

Поверхность. Рентгеновские, синхротронные и нейтронные исследования, 2020, № 5, стр. 17-30
Синтез и диагностика малых наночастиц золота и палладия на подложке CeO2 методом ИК-фурье-спектроскопии
А. А. Терещенко a, *, В. А. Поляков a, b, А. А. Гуда a, **, А. Н. Булгаков a, А. Л. Тарасов c, Л. М. Кустов c, В. В. Бутова a, b, А. Л. Тригуб d, А. В. Солдатов a
a Международный исследовательский институт интеллектуальных материалов,
Южный федеральный университет
344090 Ростов-на-Дону, Россия
b Южный научный центр РАН
344006 Ростов-на-Дону, Россия
c Институт органической химии им. Н.Д. Зелинского, Российская академия наук
119991 Москва, Россия
d Национальный исследовательский центр “Курчатовский институт”
123182 Москва, Россия
* E-mail: tereshch1@gmail.com
** E-mail: guda_sasha@mail.ru
Поступила в редакцию 29.07.2019
После доработки 28.09.2019
Принята к публикации 28.09.2019
Аннотация
Приводятся результаты синтеза и характеризации наночастиц палладия (Pd/CeO2), золота (Au/CeO2) и биметаллического образца (PdAu/CeO2), полученных методом пропитки, модифицированной тетраэтиленпентамином поверхности диоксида церия с последующим восстановлением в потоке H2/Ar. Образовавшиеся частицы, характеризуются высокой степенью дисперсии, однако их малые размеры усложняют анализ традиционными методами, такими как просвечивающая электронная микроскопия и рентгеновская порошковая дифракция. Для исследования размеров и морфологии наночастиц и подложки использовали инфракрасную фурье-спектроскопию диффузного отражения в сочетании с молекулами-пробами CO. По соотношению интенсивностей полос, относящихся к мостиковым и линейным карбонилам на центрах палладия, было подтверждено наличие высокодисперсных наночастиц в случае монометаллического образца и наночастиц большего размера в случае биметаллического образца. Полученные материалы продемонстрировали высокую каталитическую активность и стабильность: полная конверсия СО в случае Pd/CeO2 была достигнута примерно при 50°C. Зафиксировано, что каталитическая активность образцов убывала в ряду Pd/CeO2–Au/CeO2–PdAu/CeO2, что свидетельствует об отсутствии синергетического эффекта в случае биметаллического образца.
ВВЕДЕНИЕ
Нанокластеры на основе благородных металлов являются известными катализаторами различных реакций гидрирования и окисления, в частности окисления CO. Был предпринят ряд попыток [1–8] синтезировать катализатор на основе наночастиц палладия и/или золота на различных подложках, проявляющий высокую активность при низких температурах. Известно, что каталитическая активность наночастиц зависит от их размера и формы [9], материала подложки [10], функционализации [11] и состава [12], причем важнейшими факторами повышенной каталитической активности наночастиц являются их высокая дисперсность и равномерное распределение по поверхности подложки [13]. Один из способов достижения этого – использование матричных поверхностей в качестве подложек, модифицированных, например, описанным в [14] методом осаждения палладия на подложку SiO2. Силикагель синтезировали in situ с одновременной модификацией поверхности путем введения в систему 3-аминопропилтриэтоксисилана. Восстановление солей-прекурсоров проводили с помощью водного раствора борогидрида натрия. При использовании модифицированного силикагеля были получены наночастицы палладия размером 1.5–2 нм. В случае немодифицированной поверхности размер частиц оказался значительно больше (4–5 нм). Авторы связывают этот факт с более сильным взаимодействием ионов палладия с модифицированной поверхностью.
Авторы [15] использовали мезопористый углерод FDU–15 в качестве матрицы, поверхность которой была модифицирована двумя различными методами: нагревом в потоке газообразного аммиака (аммонолиз) и обработкой подложки меламином при нагревании. Восстановление солей-прекурсоров проводили с использованием борогидрида натрия. Наиболее мелкие частицы палладия были получены методом аммонолиза, а их размер варьировался от 1 до 3 нм. При использовании меламина сформировались наночастицы размером 3–4 нм.
