

Почвоведение, 2019, № 9, стр. 1063-1069
Фотохимическая деградация органических загрязняющих веществ в растворах почвенных гуминовых кислот
О. А. Трубецкой 1, *, С. В. Пацаева 2, О. Е. Трубецкая 3
1 Институт фундаментальных проблем биологии РАН
142290 Московская область, Пущино, ул. Институтская, 2, Россия
2 МГУ им. М.В. Ломоносова
119991 Москва, Ленинские горы, 1, Россия
3 Филиал Института биоорганической химии им. академиков М.М. Шемякина и Ю.А. Овчинникова РАН
142290 Московская область, Пущино, ул. Институтская, 6, Россия
* E-mail: olegi03@yahoo.com
Поступила в редакцию 31.08.2018
После доработки 23.01.2019
Принята к публикации 30.01.2019
Аннотация
С помощью сочетания препаративной эксклюзионной хроматографии низкого давления с аналитическим электрофорезом в полиакриламидном геле или многократной ультрафильтрацией получены фракции гуминовых кислот чернозема с номинальными молекулярными размерами: >100, 30–100, 5–30 и <5кДа. Исходный препарат гуминовых кислот и фракции протестированы на способность разрушать модельные ароматические соединения 2,4,6-триметилфенол и фурфурол в водном растворе при освещении полихроматическим светом в диапазоне 300–450 нм в лабораторных условиях. Обнаружено, что фракции гуминовых кислот чернозема с молекулярными размерами <5 кДа обладают максимальной способностью разлагать вышеназванные субстраты, в то время как фракции >30 кДа этой функцией практически не обладают. Полученные данные позволяют частично объяснить механизмы фотоиндуцированной трансформации многих органических загрязняющих веществ почвенными гуминовыми веществами.
ВВЕДЕНИЕ
Гуминовые вещества (ГВ), содержащиеся в природных твердофазных средах (почвах, донных отложениях, торфах, углях и т. д.), пресноводных источниках (реках, озерах, болотах), а также в морях и океанах, являются природными средствами защиты и восстановления окружающей среды. Несмотря на более чем двухсотлетнюю историю изучения ГВ, представления об их молекулярной структуре и роли в окружающей среде до конца не выяснены. Тем не менее, некоторые базисные функции ГВ в биосфере не вызывают сомнения. В отличие от подавляющего большинства индивидуальных органических соединений, поглощение солнечной энергии гуминовыми веществами увеличивается экспоненциально с уменьшением длины волны облучающего света, обеспечивая защиту живых организмов от воздействия ультрафиолетовой радиации в атмосфере, воде и на суше [1, 2, 6, 8, 11]. Другой сравнительно недавно обнаруженной базисной функцией ГВ является деградация или трансформация органических загрязняющих веществ с использованием энергии солнечного света, то есть сенсибилизированный фотолиз поллютантов [3, 9, 21, 22]. Прямой фотолиз загрязняющего вещества происходит посредством поглощения света самим веществом, вызывая его деградацию. Однако значительное число поллютантов не поддается прямому фотолизу, то есть не разрушается в результате прямого воздействия моно- или полихроматическим источником света [22]. Сенсибилизированная деградация (то есть косвенный фотолиз) загрязняющего вещества происходит путем воздействия на него активных промежуточных реакционных продуктов, образующихся при абсорбции света сенсибилизаторами. Попадая прямым смывом или через грунтовые воды из сухопутных в водные экосистемы, почвенные ГВ выполняют функцию сенсибилизатора, образуя под действием света несколько промежуточных реакционных продуктов, способных окислять органические загрязняющие вещества или уменьшать их содержание [9, 10, 21], инактивировать патогены [14, 15] и участвовать в циклах углерода в водных системах [12].
Первичным актом является поглощение молекулой ГВ кванта света и ее переход в возбужденное триплетное состояние 3ГВ*. Этот главный переходный реакционный продукт способен самостоятельно осуществлять деградацию устойчивых к фоторазложению органических загрязняющих веществ. Кроме того, возбужденная молекула 3ГВ* способна взаимодействовать с растворенным в воде молекулярным кислородом, образуя активный синглетный кислород 1O2, который в свою очередь является сильнейшим окислителем многочисленных органических загрязняющих веществ. Механизмы образования пероксида водорода H2O2, гидроксильных радикалов ·OH и некоторых других промежуточных реакционных продуктов, образующихся при освещении ГВ, до конца не ясны, однако они вносят незначительный вклад в разрушение поллютантов в растворах ГВ при освещении светом [16].
