

Поверхность. Рентгеновские, синхротронные и нейтронные исследования, 2020, № 10, стр. 65-73
Особенности формирования углеродного нанопокрытия, полученного магнетронным напылением на поверхность полиуретана
И. А. Морозов a, *, А. С. Каменецких b, М. Г. Щербань c, Р. И. Изюмов a, Д. М. Кисельков d
a Институт механики сплошных сред УрО РАН
614013 Пермь, Россия
b Институт электрофизики УрО РАН
620016 Екатеринбург, Россия
c Пермский государственный национальный исследовательский университет
614990 Пермь, Россия
d Институт технической химии УрО РАН
614013 Пермь, Россия
* E-mail: ilya.morozov@gmail.com
Поступила в редакцию 21.01.2020
После доработки 24.02.2020
Принята к публикации 27.02.2020
Аннотация
Нанесение углеродных покрытий используется для модификации свойств (механических, биомедицинских и др.) поверхностей. Интерес представляет обработка мягких материалов. В данной работе на поверхность двух упругих полимеров (полиуретанов), отличающихся фазовым составом и жесткостью, наносили углерод методом импульсного магнетронного напыления. В результате образуется неоднородное углеродсодержащее покрытие толщиной от 3 до 30 нм. На начальном этапе обработки происходит взаимодействие углерода с атомами полимера и образование гидрофобных покрытий с высокой энергией поверхности. С ростом толщины энергия поверхности покрытия снижается, а на поверхности формируется нанослой аморфного углерода. На определенном этапе нанесения покрытия на поверхности формируется хаотичная складчатая текстура, характеризующаяся спектром длин волн и амплитуд. Модуль упругости полученных покрытий не превышает нескольких ГПа. Все изученные свойства коррелируют как со свойствами исходной подложки, так и количеством импульсов углеродного напыления. Подобные покрытия благодаря их высокой активности и складчатой текстуре представляют потенциальный интерес при создании биомедицинских изделий.
ВВЕДЕНИЕ
Плазменная обработка материалов существенным образом влияет на свойства поверхности и изменяет эксплуатационные характеристики материалов. Одним из направлений исследований является создание углеродных покрытий. В зависимости от способа нанесения состояние углерода может изменяться в широком диапазоне: от аморфного полимероподобного с высокой степенью гидрогенизации до алмазоподобного [1]. Каждое из состояний обладает уникальными свойствами. В частности, пленки алмазоподобного углерода уменьшают газовую проницаемость материала [2]; допирование углерода азотом снижает твердость покрытий, кремнием – увеличивает [3], кроме этого может быть повышена коррозионная стойкость, изменены фрикционные и гидрофобные свойства. Большое внимание уделяется влиянию углеродных покрытий на биомедицинские свойства: сорбционная активность определенных протеинов (белков, способствующих росту клеток, например, альбумин, фибронектин) может быть увеличена [4], других (фибриноген, который ускоряет тромбообразование) снижена [5], улучшаются антибактериальные свойства [6], снижается тромбообразование [7] и коагуляция крови [8].
Полимерные материалы обладают сложным химическим составом. Плазменная обработка мягких полимеров (даже в условиях низкоэнергетической газовой плазмы) приводит к существенным изменениям структуры поверхности [9]: разрушаются макромолекулы, формируются новые углеродные связи (происходит карбонизация поверхности [10]), образуются свободные радикалы и высоко энергетические водородные связи. В первую очередь, это влияет на гидрофобность и свободную энергию поверхности. Увеличение интенсивности (времени) обработки ведет к формированию все более жесткого и толстого покрытия (или модифицированного слоя). Это приводит к потере устойчивости и образованию складчатой структуры поверхности. Хаотичная складчатая структура может являться основой для контролируемого роста клеток [11] и создает препятствия для адгезии бактериальных пленок [12]. При определенных свойствах покрытия его складки могут расправляться при растяжении материала, увеличивая деформативность поверхности [13].