Также для синтеза ультрамалых биметаллических наночастиц различного состава (Pt–Pd, Pd–Co, Pd–Cu, Cu–Ni и других) применяли метод электростатической адсорбции. Заряженные ионы прекурсоров металлов эффективно адсорбировались на противоположно заряженных поверхностях путем регулирования рН относительно точки нулевого заряда поверхности [16]. Силикагель пропитывали аммиачными комплексами солей-прекурсоров (хлорид гексааминникеля(II), нитрат тетрааминмеди(II), хлорид гексааминкобальта(III), гидроксид тетрааминплатины(II), хлорид тетрааминпалладия(II)). Затем полученные материалы были обработаны в токе водорода при 400°C, и в результате были получены биметаллические кластеры размером 1–1.5 нм.
Авторы [17] описали метод, согласно которому наночастицы палладия, покрытые поливинилпирролидоном или гидроксиполиамидамином, были введены в реакцию для образования in situ силикагеля из триэтоксисилана и 1,2-бис(триэтоксисилил)этана. Этот подход был использован для получения наночастиц палладия размером 1–4 нм. Также упомянуто несколько методов модификации поверхности диоксида церия, которые можно разделить на in situ и постсинтетические. В частности, к первому типу относится методика, описанная в [18], где молекулы-модификаторы (бромид гексадецилтриметиламмония, бромид цетилпиридиния и другие) были введены на стадии образования диоксида церия. В данном случае такие катионные модификаторы были выбраны из-за анионной функции промежуточного продукта окисления ионов Ce3+ – гидроксида церия. Постсинтетические модификации описаны в [19, 20]. В [19] предварительно синтезированный диоксид церия обрабатывали раствором азотной кислоты. Была получена поверхность наночастиц, содержащая нитрогруппы. В [20] описывается способ аммонолиза пленки CeO2 при различных температурах, в результате которого атомы азота заменяют поверхностные атомы кислорода наночастиц.
В [21] авторы использовали два метода синтеза наночастиц золота на диоксиде церия. Первый метод – соосаждение in situ путем смешивания водных растворов тетрахлорзолотоводородной кислоты и нитрата церия-аммония. После этого рН раствора доводили до 8, полученный осадок собирали и прокаливали при 400°С. Второй – метод пропитки готовой подложки. Золотосодержащую кислоту осаждали на уже готовую и прокаленную подложку CeO2. Образец, полученный методом соосаждения, содержал более крупные наночастицы золота ~8 нм. Материал, полученный методом пропитки, содержал частицы золота размером менее 5 нм. Авторы связывают этот эффект с процессом кальцинирования, при котором мелкие частицы спекаются в более крупные агломераты. Полученные материалы были исследованы как катализаторы конверсии смеси CO/H2O.
Авторы [3] проводили синтез наночастиц палладия и золота на различных подложках методом пропитки и восстановления в полиэтиленгликоле под действием микроволнового излучения. В данном случае полиэтиленгликоль выступает в роли эффективного поглотителя микроволнового излучения за счет своей высокой диэлектрической проницаемости. В процессе быстрого и равномерного нагрева образуется пересыщенный раствор металла. Чем выше перенасыщение, тем выше скорость нуклеации и тем меньше критические размеры ядер, необходимые для преодоления барьера нуклеации. Кроме того, быстрый нагрев микроволновым излучением обеспечивает равномерную температуру и необходимые величины концентрации ионов для зарождения и роста.
В [1] описан синтез катализатора Au/CeO2 методом пропитки готовой подложки в сверхкритических условиях. В результате образовались кластеры золота размером 1–10 нм. Также стоит отметить еще один метод получения подобных материалов. В [2] описан способ синтеза наночастиц палладия, золота и их биметаллических кластеров на подложке из диоксида церия методом лазерного испарения и контролируемой конденсации. Данный метод не подразумевает проведение химических превращений материалов, а основан на процессе испарения/конденсации готовых порошков металлов и их оксидов. Были получены наночастицы палладия и золота размером 5–10 нм. Материалы показали высокую каталитическую активность в реакции окисления СО.
