

Поверхность. Рентгеновские, синхротронные и нейтронные исследования, 2020, № 5, стр. 9-16
Теоретическое моделирование энергии связи и колебательных частот молекул CO на биметаллических наночастицах PtSn
Ю. В. Русалев a, *, А. А. Терещенко a, А. А. Гуда a, А. В. Солдатов a
a Международный исследовательский институт интеллектуальных материалов,
Южный Федеральный университет
344090 Ростов-на-Дону, Россия
* E-mail: yuri.rusalev@gmail.com
Поступила в редакцию 06.07.2019
После доработки 30.08.2019
Принята к публикации 30.08.2019
Аннотация
Адсорбция молекул СО наночастицами Pt и Pt–Sn была изучена с помощью расчетов из первых принципов. Были рассчитаны длины связей Pt–C, C–O, энергия адсорбции и частоты колебаний адсорбированных молекул на поверхности наночастиц в различных позициях. Рассчитанные значения согласуются с экспериментальными данными. Также наблюдалась сегрегация биметаллических наночастиц. Выяснено, что конфигурация наночастицы, в которой атомы платины находятся в ядре, а атомы олова на поверхности, энергетически наиболее выгодна. Было также исследовано влияние атомов Sn вблизи Pt на плотность электронных состояний платины и частоту колебания адсорбированных на ней молекул СО. Установлено, что Sn в первой координационной сфере понижает частоту колебания молекулы СО. Полученные результаты позволят проводить количественную интерпретацию инфракрасных спектров биметаллических наночастиц в ходе каталитической реакции для анализа морфологии их поверхности, построенные карты энергии адсорбции – предсказывать заселенность неэквивалентных позиций адсорбции при разных температурах и давлении газа.
ВВЕДЕНИЕ
Наночастицы платины являются эффективными катализаторами реакций окисления в гетерогенном катализе, а также наиболее эффективными электрокатализаторами, используемыми в топливных элементах с протонообменными мембранами. Тем не менее, два фактора препятствуют их дальнейшему крупномасштабному применению: высокая стоимость и склонность к отравлению остатками СО, содержащимися в газе реформинга [1–3]. Добавление атомов олова в наночастицы платины улучшает их стабильность, сохраняя при этом высокую каталитическую активность. Так, в [4] показано, что структуры типа ядро–оболочка Pt–Sn@Pt более устойчивы к отравлению угарным газом, чем обычные катализаторы на основе чистой Pt. Катализаторы Pt–Sn характеризуются также лучшей активностью, селективностью и устойчивостью к дезактивации по сравнению с аналогичными катализаторами Pt в реакциях гидрирования [5], дегидрирования [6] и окисления этанола, метанола [7] и оксида углерода [8, 9].
Для разработки новых нанокатализаторов необходимо детально понимать структуру поверхности активных фаз, участвующих в реакции, и связь структуры с каталитическими свойствами. При синтезе биметаллических наночастиц могут образовываться различные интерметаллические соединения, например, упорядоченные сплавы Pt3Sn и PtSn. Начальная форма, размер, отношение Pt/Sn, а значит, и свойства катализатора сильно зависят от методики синтеза наночастиц [10]. Авторы отмечают, что некоторые наночастицы образуют структуры ядро–оболочка, где Sn в основном изначально сегрегируется на поверхности.
Молекулы CO обычно используют в качестве молекулярного зонда при измерении рентгеновских фотоэлектронных спектров (РФЭС) [8, 11] для исследования природы и концентрации фаз на поверхности металлических наночастиц. Исследования проводят непосредственно после синтеза, вовремя и после протекания каталитической реакции для наблюдения перестройки поверхности. Было обнаружено [8, 12], что при взаимодействии наночастиц PtSn с атмосферой O2 или CO может происходить сегрегация фаз Pt и Sn, а также образовываться оксид олова. В [8] в экспериментах РФЭС было установлено, что в атмосфере водорода Pt и Sn перемешаны случайным образом. Однако при переключении на смесь СО и O2 атомы Pt и Sn становятся подвижными и образуют домены из металлической платины и оксида олова. Также было установлено, что Pt и Sn остаются в одной наночастице.