Следует отметить, что механизмы фотохимических превращений органических поллютантов в присутствии почвенных ГВ, интегральные структурные компоненты и хромофоры, осуществляющие эти реакции, не определены. Поэтому исследование механизмов фотолиза загрязняющих веществ с участием почвенных ГВ является чрезвычайно актуальной задачей, позволяющей уточнить как их роль в экосистемах, так и влияние на биосферу. Решение данной задачи позволит создать теоретическую основу для химического синтеза препаратов, аналогичных компонентам почвенных ГВ, имеющих функции фоторазрушения или фототрансформации антропогенных органических поллютантов. Данные препараты-аналоги могут получить широкое практическое применение для очистки почв и водоемов от органических загрязняющих веществ.
Одним из подходов для решения обозначенной выше проблемы является разделение почвенных ГВ на фракции, имеющие различную фотохимическую активность с последующим анализом физико-химических свойств и структуры этих фракций. Однако попытки ряда исследователей обнаружить в составе почвенных ГВ стабильные фракции, имеющие повышенную по сравнению с исходным препаратом фотохимическую активность, не принесли существенного результата [7, 13, 17].
Ранее были разработаны эффективные методы препаративного фракционировании природных ГВ различного генезиса по молекулярным размерам (МР), основанные на сочетании (1) препаративной эксклюзионной хроматографии низкого давления на сефадексе (ЭХ) и аналитического электрофореза в полиакриламидном геле (ЭПАГ) [19] и (2) многократной ультрафильтрации (МУФ) [18].
Новизна и преимущество сочетаний ЭХ-ЭПАГ и МУФ состоит в том, что 7М мочевина, разрывающая водородные связи и используемая как в процессе препаративного фракционирования методами ЭХ и МУФ, так и при анализе полученных фракций методом ЭПАГ, предотвращает взаимодействие между фракционируемыми ГВ и твердой неподвижной фазой, в которой осуществляется разделение. Одновременно происходит дезагрегация ассоциатов ГВ, состоящих из объединенных водородными связями нативных гуминовых компонентов. Таким образом, предложенные процедуры осуществляют разделение первичных структурных гуминовых компонентов, а не их агрегатов, решая одну из основных проблем, возникающих при фракционировании ГВ. Использование методов ЭХ-ЭПАГ и МУФ позволило получить из ГВ различного генезиса препаративные количества фракций, различающихся по МР, электрофоретической подвижности, гидрофобности, интенсивности флуоресценции и другим физико-химическим характеристикам [5, 18, 20].
Цель работы – сравнение фотохимической активности фракций различного МР, полученных методами ЭХ-ЭПАГ или МУФ из препарата гуминовых кислот чернозема. Данное исследование является частью комплексного изучения структуры и функции почвенных ГК, проводимое нами в течение последних 25 лет.
ОБЪЕКТЫ И МЕТОДЫ
Образец почвы был взят из гумусового гор. А чернозема типичного тяжелосуглинистого на лёссе (Курская область) (Haplic Chernozem (Loamic, Pachic)). ГВ экстрагировали несколько раз раствором 0.1 М пирофосфата натрия и 0.1 М NaOH (рН 13.0) до практически полного отсутствия окраски экстракта. ГК получали кислотным осаждением. Осадок ГК диализовали 7 дней против дистиллированной воды с двукратной сменой воды в течение суток в диализных мешках с номинальным размером пор 10 кДа и лиофилизовали. Таким образом, номинальный МР препарата ГК чернозема был более 10 кДа. Содержание углерода, водорода и азота в препарате ГК чернозема исследовали на CHNS/O-анализаторе серии II 2400 (Perkin Elmer, США), а зольность и влажность препаратов – на термальном анализаторе (Perkin Elmer, США). Элементный состав ГК в массовых процентах (в расчете на сухое беззольное вещество): С – 62.6, Н – 4.7, N – 4.3. Содержание воды и золы 8.9 и 2.5% соответственно.