Одной из проблем нанесения углеродных покрытий являются возникающие в них высокие внутренние напряжения, приводящие к отслаиванию покрытий. Предлагаются различные решения [14]: применяют плазменное осаждение углерода из газовой фазы, предварительно обрабатывают поверхность, увеличивают степень гидрогенизации углерода, проводят постобработку покрытий в плазме аргона [15]. Другим нежелательным эффектом является растрескивание жесткого покрытия на эластичной подложке в результате механических воздействий в процессе эксплуатации [16].
Полиуретан – широко распространенный синтетический полимер. Его механические свойства (в зависимости от рецептуры) могут варьироваться в широком диапазоне: от вязких жидкостей до жестких пластиков. Упругие полиуретаны применяются при изготовлении изделий биомедицинского назначения: трубок, катетеров и искусственных сосудов [17, 18], имплантатов [19‒21], клеточных матриксов [22], межфаланговых протезов [23] и т.д.
Полиуретан является двухфазным полимером: на структурном уровне состоит из жестких блоков (имеют фибриллярную или глобулярную структуру), расположенных в более мягкой матрице. В данной работе исследовали особенности нанесения углерода магнетронным распылением графитовой мишени на поверхность мягких упругих полиуретанов (модуль упругости 6 и 25 МПа), было показано существенное влияние полимерной подложки и длительности напыления на свойства покрытий.
МАТЕРИАЛЫ И МЕТОДЫ
Создание полиуретанов
В работе исследовали две полиуретановые композиции, изготовленные из коммерчески доступного полимера (уретановый фоорполимер на основе простого полиэфира) и сшивающего агента, представляющего собой смесь отвердителя (МОКА) и пластификатора (полифурит)) в соотношении 100 : 33 (материал ПУ1) и 100 : 46 (ПУ2). Расчетное количество форполимера нагревали до 80°C и вакуумировали в течение 5 мин. Затем добавляли остальные компоненты, разогретые до 80°C. Смесь вакуумировали и заливали в форму с открытой верхней поверхностью и отверждали в термошкафу при 100°C в течение 20 ч. Средняя толщина полученных пластин составляла 2 мм.
Плазменная обработка
Камеру плазменной установки откачивали до остаточного давления 3 × 10‒3 Па. Затем в камеру напускали аргон до рабочего давления 0.3 Па. Применялся плоский сбалансированный магнетрон с диаметром графитовой мишени 80 мм, который работал в импульсном модулированном режиме генерации импульсов тока разряда. Амплитуда импульсов тока составляла 40 А при длительности импульса 8 мкс, пауза между импульсами – 10 мс. Расстояние между образцами и мишенью магнетрона ‒ 100 мм. Температура образцов в процессе обработки (измерялась инфракрасным пирометром) не превышала 30°C. Количество N импульсов изменялось от 4 × 103 до 1.2 × 106 (а именно: 4, 8, 20, 40, 144, 288, 360, 1200 тыс. импульсов). Предварительные исследования показали, что при N < 4000 покрытие не образовывалось, по-видимому, в силу недостаточной поверхностной энергии подложки: структурно-механические свойства поверхностей были идентичны необработанному полимеру. Отметим, что для закрепления на полиуретане островковых углеродных покрытий, образующихся при малых N, требуется предварительная активация поверхности полимера в газовой плазме [13].
Энергия поверхности
Краевой угол смачивания определяли методом лежащей капли. В качестве тестовых жидкостей использовали воду и диэтиленгликоль (мы не использовали дийодметан, так как он, как и другие растворители, вступают в реакцию с полимером), при этом диаметры капель варьировались от 2 до 3 мм. Свободную поверхностную энергию вычисляли методом Оуэнса–Вендта–Рабеля–Каелбле как сумму дисперсионной (учитывает Ван-дер-Ваальсовые взаимодействия поверхности и тестовой жидкости) и полярной (дипольные взаимодействия и энергия водородных связей) составляющих. Известно, что энергия поверхности после плазменной обработки асимптотически уменьшается. В данной работе измерения производили спустя 7‒10 дней после плазменной обработки, когда энергия поверхности достигает равновесного значения.
Спектроскопия комбинационного рассеяния
Спектры КР были получены на дисперсионном спектрометре комбинационного рассеяния света Bruker Senterra с применением возбуждающего лазера, при этом длина волны составляла 532 нм, продолжительность накопления – 1 с, число накоплений – 1200, щелевая диафрагма имела размеры 25 × 1000 мкм. Для предотвращения разрушения мягкого полимера мощность излучения ограничивали 0.2 мВт. Базовую линию выравнивали адаптивным методом в программe Spectragryph.