Наночастицы чистой Pt и сплава Pt–Sn на подложках были также исследованы с помощью инфракрасной спектроскопии [10, 11, 13–16]. Стоит отметить, что CO адсорбируется только на атомах Pt. В ходе экспериментов было выяснено, что добавление Sn снижает частоту карбонильного пика, соответствующего адсорбции на одном атоме Pt. Также было установлено, что в присутствии Sn нет карбонильных пиков, соответствующих адсорбции между двумя атомами, а при большой степени покрытия слабо изменяется энергия адсорбции, что свидетельствует об уменьшении диполь-дипольного взаимодействия между адсорбированными молекулами СО. Это значит, что атомы Pt на поверхности находятся между атомами Sn, на котором СО не может адсорбироваться. При изменении температуры в атмосфере СО также наблюдается сдвиг пика линейных карбонилов, объясняемый сегрегацией Sn на поверхности наночастиц [11]. Таким образом, поверхность наночастицы PtSn подвергается значительным изменениям, зависящим от температуры и атмосферы во время предварительной обработки, самой реакции и постобработки [17–19].
Существует ряд теоретических работ, описывающих взаимодействие CO со сплавами PtSn [20–24]. Полученные значения энергии адсорбции лежат в интервале 0.6–2 эВ, расстояния Pt–C, C–O равны 1.7–1.9, 1.1–1.2 Å соответственно, частоты колебаний составляют 1950–2100 см–1 в случае линейных карбонилов. Оказалось, что рассчитанные значения зависят от расстояний между адсорбированными молекулами и окружения металлического центра. Описанные выше результаты были получены для периодических структур. Однако на поверхности малых нанокластеров металла присутствует множество неэквивалентных позиций с низкой координацией, учет которых необходим для правильной интерпретации экспериментальных данных, как было показано для нанокластеров чистого палладия [25]. Целью настоящей работы было моделирование из первых принципов взаимодействия CO с поверхностью биметаллических наночастиц PtSn. С помощью расчетов исследовано влияние окружения олова на частоты колебания молекул СО, адсорбированных на атомах платины, а также изучено равновесное распределение атомов олова на поверхности наночастиц и его влияние на спектр колебаний молекул СО.
МЕТОДЫ
Все расчеты были проведены в рамках теории функционала электронной плотности с помощью программного комплекса VASP 5.4 [26–28] с обменно-корреляционным функционалом PAW-rPBE [29]. Наночастицы металлов и сплавов размером 55 атомов помещали в центр кубической суперъячейки с ребром 30 Å, что обеспечивало расстояние между соседними наночастицами не менее 15 Å. Размер базиса плоских волн определялся кинетической энергией 400 эВ. Для расчета в обратном пространстве использовали одну k-точку.
Координаты атомов в нанокластере оптимизировали до тех пор, пока разница сил межатомного взаимодействия не уменьшалась до 0.05 эВ/Å. Для расчета колебаний частот молекул СО использовали метод конечных разностей. Рассматривали смещения ± 0.02 Å по трем направлениям. Замораживали все атомы металла, находящиеся на расстояниях более 3 Å от атома углерода. Энергию адсорбции СО (Eads) вычисляли по формуле:
где Etot – это энергия кластера, NCO – количество молекул СО, ECO – энергия молекулы СО, Ecl – энергия кластера с учетом дефектов поверхности, вызванных адсорбцией СО. Для того чтобы учесть эти эффекты, по окончании расчета геометрической оптимизации кластера молекулу СО удаляли и рассчитывали равновесную структуру и ее полную энергию без молекулы.РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
В качестве модели наночастиц были использованы 55-атомные кластеры диаметром ~1.1 нм. Рассматривали модели трех видов со стехиометрией Pt43Sn12: фрагмент кристалла Pt3Sn (далее Pt43Sn12/кристалл); модель оболочка–ядро, в которой атомы Pt располагаются в ядре (далее Pt43Sn12/Ptядро); модель оболочка–ядро, в которой атомы олова располагаются в ядре (далее Pt43Sn12/Snядро). В качестве модельной выбрана структура чистой наночастицы Pt55, содержащей 55 атомов платины. Каждая наночастица имеет грани (100) и (111). Расчет энергии связи после геометрической оптимизации в пересчете на один атом показывает (рис. 1), что самая устойчивая конфигурация биметаллических кластеров – это когда атомы олова расположены на поверхности, а платины – внутри.