Сочетание препаративной эксклюзионной хроматографии низкого давления на сефадексе G-75 в растворе 7М мочевины с аналитическим электрофорезом в 10% полиакриламидном геле (ЭХ-ЭПАГ) использовали для получения препаративных количеств стабильных электрофоретических фракций ГК по ранее описанной методике [19]. На колонку наносили 20 мг ГК чернозема, растворенного в 1 мл раствора 7М мочевины. Для получения препаративных количеств фракций различного МР хроматографию повторяли несколько раз. Номинальные МР полученных фракций определяли с помощью ультрафильтрации на мембранах 100, 30 и 5 кДа из регенерированной целлюлозы (Millipore, США), как описано в работе [19].
МУФ 50 мг ГК чернозема, растворенных в 50 мл 7М мочевины, проводили с использованием ультрафильтрационной ячейки объемом 50 мл (Amicon, США) с мембраной 5 кДа из регенерированной целлюлозы (Millipore, США). Номинальный МР вещества, задерживаемого данной мембраной, был более 5 кДа. Ультрафильтрацию проводили в атмосфере азота под давлением 10 p.s.i. (1 p.s.i. = 6894.76 Pa). После окончания первого шага ультрафильтрации получили 30 мл ультрафильтрата (вещества, прошедшего через ультрафильтрационную мембрану с номинальным МР < 5 кДа) и 20 мл концентрата (вещество, задержанное мембраной с МР > 5кДа). Далее концентрат разбавляли до 50 мл раствором 7М мочевины и 22 раза повторяли описанный выше процесс фильтрации через одну и ту же мембрану 5 кДа до тех пор, пока раствор ультрафильтрата не стал практически полностью прозрачным. Уменьшение содержания окрашенного вещества в ультрафильтратах с 1 по 22 не было связано с уменьшением диаметра пор мембраны, так как данный процесс повторяли несколько раз с новыми порциями ГК на той же мембране, причем в каждом отдельном эксперименте наблюдали постепенное снижение содержания окрашенного вещества в последующих ультрафильтратах по сравнению с предыдущими [18]. Следует добавить, что в процессе многократной ультрафильтрации в 7М мочевине не наблюдали снижение скорости протекания через мембрану раствора, неизбежно сопровождающего существенное уменьшение диаметра пор.
В результате эксперимента получили 22 ультрафильтрата (объем каждого составлял 30 мл) и конечный концентрат К (МР > 5 кДа) объемом 20 мл. Ультрафильтраты с 1 по 3 объединили во фракцию F1, а ультрафильтраты с 4-го по 22-й – во фракцию F2. Затем концентрат К, фракции F1 и F2 диализовали с использованием 1 кДа диализных мембран (Sigma_Aldrich, Россия) в течение семи дней и лиофилизовали.
Для проверки влияния мочевины на структуру и фотохимическую активность исходного препарата, 50 мг ГК чернозема растворяли в 50 мл 7М мочевины, выдерживали 7 дней в темноте при комнатной температуре, затем раствор диализовали и лиофилизовали в диализном мешке с номинальным размером пор 10 кДа. Как показано ранее [4, 5] элементный состав, спектры поглощения, флуоресценции, 1Н- и 13С-ЯМР препарата ГК чернозема после его обработки мочевиной существенно не изменились.
Исходный препарат ГК чернозема и обработанный мочевиной (ГКм), а также фракции различного МР, полученные с помощью ЭХ-ЭПАГ и МУФ, хранили в сухом виде в плотно закрывающихся стеклянных баночках в темноте при комнатной температуре и использовали для дальнейших анализов.
Фосфатный буфер с заданным значением рН 6.5 готовили путем смешивания трех объемов раствора 6.6 мМ Na2HPO4 (рН 9.8) и семи объемов раствора 6.6 мМ NaH2PO4 (рН 4.5). Для достижения полного растворения сухие препараты ГК и фракций сначала растворяли в щелочном буфере (3 объема), затем постепенно добавляли 7 объемов кислого буфера.
Спектры поглощения в ультафиолетовой и видимой областях измеряли на спектрофотометре Cary 3 (Varian) в кварцевой кювете длиной 1 см в растворе 6.6 мМ фосфатного буфера (рН 6.5). На основании полученных спектров рассчитывали соотношения А270/А400 для ГК и фракций различного МР. Спектры флуоресценции концентрата К и 22 ультрафильтратов измеряли на спектрофлуориметре Cary Eclipse (Varian) в кварцевой четырехсторонней кювете длиной 1 см при длине волны возбуждения 270 нм как в работе [18].
