volkov I.I., Neretin L.N. Hydrogen sulfide in the Black Sea // In: Kostianoy A.G., Kosarev A.N. (Eds.). The Black Sea environment. Berlin, Heidelberg, New York: Springer-Verlag, 2008. P. 309–331.
Wakeham S.G., Amann R., Freeman K.H. et al. Microbial ecology of the stratified water column of the Black Sea as revealed by a comprehensive biomarker study // Organic Geochemistry. 2007. V. 38. P. 2070–2097.
Yakushev E., Pakhomova S., Sørenson K. et al. Importance of the different manganese species in the formation of water column redox zones: Observations and modeling // Marine Chem. 2009. V. 117. P. 59–70.
Zhang J-Z., Millero F.J. The chemistry of the anoxic waters in the Cariaco Trench // Deep-Sea Research. 1993a. V. 40. № 5. P. 1023–1041.
Zhang J-Z., Millero F.J. The products from the oxidation of H2S in seawater Geochim. Cosmochim. Acta. 1993b. V. 57. P. 1705–1718.
Zopfi J., Ferdelman T.G., Fossing H. Distribution and fate of sulfur intermediates – sulfite, tetrathionate, thiosulfate, and elemental sulfur – in marine sediments // In: Amend J.P. (Eds.). Sulfur biogeochemistry—Past and present. Boulder, Colorado, Geological Society of America, 2004. V. 379. P. 97–116.
Дополнительные материалы отсутствуют.