Рис. 1.
55-Атомные нанокластеры Pt55 (а) и Pt43Sn12: б – Pt43Sn12/кристалл; в – Pt43Sn12/Ptядро; г – Pt43Sn12/Ptоболочка. Светлые шарики – атомы Pt, темные – атомы Sn. Цифры – значения энергии связи на атом в эВ.

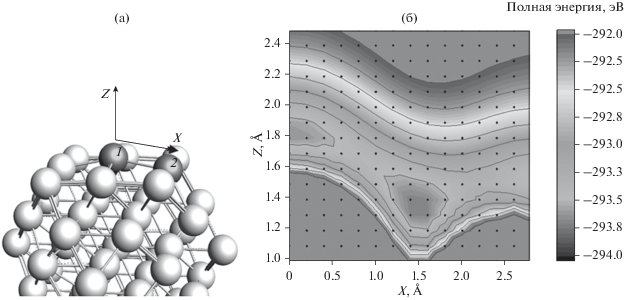
На поверхности наночастицы Pt55 было построено сечение полной энергии при различных положениях адсорбированной молекулы CO (рис. 2). Молекула СО перемещалась по сетке из 254 точек в плоскости над поверхностью наночастицы. В каждой точке рассчитывали полную энергию системы. На рис. 2 изображена карта энергии. На этой карте наблюдаются два минимума полной энергии системы с координатами (0, 1.8) и (1.5, 1.35), которые соответствуют адсорбции на одном атоме Pt (линейный карбонил) и между двух атомов Pt (позиция “мостик”). Первый минимум энергии более доступен при адсорбции молекулы СО из газовой фазы, так как он находится выше над поверхностью наночастицы. Второй минимум будет занят при больших степенях покрытия, так как он менее доступен для молекул в газе, и молекуле СО требуется преодолеть потенциальный барьер, чтобы его занять.
Рис. 2.
Расчет полной энергии системы: а – плоскость XZ, в которой производился расчет, серым цветом выделены атомы с координатами (0, 0) и (2.4, –0.2), лежащие в этой плоскости; б – сечение полной энергии системы Pt55CO1 в плоскости XZ, по осям – координаты молекулы СО, черные точки обозначают сетку, по которой перемещалась молекула СО.
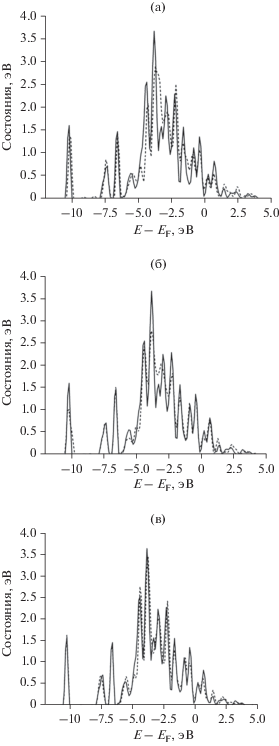
В центр поверхности (100) оптимизированного кластера Pt55 была помещена одна молекула СО (рис. 3а) в позиции линейной адсорбции на одном атоме Pt. Для нее была проведена геометрическая оптимизация и рассчитана частота колебаний. Для того чтобы смоделировать влияние Sn в окружении Pt на частоту и энергию адсорбции молекулы СО, были созданы следующие три модели кластеров: Pt54Sn1угол, Pt54Sn1снизу, Pt54Sn1край (рис. 3б–3г). В этих моделях атом Pt в первой, второй и третьей координационной сферах с адсорбированной молекулой СО заменен на атом Sn. Длины связей Pt–C, C–O, энергия адсорбции (Eads) и частота колебаний молекулы СО в зависимости от расстояния Pt–Sn приведены в табл. 1. Из таблицы видно, что при введении атома Sn длины связей Pt–C, C–O остаются практически неизменными и равны 1.81 и 1.17 Å соответственно. В зависимости от расположения атома Sn частота и энергия адсорбции молекулы СО могут как увеличиваться, так и уменьшаться относительно чистой Pt. В экспериментальных ИК-фурье-спектрах [11] частота колебаний связи С–О на катализаторе Pt–Sn ниже, чем на чистой Pt. Это позволяет предположить, что Sn находится в первой координационной сфере Pt. В окружении Sn меняется плотность d-состояний Pt, что влияет на энергию адсорбции CO. Плотность d-состояний (рис. 4) поверхностных атомов Pt уменьшается у поверхности Ферми, центр d-зоны удаляется от нее, что соответствует снижению энергии связи [24]. Из табл. 1 видно, что Sn в первой координационной сфере снижает энергию адсорбции, что согласуется с экспериментальными данными [11].