Для определения фотохимической активности каждый образец ГК и фракций разбавляли в отдельной пробирке до оптической плотности 0.1 ± 0.005 при длине волны 300 нм в 3.3 мМ фосфатном буфере (рН 6.5), содержащем 0.1 мМ 2,4,6-триметилфенола или 0.1 мМ фурфурола (2‑фуранкарбальдегида). Субстрат триметилфенол используют для проверки фотохимической активности ГК в возбужденном триплетном состоянии (3ГК*), а субстрат фурфурол – для оценки способности 3ГК* стимулировать образование активного синглетного кислорода 1O2 [16, 21]. Стеклянный реактор с 10 мл реакционной смеси помещали в центр алюминиевого цилиндра, оснащенного шестью ксеноновыми лампами, излучающими свет в диапазоне длин волн от 300 до 450 нм с максимумом при 365 нм. Оба субстрата не поглощают излучения, испускаемого лампами, и не подвергаются разрушению в отсутствии ГК. Кинетику реакции разложения субстратов в присутствии препаратов ГК при освещении определяли на основании анализа изменения концентрации субстрата в реакционной смеси через определенные промежутки времени от начала эксперимента: 0, 10, 20, 30, 60, 90, 120, 150 и 180 мин. Концентрацию субстрата в отобранных пробах определяли методом высокоэффективной жидкостной хроматографии на обратно-фазовой колонке Spherisorb ODS-2 с детекцией содержания триметилфенола или фурфурола при 220 нм. Элюцию субстрата с колонки проводили смесью воды (80%) и метанола (20%) при скорости потока 1 мл/мин. Элюирующий раствор был подкислен 1%-ной фосфорной кислотой. Все карбоксильные группы добавленных в реакционную смесь ГВ в кислой среде протонированы, поэтому в процессе хроматографии они необратимо адсорбировались на колонке и не мешали определению концентрации субстратов. Рассчитывали кажущиеся константы скорости реакции первого порядка деградации обоих субстратов в присутствии исследованных препаратов ГК и фракций на основании соотношения K = dC/dТ C0, где K – константа скорости реакции первого порядка, dC – разность концентраций субстрата за период времени dТ, C0 – начальная концентрация субстрата.
В контрольном эксперименте, когда реакционная смесь, содержащая исследуемый образец и субстрат, находилась в темноте в течение 180 мин (время проведения эксперимента), концентрация субстратов была такой же, как и в начале эксперимента, что указывает на отсутствие сорбции субстрата на ГК или фракциях в течение эксперимента.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
С помощью сочетания ЭХ-ЭПАГ из ГК чернозема получены индивидуальные электрофоретические фракции А, В и C + D, различающиеся по времени выхода с колонки, то есть по МР. Было показано, что МРА > МРВ > МРC + D. С помощью метода ультрафильтрации обнаружено, что фракция А имеет МР, сравнимые размерами глобулярных белков с молекулярной массой более 100 кДа, фракция В – 30–100 кДа, фракция C + D – 5–30 кДа.
Для получения фракций ГК с МР менее 5 кДа использовали недавно разработанную методику многократной ультрафильтрации [18]. Были получены концентрат ГК с МР более 5 кДа (К > > 5 кДа) и 22 ультрафильтрата с МР < 5 кДа. Несмотря на сходные номинальные МР ультрафильтратов, они различались по максимумам флуоресцентной эмиссии, что может быть связано с неоднородностью их химического состава и возможных различиях в фотохимической активности. Для проверки данного предположения ультрафильтраты 1–3 шагов объединили во фракцию F1 (λмакс < 490 нм), а ульрафильтраты 4–22 шагов – во фракцию F2 (λмакс > 490 нм). Следует отметить, что концентрат К > 5 кДа не обладал какой-либо существенной флуоресценцией.