Атомно-силовая микроскопия
Использовали атомно-силовой микроскоп (АСМ) Ntegra Prima в полуконтактном режиме (исследование микрорельефа), проводили наноиндентирование и наномеханическое картирование (режим Hybrid). Во всех случаях использовали зонды калиброванной жесткости (методом Садера и тепловых колебаний) и геометрии острия (с использованием стандартного образца Tip Check).
Толщину покрытий определяли наноиндентированием. Использовали жесткие зонды (жесткость кантилевера ~35 Н/м). Рельеф поверхностей до и после индентирования исследовали в полуконтактном режиме. Было обнаружено, что после индентирования со значительным усилием F (от 80 до 600 нН, в зависимости от образца) на покрытиях остаются следы от уколов. В то же время, после индентирования необработанного полимера на ту же глубину, поверхность упруго восстанавливалась. Таким образом, по глубине следа от укола можно оценить толщину покрытия.
Наномеханическое картирование использовали для структурно-механического исследования свойств необработанных полимеров: одновременно со сканированием рельефа происходит индентирование поверхности и определение механических характеристик, в частности, модуля упругости. Использовали зонды ScanAsyst Air с жесткостью 0.4 Н/м и радиусом острия 5 нм. Частота индентации – 0.5 кГц, максимальное усилие (0.25‒0.5 нН) задавали из условия, чтобы глубина индентирования не превышала 20 нм. Приведенный модуль упругости E/(1 ‒ ν2) определяли встроенной в программное обеспечение обработкой силовых кривых при помощи модели Джонсона–Кендалла–Робертса, где E – модуль упругости, ν – коэффициент Пуассона.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Рельеф поверхности и карта модуля упругости необработанного ПУ1 представлена на рис. 1а. Среднеквадратическая шероховатость для базовой длины 3 × 3 мкм составила 3 нм. Жесткая фаза материала ПУ1 имеет спутанную фибриллярную структуру (рис. 1a, вставка крупного плана на карте модуля). Ее распределение неоднородно. На поверхностях видны агломераты, представляющие собой участки высокой концентрации жесткой фазы. Анализ рельефа при помощи преобразования Фурье показал пики в области 200‒400 нм: характерные расстояния между агломератами жесткой фазы полимера. Распределение модуля упругости (рис. 1в) поверхности ПУ1 имеет пологий наклон в область малых значений, что соответствует участкам поверхности с малой концентрацией жесткой фазы (темные области на карте модуля, рис. 1а).
Рис. 1.
АСМ-изображения рельефов и карт модуля упругости поверхности необработанных материалов: a ‒ ПУ1, б ‒ ПУ2. На рис. (в) показаны плотности распределения модуля упругости поверхностей ПУ1 и ПУ2.

Рельеф материала ПУ2 значительно более гладкий (шероховатость < 1 нм). Жесткая фаза распределена более однородно (плотность распределения модуля упругости имеет гауссовый вид, график на рис. 1в) и обладает наноразмерной глобулярной структурой (рис. 1б).
Характерный рельеф обработанных поверхностей представлен на рис. 2. До определенного количества импульсов рельефы обработанных покрытий качественно не отличаются от необработанного материала. Затем происходит потеря устойчивости покрытия, и на поверхности появляются складки. Свойства покрытий подробно обсуждаются далее. Они зависят как от материала, так и количества импульсов напыления.
Рис. 2.
Рельефы 20 × 10 мкм обработанных поверхностей: ПУ1 – верхний ряд; ПУ2 ‒ нижний. Количество импульсов: 8 × 103 (a), 4 × 104 (б), 2.88 × 105 (в) и 2.16 × 106 (г). Размер вставок на (г) – 2.5 × 2.5 мкм. Комментарии к выделенным областям в тексте.