Рис. 3.
55-атомные нанокластеры Pt55 (а) и Pt54Sn1 с различным расположением атома Sn и одной молекулой СО в центре поверхности (100): б – Pt54Sn1угол; в – Pt54Sn1снизу; г – Pt54Sn1край.
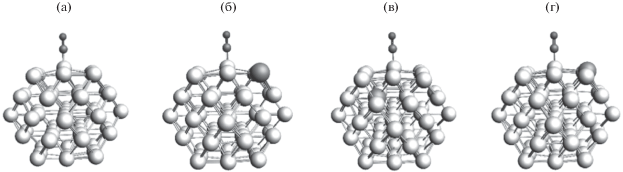
Таблица 1.
Длины связей Pt–C, C–O, энергия адсорбции (Eads) и частота колебаний молекул СО (ν) в зависимости от расстояния Pt–Sn, рассчитанные для моделей Pt55, Pt44Sn1угол, Pt44Sn1снизу, Pt44Sn1край (рис. 3)
| Структура | Pt–Sn, Å | Pt–С, Å | C–O, Å | Eads, эВ | νCO, см–1 |
|---|---|---|---|---|---|
| Pt44Sn1край | 3.02 | 1.82 | 1.17 | –2.10 | 2011.03 |
| Pt44Sn1снизу | 3.35 | 1.81 | 1.17 | –2.08 | 2026.08 |
| Pt44Sn1угол | 3.98 | 1.81 | 1.17 | –2.25 | 2028.09 |
| Pt55 | – | 1.81 | 1.17 | –2.20 | 2023.21 |
Рис. 4.
Плотность d-состояний атома Pt, на котором адсорбирована молекула СО: а – Pt55 (сплошная линия) и Pt44Sn1край (штриховая линия); б – Pt55 (сплошная линия) и Pt44Sn1снизу (штриховая линия); в – Pt55 (сплошная линия) и Pt44Sn1угол (штриховая линия).
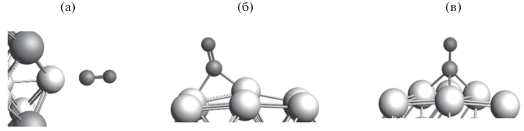
Дальнейшее моделирование адсорбции CO проведено для трех различных моделей наночастиц. На каждую из трех наночастиц Pt43Sn12/кристалл, Pt43Sn12/Ptядро, Pt43Sn12/Snядро была посажена одна молекула СО в различные позиции. Результаты расчетов длин связей Pt–C, C–O, энергии адсорбции и частот колебаний приведены в табл. 2. На данном кластере возможны три различные координации молекулы СО: линейная – адсорбция на одном атоме Pt, “мостик” – адсорбция между двумя атомами Pt, “ямка” – адсорбция между тремя и более атомами Pt (рис. 5). В присутствии Sn в случае первой и третьей структур наблюдается изменение начальной координации молекулы СО. Из позиций “мостик” и “ямка” она стремится перейти в линейное положение. Такое поведение наблюдается и в экспериментальных ИК-фурье-спектрах [13] – исчезают пики, соответствующие молекуле СО, адсорбированной между двух и более атомов.
Таблица 2.