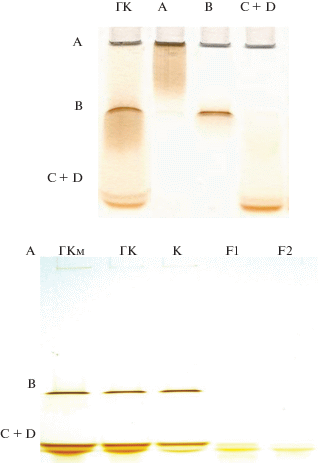
На рис. 1 представлены электроферограммы исходного ГК, препарата, обработанного мочевиной (ГКм), а также фракций А, В, C + D, F1, F2 и концентрата К. Все образцы формировали на геле естественно окрашенные коричневые или желтоватые зоны различной электрофоретической подвижности, причем фракция А наибольшего МР формировала зону на старте, фракция В – в средней части геля, фракция C + D - несколько полос близкой электрофоретической подвижности в нижней части геля. Концентрат К содержит зоны А, В и C + D, в то время как ультрафильтраты F1 и F2 формируют на геле светлые желтоватые зоны с электрофоретической подвижностью, как и у фракции C + D. Препарат ГКм имеет сходную электрофореграмму по сравнению с необработанным мочевиной исходным образцом ГК. Принимая во внимание тот факт, что все физико-химические свойства препарата ГКм, а также его фотохимическая активность не отличались от таковых для исходного препарата ГК чернозема, можно с большой долей вероятности предположить, что мочевина не вносит существенного изменения в структуру хромофоров, осуществляющих фотохимические превращения субстратов.
Рис. 1.
Электрофорез в 10%-ном ПАГ 0.1 мг ГК чернозема и обработанного мочевиной препарата (ГКм), фракций А, В, C + D, полученных с помощью ЭХ-ЭПАГ (верхняя электрофореграмма), а также концентрата К и ультрафильтратов F1 и F2, полученных методом МУФ (нижняя электрофореграмма).
Для исходного препарата ГК чернозема, а также фракций А, В, C + D, F1, F2 и концентрата К были измерены спектры поглощения, монотонно убывающие с увеличением длины волны как в ультрафиолетовой, так и в видимой областях света и не имели характерных максимумов. Тем не менее, спектры существенно отличались наклоном кривой. Данное различие отражено в виде соотношения А270/А400 (табл. 1), наибольшие значения которого наблюдали для фракций F1, F2 и C + D (4.08, 3.61 и 3.16 соответственно). В то же время концентрат К, а также фракции А и В наибольшего МР имели достоверно более низкие значения соотношения А270/А400 (2.73, 2.75 и 2.71 соответственно). Для исходного препарата ГК величина А270/А400 (2.87) была средней между фракциями наибольшего и наименьшего МР.
Таблица 1.
Номинальные молекулярные размеры (МР), отношения оптических плотностей (А270/А400) и константы скоростей разложения триметилфенола (К1) и фурфурола (К2) в присутствии исходного препарата ГК чернозема и фракций различного МР, полученных с помощью сочетания ЭХ-ЭПАГ или МУФ
| Метод фракционирования | Образец | МР, кДа | А270/А400 | K1 | K2 |
|---|---|---|---|---|---|
| ×10–4 с–1 | |||||
| Нефракционированный исходный препарат | ГК | >10 | 2.87 ± 0.03 | 2.15 ± 0.15 | 0.34 ± 0.01 |
| ЭХ-ЭПАГ в 7М мочевине | Фракция А | >100 | 2.75 ± 0.02 | 0.77 ± 0.04 | 0.06 ± 0.001 |
| Фракция В | 30–100 | 2.71 ± 0.02 | 0.66 ± 0.15 | 0.05 ± 0.006 | |
| Фракция C + D | 5–30 | 3.16 ± 0.04 | 2.32 ± 0.21 | 0.77 ± 0.02 | |
| Многократная ультрафильтрация в 7М мочевине | Ультрафильтрат F1 | <5 | 4.08 ± 0.03 | 5.93 ± 0.30 | 1.23 ± 0.05 |
| Ультрафильтрат F2 | <5 | 3.61 ± 0.02 | 4.58 ± 0.41 | 0.82 ± 0.32 | |
| Концентрат К | >5 | 2.73 ± 0.02 | 0.65 ± 0.06 | 0.06 ± 0.07 | |
Для препаратов ГК чернозема и фракций различного МР, полученных с помощью сочетания ЭХ-ЭПАГ или МУФ, была исследована фотохимическая активность (рис. 2) с использованием модельных соединений триметилфенола и фурфурола.
Рис. 2.
Константы скорости разложения триметилфенола (А) и фурфурола (Б) в присутствии исходного препарата ГК чернозема, фракций А, В, C + D, полученных с помощью ЭХ-ЭПАГ, а также концентрата К и ультрафильтратов F1 и F2, полученных с помощью МУФ.