Спектроскопия комбинационного рассеяния обработанных поверхностей (рис. 3а) позволила установить, что пик, соответствующий углероду в аморфном состоянии [1] возникает только после обработки 1.2 × 106 имп. (толщина покрытия – 30 нм). В остальных случаях, при более короткой обработке, возрастает интенсивность C‒N, C‒H, C‒C-связей (пики интенсивности в области 1800–2800 1/см) [24, 25]. Это означает, что в процессе плазменной обработки идет испарение атомов с поверхности полимера, перемешивание их с напыляемым углеродом и образование новых поверхностных углеродсодержащих соединений. Более детальный достоверный анализ спектров затруднителен в силу малой толщины покрытий, а также из-за того, что полиуретан сам по себе содержит углеродные соединения.
Рис. 3.
Спектры комбинационного рассеяния (а) и энергия поверхности (б) материалов. Стрелкой на (а) показан пик, соответствующий аморфному углероду.
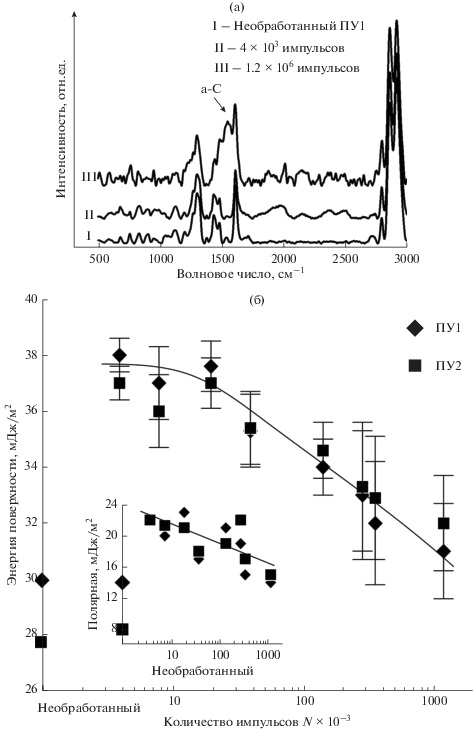
Как следует из рис. 3б, поверхностная энергия покрытий убывает с увеличением количества импульсов. За уменьшение полной энергии поверхности отвечает ее полярная составляющая (вставка на рис. 3б), дисперсионная компонента существенно не изменяется. Это связано с изменением водородных связей полиуретана [26]: на начальном этапе обработки под действием плазмы происходит перестройка структуры полимера, образуются свободные радикалы и высокоэнергетические водородные связи. Дальнейшее напыление ведет к насыщению связей, формированию все менее активного углеродсодержащего слоя, переходящего в “чистый” углерод при увеличении времени обработки. Смачиваемость обработанных поверхностей уменьшается, о чем свидетельствует снижение контактного угла смачивания водой (здесь не показан).
Энергия поверхности необработанного полиуретана (рис. 3б) влияет на формирование покрытия при малом числе импульсов. Углероду сложнее закрепиться на менее активной поверхности ПУ2 и толщина покрытия (рис. 4а) в два раза меньше, чем у ПУ1 при обработке N < 8000; затем толщины покрытий выравниваются. Более того, локальная энергия поверхности жесткой фазы полиуретана меньше энергии мягкой фазы [18]. Это также оказывает определенное влияние на закрепление покрытия и отражается в существенном разбросе их толщин.
Рис. 4.
Толщина (а) и деформация (б) покрытий. Стрелками показан момент перехода к складчатому рельефу.
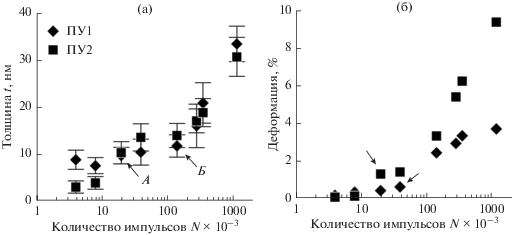
Рост толщины покрытия замедляется с увеличением числа импульсов: при N < 3.6 × 104 покрытие достигает толщины в 20 нм; для N = 1.2 × 106 ‒ 30 нм. Это является еще одним свидетельством того, что начальное покрытие формируется в значительной степени из атомов полимерной подложки.