Координация молекулы СО до и после геометрической оптимизации, длины связей Pt–C, C–O, энергия адсорбции и частоты колебаний молекул СО на поверхностях различных кластеров
| № структуры | Начальная координация | Конечная координация | Pt–C, Å | C–O, Å | Eads, эВ | νCO, см–1 |
|---|---|---|---|---|---|---|
| 1.1 | Мостик | Линейная | 1.86 | 1.17 | –2.17 | 1981.75 |
| 1.2 | Мостик | Мостик | 2.03 | 1.19 | –1.75 | 1813.15 |
| 1.3 | Мостик | Линейная | 1.85 | 1.17 | –1.93 | 1995.44 |
| 1.4 | Мостик | Мостик | 1.96 | 1.19 | –2.15 | 1826.49 |
| 1.5 | Линейная | Линейная | 1.86 | 1.17 | –1.68 | 1974.16 |
| 1.6 | Линейная | Линейная | 1.87 | 1.17 | –2.14 | 1997.76 |
| 1.7 | Мостик | Мостик | 1.99 | 1.19 | –2.04 | 1810.86 |
| 1.8 | Мостик | Линейная | 1.87 | 1.17 | –2.13 | 1985.72 |
| 1.9 | Ямка | Ямка | 2.05 | 1.21 | –1.88 | 1684.64 |
| 1.10 | Ямка | Линейная | 1.86 | 1.17 | –2.17 | 1983.53 |
| 1.11 | Ямка | Ямка | 2.1 | 1.20 | –1.77 | 1720.37 |
| 1.12 | Ямка | Мостик | 2.03 | 1.19 | –1.76 | 1811.14 |
| 1.13 | Линейная | Линейная | 1.85 | 1.17 | –2.02 | 1992.41 |
| 1.14 | Линейная | Линейная | 1.83 | 1.17 | –2.14 | 2016.77 |
| 1.15 | Линейная | Линейная | 1.86 | 1.17 | –2.17 | 1981.82 |
| 2.1 | Мостик | Мостик | 1.96 | 1.19 | –2.52 | 1822.27 |
| 2.2 | Мостик | Мостик | 2.08 | 1.21 | –1.94 | 1687.95 |
| 2.3 | Линейная | Линейная | 1.85 | 1.17 | –2.33 | 2006.75 |
| 2.4 | Мостик | Мостик | 1.98 | 1.19 | –2.15 | 1811.44 |
| 2.5 | Линейная | Линейная | 1.83 | 1.17 | –2.22 | 2003.2 |
| 2.6 | Ямка | Ямка | 1.94 | 1.20 | –2.03 | 1776.35 |
| 2.7 | Ямка | Ямка | 2.14 | 1.20 | –1.43 | 1730.35 |
| 2.8 | Линейная | Линейная | 1.81 | 1.17 | –2.20 | 2023.21 |
| 3.2 | Мостик | Линейная | 1.85 | 1.17 | –1.74 | 1992.19 |
| 3.3 | Мостик | Линейная | 1.86 | 1.17 | –2.19 | 1982.13 |
| 3.4 | Мостик | Линейная | 1.85 | 1.17 | –2.10 | 1985.84 |
| 3.8 | Линейная | Линейная | 1.86 | 1.17 | –2.23 | 1999.31 |
| 3.12 | Ямка | Мостик | 2.03 | 1.19 | –2.00 | 1819.24 |
| 3.13 | Ямка | Мостик | 2.03 | 1.19 | –1.93 | 1826.03 |
| 3.14 | Ямка | Линейная | 1.85 | 1.17 | –0.69 | 1982.97 |
| 3.17 | Линейная | Линейная | 1.83 | 1.17 | –2.34 | 2007.45 |
| 3.18 | Линейная | Линейная | 1.84 | 1.17 | –1.76 | 1998.57 |
| 3.19 | Линейная | Линейная | 1.86 | 1.17 | –2.20 | 1983.22 |
| 4.2 | Мостик | Мостик | 2.03 | 1.19 | –1.73 | 1826.57 |
| 4.6 | Ямка | Ямка | 2.08 | 1.21 | –1.78 | 1722.51 |
| 4.8 | Линейная | Линейная | 1.81 | 1.17 | –0.56 | 1997.23 |
Рис. 5.