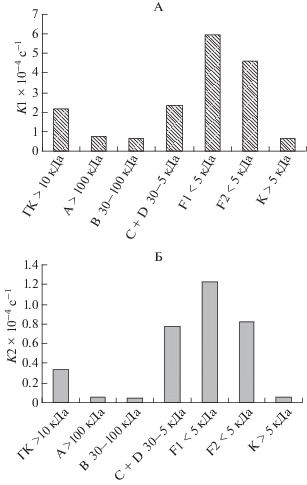
Фотоиндуцированная деградация триметилфенола (применяется для оценки фотохимической активности препаратов ГВ, переходящих в возбужденное триплетное состояние (3ГВ*) после поглощения кванта света) фракциями A и B наибольшего МР, а также концентратом К была в 2.8–3.3 раза меньше, чем в присутствии исходного препарата ГК. Наибольшую скорость разложения данного субстрата наблюдали для фракций F1 и F2 наименьшего МР (5.93 × 10–4 и 4.58 × 10–4 с–1 соответственно), причем у фракции F1 фотохимическая эффективность деградации триметилфенола была на 30% больше по сравнению с F2. Следует отметить, что фракция C + D также имела константу скорости фотоиндуцированной трансформации триметилфенола, превышающую таковую для исходной ГК, однако была в 2–2.5 раза ниже, чем у ультрафильтрационных фракций F1 и F2. Изменения значений константы скорости фотохимической деградации триметилфенола совпадают с изменением соотношений А270/А400, наибольшие значения которых наблюдали для ультрафильтрационных фракций F1 и F2, а также для фракции C + D. Следует добавить, что, несмотря на сходный кажущийся МР (<5 кДа) фракций F1 и F2, они различались по максимумам флуоресцентной эмиссии, что, по-видимому, связано с наличием в этих фракциях различных фотохимически активных компонентов.
Следующий комплекс экспериментов был выполнен с использованием субстрата фурфурола, позволяющего оценить способность ГВ индуцировать образование синглетного кислорода 1O2. Фракции A и B наибольшего МР, а также концентрат К практически не принимают участие в данной реакции – константа скорости трансформации субстрата была в 5–6 раз меньше, чем у исходного препарата ГК. Наибольшей способностью индуцировать образование 1O2 обладали фракция F1, а также фракции C + D и F2, причем эффективность фракции F1 была в полтора раза выше по сравнению с двумя последними образцами. Если в случае с триметилфенолом фракции наибольшего МР обладали более заметной активностью, то, возможно, стимуляция образования синглетного кислорода принадлежит, главным образом, фракциям наименьшего МР.
Вышеизложенные результаты исследований показывают, что методы сочетания ЭХ-ЭПАГ и МУФ, использованные для разделения почвенных ГК, позволяют получать в препаративных количествах фракции разного МР, обладающие различными спектральными и фотохимическими свойствами. Фракции наименьшего МР являются наиболее перспективными для дальнейшего субфракционирования с целью идентификации фотохимически активных хромофоров почвенных ГК, так как только они обладают выраженными фотохимическими свойствами. Особенно важно подробнее рассмотреть фракции с МР менее 5 кДa, которые можно получить методом МУФ в растворе 7М мочевины.
Идентификация в почвенных ГВ отдельных фракций со специфическими ярко выраженными фотохимическими свойствами с функцией разрушения и/или трансформации органических поллютантов открывает широкие перспективы для создания нового поколения экологически чистых фотодеструкторов на основе природных почвенных ГВ.
Список литературы
Дроздова А.Н., Пацаева С.В., Хунджуа Д.А. Флуоресценция растворенного органического вещества как маркер распространения пресных вод в Карском море и заливах архипелага Новая Земля // Океанология. 2017. Т. 57. С. 49–56.
Кононова М.М. Органическое вещество почв. М.: Изд-во АН СССР, 1963. 314 с.
Соколова И.В., Чайковская О.Н. Влияние гуминовых кислот на фотопроцессы в водных средах // Вестник ТГПУ. 2008. Т. 78. С. 42–46.
Трубецкой О.А., Демин Д.В., Трубецкая О.Е. Флуоресцентные свойства низкомолекулярных фракций гуминовых кислот чернозема // Почвоведение. 2013. № 10. С. 1222–1227.
Трубецкой О.А., Трубецкая О.Е. Обратно-фазовая высокоэффективная жидкостная хроматография стабильных электрофоретических фракций почвенных гуминовых кислот // Почвоведение. 2015. № 2. С. 166–174.
Alberts J.J., Takacs M. Total luminescence spectra of IHSS standard and reference fulvic acids, humic acids and natural organic matter: comparison of aquatic and terrestrial source terms // Org. Geochem. 2004. V. 35. P. 243–256.