В настоящем исследовании представлены результаты синтеза наночастиц палладия, золота и их биметаллического соединения на модифицированной поверхности диоксида церия. Данный материал представляет собой активную подложку, способную поставлять кислород из кристаллической решетки для облегчения реакций окисления [22]. Наночастицы диоксида церия обрабатывали водным раствором тетраэтиленпентамина (ТЭПА), содержащим в своем составе первичные и вторичные аминогруппы. Полученная поверхность обладает электронно-донорными свойствами и лучше сорбирует положительно заряженные ионы благородных металлов.
В качестве прекурсора палладия был выбран хлорид тетраамминпалладия(II). Этот выбор был сделан по двум причинам: во-первых, аммиачные комплексные соли благородных металлов хорошо растворимы в воде и не подвергаются гидролизу, в отличие от некомплексных хлоридов; во-вторых, аммиачные комплексы имеют более высокое сродство к аминогруппам. Эти факторы способствуют эффективной адсорбции ионов благородных металлов на поверхности, в то время как большое количество центров нуклеации приводит к уменьшению размеров, получаемых наночастиц. В отличие от прекурсора палладия в качестве прекурсора золота был выбран комплексный анионный хлорид – тетрахлораурат(III) натрия. Данный выбор продиктован нестабильностью катионных аммиачных комплексов золота(III) в водной среде, а тетрахлораурат-ион весьма стабилен и не подвергается гидролизу даже при повышенных температурах.
Основные методы, которые обычно применяют для характеризации благородных металлов, такие как рентгеновская порошковая дифракция и просвечивающая электронная микроскопия, имеют некоторые ограничения при исследовании наночастиц на подложках с высоким зарядовым числом, что характерно для диоксида церия (низкий Z-контраст). В работе показано, что инфракрасная (ИК) фурье-спектроскопия с использованием СО в качестве молекул-проб [23–25] является эффективной методикой для характеризации подобных образцов.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Материалы
Для приготовления подложек и катализаторов брали реактивы: церия(III) нитрат гексагидрат 99.5% (Alfa Aesar); палладия(II) хлорид 99.999% (Alfa Aesar); натрия тетрахлораурат(III) дигидрат 99.99% (Alfa Aesar); аммония гидроксид 28–30% NН3 (Alfa Aesar); тетраэтиленпентамина пентагидрохлорид (ТЭПАПГХ) 98% (Alfa Aesar); натрия хлорид 99% (Alfa Aesar); натрия карбонат безводный 99.5% (Alfa Aesar); серебро азотнокислое 99.9995% (Alfa Aesar); метанол был использован без дополнительной очистки (J.T. Baker Ultra Pure).
Синтез модифицированной подложки
Синтез наночастиц диоксида церия проводили по методике [26]. Для модификации поверхности синтезированных наночастиц диоксида церия был приготовлен 10%-раствор ТЭПАПГХ путем растворения 1.11 г ТЭПАПГХ в 10 мл дистиллированной воды. К полученному раствору по каплям добавляли концентрированный раствор аммиака до рН 7–8. Затем суспендировали 1 г синтезированного диоксида церия в полученном растворе ТЭПА. Суспензию тщательно перемешивали в течение 2 ч и затем четыре раза промывали дистиллированной водой с помощью центрифугирования при 15 000 об./мин. Полученный материал был высушен на воздухе при комнатной температуре. Далее модифицированная подложка диоксида церия обозначена как CeO2/TЭПA.