Иллюстрация различных координаций молекулы СО на кластере: а – линейная; б – мостик; в – ямка.
Расчеты показывают, что зависимость энергии адсорбции от частоты колебаний молекулы СО нелинейная, однако наблюдается кластеризация рассчитанных значений. Так, на рис. 6а изображены частоты в зависимости от координации СО. Хорошо видно, что группа линейных карбонилов занимает диапазон 1970–2025 см–1, частоты мостиковых карбонилов в основном находятся в диапазоне 1810–1830 см–1, а карбонилов в ямке – в интервале 1680–1780 см–1. На рис. 6б изображена энергия адсорбции в зависимости от координации СО. Рассчитанные значения энергии адсорбции в основном лежат в диапазоне 1.6–2.4 эВ и не зависят от координации СО. Из рис. 6в видно, что две точки линейных карбонилов выбиваются из общего тренда, их энергия адсорбции ~0.5 эВ. В этих расчетах сильно изменилась начальная структура кластера при добавлении СО. Произошла сегрегация олова на поверхности, поэтому молекулы СО в этих двух случаях слабо адсорбированы. Помимо этого, наблюдается снижение частоты колебаний линейных карбонилов на кластерах со Sn по сравнению с Pt55. Максимальная разница между кластером со Sn и без него составляет 49 см–1.

ЗАКЛЮЧЕНИЕ
Были рассчитаны энергия адсорбции молекул CO, длины связей Pt–C, C–O, частоты колебаний связи С–О на поверхности моно- и биметаллических наночастиц Pt и PtSn. Рассчитанные значения для чистой платины хорошо согласуются с имеющимися экспериментальными и теоретическими данными. В случае биметаллических наночастиц было обнаружено, что в присутствии Sn частота колебания молекулы СО на кластере Pt может снижаться максимум на 49 см–1. Частоты колебания молекул СО зависят от окружения атомов Pt, на которых они адсорбированы. Моделирование влияния Sn в окружении Pt показало, что частота уменьшается на 12 см–1, когда Sn находится в первой координационной сфере. Также Sn снижает энергию связи на 0.1 эВ, при этом изменяются d-состояния Pt. Молекула СО слабее связана с наночастицей и может легко взаимодействовать с другими адсорбированными группам. Моделирование различных конфигураций биметаллических наночастиц показало, что энергетически выгодными являются частицы со структурой, в которой атомы платины располагаются в ядре, а олова – на поверхности. Этот факт хорошо согласуется с экспериментальным наблюдением сегрегации Sn на поверхности наночастиц.
Список литературы
Crabb E. M., Marshall R., Thompsett D. // J. Electrochem. Soc. 2000. V. 147. № 12. P. 4440. https://doi.org/10.1149/1.1394083
Kim H. T., Yoo J. S., Joh H.-I. et al. // Int. J. Hydrogen En. 2011. V. 36. № 2. P. 1606. https://doi.org/10.1016/j.ijhydene.2010.10.075
Lim D.-H., Lee W.-D., Choi D.-H. et al. // Appl. Catal. B. 2010. V. 94. № 1. P. 85. https://doi.org/10.1016/j.apcatb.2009.10.024
Liu Z., Jackson G.S., Eichhorn B.W. // Ang. Chem. Int. Ed. 2010. V. 49. № 18. P. 3173. https://doi.org/10.1002/anie.200907019
Merlo A.B., Vetere V., Ruggera J.F. et al. // Catal. Commun. 2009. V. 10. № 13. P. 1665. https://doi.org/10.1016/j.catcom.2009.05.005