Определим деформацию поверхности как ε = = ls/(l0 ‒ 1), где ls – средняя длина волнистого профиля поверхности АСМ-изображения; l0 – длина стороны АСМ-изображения. Началу потери устойчивости соответствует резкое возрастание деформации (отмечены стрелками на рис. 4). Для более мягкой подложки этот момент наступает при меньшем количестве импульсов напыления. Отличается и характер изменения (подобным образом ведет себя и шероховатость): деформация более жесткой подложки выходит на асимптотическое значение ~4%, мягкой – неограниченно возрастает. В последнем случае это указывает на отслоение покрытия при максимальном числе импульсов: в вершинах складок видны острые заломы с трещинами (отмечены овалами на вставке рис. 2), т.е. критическая деформация такого покрытия <10%.
Длина λ волны (строго говоря, рельеф не периодический) и высота h (удвоенная амплитуда) складок представлены на рис. 5. При малом количестве импульсов обработки складки возникают в отдельных частях поверхности (показаны стрелками на рис. 2). Это связано с неоднородной жесткостью поверхности исходного полимера и/или неоднородными свойствами нанесенного слоя. Далее происходит резкий переход к поверхности, полностью покрытой складками.
Рис. 5.
Плотности распределения длин и высот волн покрытий материалов ПУ1 (а) и ПУ2 (б). На рис. (б) выделен типичный профиль распределения высоты волны ПУ2.
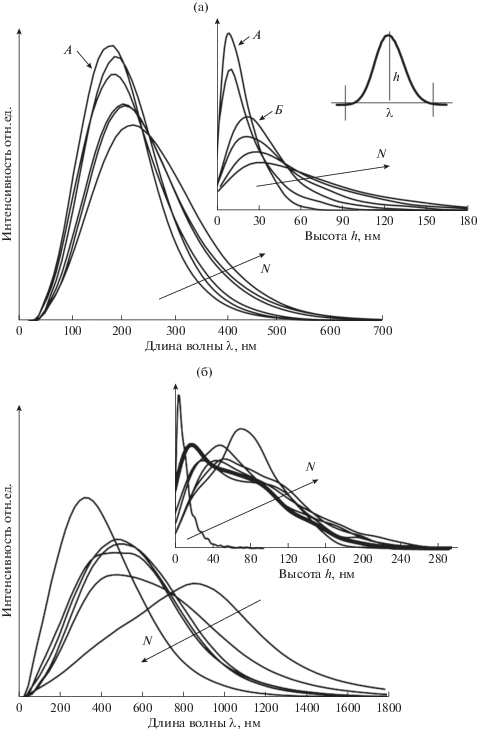
Модальное значение длины волны материала ПУ1 составляет ~200 нм (рис. 5а), с ростом числа импульсов напыления распределения смещаются вправо: появляются более широкие волны. Высота волны изменяется скачкообразно при малом числе импульсов (вставка на рис. 5а) от 10 до 30 нм, а затем монотонно возрастает.
На поверхности материала ПУ2 возникают отдельные глубокие складки, окружающие области с менее выраженными складками. Так как поверхность ПУ2 гладкая, то такая потеря устойчивости связана с механическими неоднородностями материала вблизи поверхности. Длина волны максимальна при малом числе импульсов, а затем уменьшается (рис. 5б), т.е. складки дробятся на более мелкие. Окрестность модального значения длины волны имеет широкое плато (рис. 5б), т.е. поверхность материала ПУ2 обладает целым спектром складок различной длины волны и амплитуды. Так же, как и в материале ПУ1, амплитуды складок в ПУ2 возрастают с увеличением числа импульсов обработки. Однако, в отличие от ПУ1, распределение складок на ПУ2 обладает двумя пиками (выделенное распределение на рис. 5б), которые соответствуют областям больших и малых складок (нижний ряд на рис. 2).
Линейная теория потери устойчивости жесткого покрытия на упругом основании позволяет определить модуль упругости покрытия [27] Ef (предполагается синусоидальный профиль складок): ${{E}_{f}} = 3{{E}_{s}}{{\left( {\frac{\lambda }{{2\pi t}}} \right)}^{3}}$. Расчетным путем установили, что на начальном этапе образования складок, когда амплитуда мала, модуль упругости покрытий составляет порядка 2 ГПа. Дальнейшие вычисления дают нефизичные результаты: полученные значения λ/t приводят к снижению модуля. Это можно объяснить, во-первых, неоднородностями подложки и покрытия, во-вторых, потеря устойчивости может происходить непосредственно при нанесении покрытия, и оставшееся напыление ложится уже на волнистый рельеф.