Синтез наночастиц благородных металлов
Хлорид тетрааминпалладия(II) был синтезирован из PdCl2. Хлорид палладия (II) растворяли в насыщенном водном растворе хлорида натрия; к полученному красно-коричневому раствору добавляли 5% аммиака для получения рН 10–11. Образовывались желтые кристаллы хлорида диамина палладия(II):
(1)
$\begin{gathered} {\text{N}}{{{\text{a}}}_{2}}[{\text{PdC}}{{{\text{l}}}_{4}}] + 2{\text{N}}{{{\text{H}}}_{3}}\,\cdot\,{{{\text{H}}}_{2}}{{{\text{O}}}_{{{\text{разбавл}}}}} = \\ = [{\text{Pd(N}}{{{\text{H}}}_{{\text{3}}}}{{{\text{)}}}_{{\text{2}}}}{\text{C}}{{{\text{l}}}_{2}}]{\kern 1pt} \downarrow + \,2{{{\text{H}}}_{{\text{2}}}}{\text{O}} + 2{\text{NaCl}}{\kern 1pt} . \\ \end{gathered} $(2)
$\begin{gathered} \text{[}{\text{Pd(N}}{{{\text{H}}}_{3}}{{)}_{2}}{\text{C}}{{{\text{l}}}_{2}}] + 2{\text{N}}{{{\text{H}}}_{3}}\,\cdot\,{{{\text{H}}}_{2}}{{{\text{O}}}_{{{\text{конц}}}}} = \\ = [{\text{Pd(N}}{{{\text{H}}}_{3}}{{)}_{4}}]{\text{C}}{{{\text{l}}}_{2}} + 2{{{\text{H}}}_{2}}{\text{O}}. \\ \end{gathered} $Навеску прекурсора металла растворяли в 12.5 мл дистиллированной воды. Раствор нагревали до 60°C при перемешивании, затем добавляли CeO2/TЭПА. Полученную суспензию перемешивали в течение 30 мин, затем добавляли 0.2 M раствора карбоната натрия до рН 8. Смесь перемешивали в течение 1 ч, затем центрифугировали и трижды промывали дистиллированной водой до отрицательной реакции на хлорид-ионы в промывных водах (AgNO3-тест). Промытый материал сушили в течение ночи при температуре 60°С. Затем материал помещали в трубчатую печь в фарфоровых лодочках и нагревали до 350°С в потоке чистого аргона (50 мл/мин). Далее заменяли поток аргона смесью 5% H2 и 95% Ar, сохраняя общую величину потока. В таких условиях материал выдерживали в течение 1 ч, затем систему охлаждали до комнатной температуры в потоке чистого аргона (50 мл/мин). Теоретическое содержание металла составляло 4 мас. %.
Аналогичным образом были синтезированы золотосодержащий образец с использованием дигидрата тетрахлораурата(III) натрия в качестве прекурсора и теоретической загрузкой золота 4 мас. % и биметаллический (PdAu) образец, в котором в качестве прекурсоров использовали хлорид тетрааминпалладия(II) и дигидрат тетрахлораурата(III) натрия (расчетная загрузка благородных металлов по 2 мас. % золота и палладия). Указанные материалы обозначены как на Pd/CeO2, Au/CeO2 и PdAu/CeO2. Используемые количества реагентов приведены в табл. 1.
Методики анализа
Рентгенофлуоресцентные спектры измеряли на спектрометре M4 Tornado (Bruker, Billerica, MA, USA), оборудованном детектором XFlash 430 в диапазоне значений энергии от 0 до 24 кэВ. Рентгеновские порошковые дифрактограммы были получены на лабораторном дифрактометре D2 PHASER (Bruker, Billerica, MA, USA) в геометрии Брэгга–Брентано с использованием CuKα-излучения (λ = 1.5406 Å), диапазон 2θ от 5° до 90°, шаг 0.02°, время выдерживания в каждой точке 0.1 с. Полученные пики были аппроксимированы функцией Лоренца с использованием программы Jana2006 [27] с целью определения их точного положения.
Материалы охарактеризовывали с использованием микроскопа FEI Tecnai G2 F20 (FEI Company, Hillsboro, OR, USA) с установленной на нем энергодисперсионной приставкой EDAX (модель Apollo XLT) и детектором Fischione Instruments Model 3000 методом просвечивающей электронной микроскопии высокого разрешения (ПЭМВР) при ускоряющем напряжении 80–200 кВ и методом растровой просвечивающей электронной микроскопии (РПЭМ). Распределение частиц по размерам определяли с помощью программы ImageJ [28]. Статистика была собрана по 500 частицам.