Burch R., Garla L.C. // J. Catal. 1981. V. 71. № 2. P. 360. https://doi.org/10.1016/0021-9517(81)90239-6
Neto A.O., Dias R.R., Tusi M.M. et al. // J. Power Sources. 2007. V. 166. № 1. P. 87. https://doi.org/10.1016/j.jpowsour.2006.12.088
Michalak W.D., Krier J.M., Alayoglu S. et al. // J. Catal. 2014. V. 312. P. 17. https://doi.org/10.1016/j.jcat.2014.01.005
Schubert M.M., Kahlich M.J., Feldmeyer G. et al. // Phys. Chem. Chem. Phys. 2001. V. 3. № 6. P. 1123. https://doi.org/10.1039/B008062O
Wang X., Altmann L., Stöver J. et al. // Chem. Maters. 2013. V. 25. № 8. P. 1400. https://doi.org/10.1021/cm302077w
Moscu A., Schuurman Y., Veyre L. et al. // Chem. Commun. 2014. V. 50. № 62. P. 8590. https://doi.org/10.1039/C4CC03208J
Uemura Y., Inada Y., Bando K.K. et al. // J. Phys. Chem. C. 2011. V. 115. № 13. P. 5823. https://doi.org/10.1021/jp111286b
Arrahli A., Kherbeche A., Bianchi D. // Res. Chem. Intermediates. 2019. V. 45. № 3. P. 1421. https://doi.org/10.1007/s11164-018-3686-4
Arteaga G.J., Anderson J.A., Rochester C.H. // J. Catal. 1999. V. 184. № 1. P. 268. https://doi.org/10.1006/jcat.1999.2433
De Ménorval L.-C., Chaqroune A., Coq B. et al. // J. Chem. Soc. Faraday Transac. 1997. V. 93. № 20. P. 3715. https://doi.org/10.1039/A702174G
Passos F.B., Schmal M., Vannice M.A. // J. Catal. 1996. V. 160. № 1. P. 106. https://doi.org/10.1006/jcat.1996.0128
Renzas J.R., Huang W., Zhang Y. et al. // Phys. Chem. Chem. Phys. 2011. V. 13. № 7. P. 2556. https://doi.org/10.1039/C0CP01858A
Tao F., Grass M.E., Zhang Y. et al. // J. Am. Chem. Soc. 2010. V. 132. № 25. P. 8697. https://doi.org/10.1021/ja101502t
Zheng F., Alayoglu S., Pushkarev V.V. et al. // Catal. Today. 2012. V. 182. № 1. P. 54. https://doi.org/10.1016/j.cattod.2011.10.009
Liao M.-S., Cabrera C.R., Ishikawa Y. // Surf. Sci. 2000. V. 445. № 2. P. 267. https://doi.org/10.1016/S0039-6028(99)01075-4
Oka T., Mizuseki H., Kawazoe Y. // Mater. Transac. 2007. V. 48. № 7. P. 1907. https://doi.org/10.2320/matertrans.N-MRA2007858
Shubina T.E., Koper M.T.M. // Electrochim. Acta. 2002. V. 47. № 22. P. 3621. https://doi.org/10.1016/S0013-4686(02)00332-8
Sümer A., Aksoylu A.E. // Surf. Sci. 2008. V. 602. № 9. P. 1636. https://doi.org/10.1016/j.susc.2008.02.035
Wang X., Li X., Liao S. et al. // Comput. Mater. Sci. 2018. V. 149. № P. 107. https://doi.org/10.1016/j.commatsci.2018.03.014
Zeinalipour-Yazdi C.D., Willock D.J., Thomas L. et al. // Surf. Sci. 2016. V. 646. P. 210. https://doi.org/10.1016/j.susc.2015.07.014
Kresse G., Furthmüller J. // Comput. Mater. Sci. 1996. V. 6. № 1. P. 15. https://doi.org/10.1016/0927-0256(96)00008-0
Kresse G., Furthmüller J. // Phys. Rev. B. 1996. V. 54. № 16. P. 11169. https://doi.org/10.1103/PhysRevB.54.11169
Kresse G., Hafner J. // Phys. Rev. B. 1993. V. 47. № 1. P. 558. https://doi.org/10.1103/PhysRevB.47.558
Hammer B., Hansen L.B., Nørskov J.K. // Phys. Rev. B. 1999. V. 59. № 11. P. 7413. https://doi.org/10.1103/PhysRevB.59.7413
Дополнительные материалы отсутствуют.
Инструменты
Поверхность. Рентгеновские, синхротронные и нейтронные исследования