Belin C., Quellec C., Lamotte M., Ewald M., Simon Ph. Characterization by fluorescence of the dissolved organic matter in natural water: application to fractions obtained by tangential ultrafiltration and XAD resin isolation // Environ. Technol. 1993. V. 14. P. 1131–1144.
Boyle E.S., Guerriero N., Thiallet A., Vecchio, R.D., Blough N.V. Optical properties of humic substances and CDOM: Relation to structure // Environ. Sci. Technol. 2009. V. 43. P. 2262–2268.
Canonica S., Hellrung B., Wirz J. Oxidation of phenols by triplet aromatic ketones in aqueous solution // J. Phys. Chem. A. 2000. V. 104. P. 1226–1232.
Canonica S., Hellrung B., Muller P., Wirtz J. Aqueous oxidation of phenylurea herbicides by triplet aromatic ketones // Environ. Sci. Technol. 2006. V. 40. P. 6636–6641.
Dahlen J., Bertilsson S., Pettersson C. Effects of UV-A irradiation on dissolved organic matter in humic surface waters // Environ. Int. 1996. V. 22. P. 501–506.
Gao H., Zepp R.G. Factors influencing photoreactions of dissolved organic matter in a coastal river of the southeastern United States // Environ. Sci. Technol. 1998. V. 32. P. 2940–2946.
Haag W.R., Hoigne J. Singlet oxygen in surface waters. 3. Photochemical formation and steady-state concentrations in various types of waters // Environ. Sci. and Technol. 1986. V. 20. P. 341–348.
Jasper J.T., Nguyen M.T., Jones Z.L., Ismail N.S., Sedlak D.L., Sharp J.O., Luthy R.G., Horne A.J., Nelson K.L. Unit process wetlands for removal of trace organic contaminants and pathogens from municipal wastewater Effluents // Environ. Eng. Sci. 2013. V. 30. P. 421–436.
Kadir K., Nelson K.L. Sunlight mediated inactivation mechanisms of Enterococcus faecalis and Escherichia coli in clear water versus waste stabilization pond water // Water Res. 2014. V. 50. P. 307–317.
McKay G., Couch K.D., Mezyk S.P., Rosario-Ortiz F.L. Investigation of the coupled effects of molecular weight and charge-transfer interactions on the optical and photochemical properties of dissolved organic matter // Environ. Sci. Technol. 2016. V. 50. P. 8093–8102.
Schmitt-Kopplin P., Hetkorn N., Shulten H.R., Kettrup A. Structural changes in dissolved soil humic acid during photochemical degradation processes under O2 and N2 atmosphere // Environ. Sci. Technol. 1998. V. 32. P. 2531–2541.
Trubetskaya O., Shaloiko L., Demin D., Marchenkov V., Proskuryakov I., Coelho C., Trubetskoj O. Combining electrophoresis with detection under ultraviolet light and multiple ultrafiltration for isolation of humic fluorescence fractions // Analytica Chimica Acta. 2011. V. 690. P. 263–268. https://doi.org/10.1016/j.aca.2011.02.016
Trubetskoj O.A., Trubetskaya O.E., Afanas’eva G.V., Reznikova O.I., Saiz-Jimenez C. Polyacrylamide gel electrophoresis of soil humic acid fractionated by size exclusion chromatography and ultrafiltration // J. Chromatography A. 1997. V. 767. P. 285–292.
Trubetskoj O.A., Richard C., Voyard G., Marchenkov V.V., Trubetskaya O.E. Molecular size distribution of fluorophores in aquatic natural organic matter: application of HPSEC with multi-wavelength absorption and fluorescence detection following LPSEC-PAGE fractionation // Environ. Sci. Technol. 2018. V. 52. P. 5287–5295. https://doi.org/10.1021/acs.est.7b03924
Wenk J., Aeschbacher M., Sander M., Gunten U.V., Canonica S. Photosensitizing and inhibitory effects of ozonated dissolved organic matter on triplet-induced contaminant transformation // Environ. Sci. Technol. 2015. V. 49. P. 8541–8549.
Zepp R.G., Schlotzhauer P.F., Sink R.M. Photosensitized transformations involving electronic energy transfer in natural waters: role of humic substances // Environ. Sci. Technol. 1985. V. 19. P. 74–81.
Дополнительные материалы отсутствуют.