Спектры ближней тонкой структуры рентгеновского поглощения (XANES) на K-крае палладия были измерены для синтезированного Pd/CeO2 на станции Structural Materials Science [29] Курчатовского источника синхротронного излучения (Москва, Россия). В качестве источника излучения использовано накопительное кольцо с энергией электронного пучка 2.5 ГэВ и током 80–100 мА. Измерения проводили в атмосфере и без активации в водороде. Для этого 40 мг порошка катализатора помещали в кювету толщиной 3 мм с прозрачными в рентгеновском диапазоне окнами из каптона. Палладиевую фольгу использовали в качестве образца сравнения и для калибровки. Спектры регистрировали в режиме флуоресценции с помощью монохроматора Si(111) и твердотельного детектора, а палладиевую фольгу измеряли в режиме пропускания. Полученные экспериментальные спектры были проанализированы с использованием программы Athena программного пакета Demeter [30].
ИК-спектры регистрировали с помощью спектрометра Vertex 70 (Bruker, Billerica, MA, USA), оснащенного высокочувствительным детектором из теллурида ртути, охлаждаемым с помощью жидкого азота. Для измерений методом ИК-фурье-спектроскопии диффузного отражения использовали установленную в спектрометре низкотемпературную реакционную камеру Praying Mantis (Harrick Scientific Products Inc., Нью-Йорк, США). Все спектры были записаны с разрешением 1 см–1 в ходе 64 сканирований и преобразованы в спектры поглощения с использованием функции Кубелки–Мунка.
Для управления газовыми потоками (Ar, H2 и CO, проходящими через ячейку и смешивающимися) применяли внешнюю газовую систему, оснащенную регуляторами массового расхода (EL-FLOW, Bronkhorst High-Tech B.V., Ruurlo, Нидерланды). Экспериментальная установка позволяла контролировать температуру образца и газовый поток, а также содержала вакуумный насос, позволяющий достигать давления внутри объема реакционной камеры (образец порошка внутри) вплоть до 5 × 10–3 мбар. Экспериментальная процедура состояла из двух этапов: активации образца и последующей низкотемпературной адсорбции/десорбции CO. На первом этапе образцы нагревали в процессе вакуумирования до 200°С для удаления воды и других гостевых молекул, затем через образец пропускали поток водорода (5 мл/мин), смешанный с аргоном (45 мл/мин) в течение 30 мин для удаления кислорода и возможных остатков хлора в случае палладийсодержащих образцов. Наконец, образцы постепенно охлаждали до –140°C (с использованием жидкого азота в качестве хладагента) во время непрерывного вакуумирования (3 ч). Затем при этой температуре образец продували потоком СО (5 мл/мин) в течение 10 мин. После этой процедуры образцы вакуумировали, плавно уменьшая давление СО внутри реакционной камеры, и, соответственно, покрытие CO на исследуемом материале. ИК-спектры измеряли в течение всего процесса десорбции. Последний спектр, измеренный непосредственно перед адсорбцией CO при низкой температуре, регистрировали в качестве фона, который вычитали из последующих спектров для исключения каких-либо фоновых признаков, не связанных с процессом адсорбции CO. Для получения информации об устойчивости образца Pd/CeO2 после функционирования в реакционных условиях для него в течение 2 ч создавали условия реакции. Активированный образец помещали в упомянутую выше реакционную камеру при температуре 50°C (температура выше максимальной наблюдаемой конверсии), а смесь 16.9% O2, 62.4% Ar и 20.7% CO пропускали через нее со скоростью потока 50 мл/мин. После этого активацию проводили снова с последующей адсорбцией CO.
Испытания каталитической активности осуществляли в проточном микрореакторе (стеклянной трубке с внутренним диаметром 0.8 мм), в который подавали смесь воздуха и CO (16.9% O2 + + 62.4% N2 + 20.7% CO) при давлении 1 атм и расходе 20 мл/мин. Объемная скорость подачи 120 000 ч–1. Молярное соотношение O2/CO составляло 0.82. Загрузка катализатора 10 мг. Реактор нагревали с помощью электропечи, а температуру устанавливали и контролировали с помощью термостата Термодат-17 и термопары, размещенной на внешней стенке микрореактора. Катализатор активировали путем нагрева в потоке указанной выше смеси до 300°C (30 мин), затем охлаждали до комнатной температуры, прекратив подачу газов. Далее катализатор нагревали со скоростью 10 град/мин и анализировали газообразные продукты при фиксированных температурах. Измерение было повторено три раза для анализа стабильности образца Pd/CeO2 и однократно для всех остальных.