Наши расчеты методами конечных элементов [28] образования складок на материале подобной жесткости с механическими неоднородностями показали, что модуль упругости подобных покрытий не превышает нескольких ГПа: более жесткие покрытия должны отслаиваться от мягкой подложки без образования складок.
ЗАКЛЮЧЕНИЕ
В работе исследовали особенности роста углеродного покрытия, полученного магнетронным напылением, на двух полиуретановых подложках с модулем упругости 6 и 25 МПа. Полиуретан – блочный сополимер, структура которого разделяется на жесткую и мягкую фазы. В более мягком материале жесткая фаза имела вид наноразмерной глобулярной структуры; в жестком – неоднородной фибриллярной структурой. В зависимости от количества импульсов (от 4 до 1200 тыс.) магнетронного углеродного напыления на полимерах были получены неоднородные (обусловлено структурой необработанного полимера) углеродные или углеродсодержащие покрытия толщиной от 3 до 30 нм. На начальном этапе плазменной обработки покрытие формируется преимущественно за счет осаждения на поверхность соединений углерода и испарившихся атомов полимера. Гидрофобность и энергия поверхности таких покрытий максимальна. Затем происходит постепенное формирование слоя аморфного углерода (по данным КР-спектроскопии) меньшей активности, а на поверхности возникает структура складок – результат потери устойчивости жесткого слоя на мягкой подложке.
Особенности исходного полимера влияют как на свойства (толщину, активность) начальных покрытий, так и на структуру и формирование складок. Модуль упругости полученных покрытий не превышает нескольких ГПа (при больших значениях покрытие отслаивались бы от подложки), столь невысокий модуль обусловлен соединением углерода с атомами подложки. Невысокая жесткость покрытий улучшает их деформативность, а повышенная активность поверхности обладает потенциалом в биомедицинских приложениях.
Список литературы
Chu P.K., Li L. // Mater. Chem. Phys. 2006. V. 96. № 2–3. P. 253. https://doi.org/10.1016/j.matchemphys. 2005.07.048
Ray S.C., Mukherjee D., Sarma S., Bhattacharya G., Mathur A., Roy S.S., McLaughlin J.A. // Diam. Relat. Mater. 2017. V. 80. P. 59. https://doi.org/10.1016/j.diamond.2017.09.001
Ashtijoo P., Bhattacherjee S., Sutarto R., Hu Y., Yang Q. // Surf. Coat. Tech. 2016. V. 308. P. 90. https://doi.org/10.1016/j.surfcoat.2016.06.090
Berlind T., Tengvall P., Hultman L., Arwin H. // Acta Biomater. 2011. V. 10. P. 1369. https://doi.org/10.1016/j.actbio.2010.10.024
Alekhin A.P., Boleiko G.M., Gudkova S.A., Markeev A.M., Sigarev A.A., Toknova V.F., Kirilenko A.G., Lapshin R.V., Kozlov E.N., Tetyukhin D.V. // Nanotechnol. Russia. 2010. V. 5. P. 696. https://doi.org/10.1134/S1995078010090144
Maas M. // Materials. 2016. V. 9. P. 617. https://doi.org/10.3390/ma9080617
Stüber M., Niederberger L., Danneil F., Leiste H., Ulrich S., Welle A., Marin M., Fischer H. // Adv. Eng. Mater. 2007. V. 9. P. 1114. https://doi.org/10.1002/adem.200700224
Hauert R., Thorwarth K., Thorwarth G. // Surf. Coat. Tech. 2013. V. 233. P. 119. https://doi.org/10.1016/j.surfcoat.2013.04.015