Состав исходного газа и газовую смесь на выходе из ловушки анализировали на хроматографе модели 3700 производства научно-производственного объединения “Гранат” (Санкт-Петербург, Россия) на двух колонках, заполненных молекулярными ситами NaX (Х = O2, N2, CO) и HayeSep-Q (воздух + CO и CO2). Проба газа заполняла два контура фиксированного объема, и с помощью шестиходовых кранов ее подавали для анализа в режиме реального времени. Детектор теплопроводности применяли при температуре колонки 60°С. В качестве газа-носителя использовали гелий.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Элементный состав
Рентгеноспектральный флуоресцентный анализ подтвердил наличие палладия и/или золота в синтезированных образцах. Измеренные концентрации приведены в табл. 2. На рис. 1 и 2 представлены изображения и распределения благородных металлов Pd и Au на поверхности диоксида церия, полученные с помощью РПЭМ и энергодисперсионного рентгеновского спектрального анализа для монометаллических и биметаллических образцов соответственно. В случае образца Pd/CeO2 (рис. 1б) наблюдается наиболее равномерное распределение палладия по подложке, крупные агломераты отсутствуют. В образце Au/CeO2, напротив, распределение частиц по размерам значительно шире, и были обнаружены наночастицы золота большего размера (рис. 1г). В случае биметаллического образца PdAu/CeO2 наблюдалось равномерное распределение палладия (рис. 2б), как в образце Pd/CeO2, но отмечены агломераты золота (рис. 2в), хоть и меньшего размера, чем в образце Au/CeO2.
Таблица 2.
Результаты рентгеноспектрального флуоресцентного анализа
| Образец | Теоретическая концентрация, мас. % | Измеренная концентрация, мас. % | ||
|---|---|---|---|---|
| Pd | Au | Pd | Au | |
| Pd/CeO2 | 4 | – | 2.8 | – |
| Au/CeO2 | – | 4 | – | 4.4 |
| PdAu/CeO2 | 2 | 2 | 1.7 | 3.1 |


Структура и размер частиц
Наличие кристаллической подложки CeO2 было подтверждено большими пиками на всех рентгеновских порошковых дифрактограммах (рис. 3). Для материала подложки CeO2 все дифракционные пики могут быть отнесены к гранецентрированной кубической решетке флюорита (пространственная группа Fm$\bar {3}$m, JCPDS № 34-0394), средний размер частиц 10.8 ± 2.3 нм оценен по дифракционным пикам с применением уравнения Дебая–Шеррера. Однако ни рентгеновские порошковые дифрактограммы, ни исходные ПЭМВР-изображения не позволили идентифицировать наночастицы палладия как в случае монометаллического образца Pd/CeO2, так и биметаллического PdAu/CeO2. Присутствие фаз Pd или PdO на дифракционных картинах Pd/CeO2 не было обнаружено даже после очень длительной регистрации сигнала (более 3 ч в узкой области 2θ от 35° до 45° вблизи ожидаемого положения наиболее интенсивного рефлекса Pd 111). В то же время более крупные частицы золота на диоксиде церия были хорошо видны как в случае Au/CeO2, так и PdAu/CeO2, что согласуется с данными РПЭМ. Это явление может быть объяснено малым размером частиц палладия, что привело к значительному уширению дифракционных пиков, характерному для наночастиц размером менее 4–5 нм [31, 32], или высокой дисперсностью палладия [33].
Рис. 3.
Рентгеновские порошковые дифрактограммы исследуемых образцов: 1 – PdAu/CeO2; 2 – Au/CeO2; 3 – Pd/CeO2; 4 – материала подложки CeO2.