Wilson D.J., Rhodes N.P., Williams R.L. // Biomaterials. 2003. V. 24. P. 5069. https://doi.org/10.1016/s0142-9612(03)00423-x
Calcagno L., Compagnini G., Foti G. // Nucl. Instrum. Meth. B. 1992. V. 65. P. 413. https://doi.org/10.1016/0168-583X(92)95077-5
Asakawa R., Nagashima S., Nakamura Y., Hasebe T., Suzuki T., Hotta A. // Surf. Coat. Tech. 2011. V. 206. P. 676. https://doi.org/10.1016/j.surfcoat.2011.02.064
Morozov I.A., Mamaev A.S., Osorgina I.V., Lemkina L.M., Korobov V.P., Belyaev A.Yu., Porozova S.E., Sherban M.G. // Mat. Sci. Eng. C-Mater. 2016. V. 62. P. 242. https://doi.org/10.1016/j.msec.2016.01.062
Morozov I.A., Kamenetskikh A.S., Scherban M.G., Izumov R.I., Kiselkov D.M. // Appl. Surf. Sci. 2019. V. 497. P. 143 706. https://doi.org/10.1016/j.apsusc.2019.143706
Wang Y., Ye Y., Li H., Ji L., Chen J., Zhou H. // Appl. Surf. Sci. 2011. V. 257. P. 1990. https://doi.org/10.1016/j.apsusc.2010.09.040
Bilek M.M.M., McKenzie D.R. // Surf. Coat. Tech. 2006. V. 200. P. 4345. https://doi.org/10.1016/j.surfcoat.2005.02.161
Tsubone D., Hasebe T., Kamijo A., Hotta A. // Surf. Coat. Tech. 2007. V. 201. P. 6423. https://doi.org/10.1016/j.surfcoat.2006.12.008
Kawamoto Y., Nakao A., Ito Y., Wada N., Kaibara M. // J. Mater. Sci.-Mater. M. 1997. V. 8. P. 551. https://doi.org/10.1023/a:1018598714996
Takahashi A., Kita R., Kaibara M. // J. Mater. Sci.-Mater. M. 2002. V. 13. P. 259. https://doi.org/10.1023/A:1014054716444
Dulińska-Molak I., Lekka M., Kurzydłowski K.J. // Appl. Surf. Sci. 2013. V. 270. P. 553. https://doi.org/10.1016/j.apsusc.2013.01.085
Marzec M., Kucińska-Lipka J., Kalaszczyńska I., Janik H. // Mat. Sci. Eng. C-Mater. 2017. V. 80. P. 736. https://doi.org/10.1016/j.msec.2017.07.047
Осоргина И.В., Порозова С.Е., Плаксин С.А., Морозов И.А. // Медицинская техника. 2016. С. 45.
Asefnejad A., Khorasani M.T., Behnamghader A., Farsadzadeh B., Bonakdar S. // Int. J. Nanomed. 2011. V. 6. P. 2375. https://doi.org/10.2147%2FIJN.S15586
Beliaev A., Svistkov A., Iziumov R., Osorgina I., Kondyurin A., Bilek M., McKenzie D. // IOP Conf. Ser.-Mat. Sci. 2016. V. 123. P. 012001. https://doi.org/10.1088/1757-899X/123/1/012001
Miller C.E., Archibald D.D., Myrick M.L., Angel S.M. // Appl. Spectrosc. 1990. V. 44. P. 1297. https://doi.org/10.1366/000370290789619577
Kuptsov A.H., Zhizhin G.N. Handbook of Fourier Transform Raman and Infrared Spectra of Polymers / Elsevier Science, 1998.
Kuang P., Constant K. Increased Wettability and Surface Free Energy of Polyurethane by Ultraviolet Ozone Treatment // Wetting and Wettability / ed. Aliofkhazraei M. InTech, 2015. https://doi.org/10.5772/60798
Chung J.Y., Nolte A.J., Stafford C.M. // Adv. Mater. 2011. V. 23. P. 349. https://doi.org/10.1002/adma.201001759
Morozov I.A., Beliaev A.Y., Izyumov R.I. // Key Eng. Mat. 2018. V. 773. P. 3. https://doi.org/10.4028/www.scientific.net/KEM.773.3
Дополнительные материалы отсутствуют.
Инструменты
Поверхность. Рентгеновские, синхротронные и нейтронные исследования