ПЭМВР-изображения представлены на рис. 4а–4в. Они позволили увидеть нанокристаллическую структуру CeO2, однако в случае Pd/CeO2 и PdAu/CeO2 не удалось отличить наночастицы палладия от материала подложки из-за низкого Z-контраста (Z(Pd) = 46 и Z(Ce) = 58) [34]. При использовании программного обеспечения ImageJ [28] с целью получения информации о фазовом составе исследуемых образцов было выполнено фурье-преобразование всех изображений, по которым в дальнейшем были проведены индицирование рефлексов и идентификация фаз. Исходные и преобразованные изображения с наиболее отчетливыми рефлексами, через которые были проведены окружности с радиусами, соответствующими межплоскостным расстояниям, представлены на рис. 4г–4е для всех исследуемых образцов. В случае образца Pd/CeO2 (рис. 4г) фурье-преобразование позволило обнаружить слабые рефлексы, связанные с дифракцией на плоскостях (111) Pd, d = 0.22(9) нм (JCPDS № 46-1043), а также (110) PdO, d = 0.21(1) нм, и (121) PdO, d = 0.13(1) нм (JCPDS № 41-1107). Все остальные более яркие пятна соответствовали материалу подложки CeO2 (JCPDS № 34-0394). Аналогичным образом для образца Au/CeO2 (рис. 4д) был обнаружен (помимо сильного сигнала от материала подложки) рефлекс, связанный с дифракцией на плоскостях (111) Au, d = 0.23(0) нм (JCPDS № 04-0784). На дифрактограмме биметаллического образца PdAu/CeO2 (рис. 4е) сигнал от наночастиц золота и палладия имел низкую интенсивность по сравнению с сигналом от диоксида церия (что объяснимо в связи с меньшей загрузкой каждого металла в отдельности (табл. 2)), тем не менее, были обнаружены слабые рефлексы 111 Pd, d = 0.21(9) нм, и Au, d = 0.24(1) нм.
Рис. 4.
Исходные ПЭМВР-изображения образцов Pd/CeO2 (а), Au/CeO2 (б), PdAu/CeO2 (в) и их фурье-преобразование (г–е). Круги проведены через дифракционные рефлексы, связанные с фазами Pd, PdO и Au (фаза CeO2 не отмечена).

На рис. 5а показаны спектры XANES, измеренные для образца Pd/CeO2. В качестве объемного образца сравнения выступала палладиевая фольга. Для них были выполнены преобразования Фурье k2χ(k) (рис. 5б). Интенсивный пик при 1.5 Å (без фазовой коррекции) соответствует связям Pd–O. Наличие связей Pd–O коррелирует с результатами ПЭМВР, когда наблюдались слабые, но различимые дифракционные пятна фазы PdO. Два максимума при 2.5 и 3.0 Å соответствуют связям Pd–Pd, которые присутствовали как в неокисленном ядре наночастиц катализатора, так и на окисленной поверхности. Качественно наблюдалось значительное снижение амплитуды пика Pd–Pd в фурье-преобразованном спектре Pd/CeO2 по сравнению с металлической фольгой, что можно объяснить меньшим координационным числом во второй координационной сфере. Другой эффект связан с наблюдаемым сдвигом максимумов Pd–Pd в Pd/CeO2 к более коротким длинам связей. Оба наблюдения указывают на малый размер наночастиц в исследуемом образце. Действительно, теоретическое моделирование [35] предсказывает, что расстояния Pd–Pd уменьшаются вместе с размером наночастиц из-за поверхностной релаксации, а уменьшение координационных чисел происходит за счет большего поверхностного вклада ненасыщенных связей Pd–Pd.

Характеристика активных центров
Использование ИК-фурье-спектроскопии диффузного отражения в сочетании с адсорбцией молекул-проб СО позволило устранить нехватку информации о доступных активных центрах и размерах наночастиц в образцах PdAu/CeO2 и Pd/CeO2. Аналогичные измерения образца Au/CeO2 и материала подложки CeO2 проводили для сравнения и исключения возможного влияния адсорбции CO на материале подложки. Для уменьшения образования поверхностных карбонатов, вызывающего некоторые электронные эффекты при адсорбции на катионах [36], адсорбцию CO на всех образцах осуществляли при ‒140°C после их восстановления в потоке H2/Ar. В настоящем исследовании для анализа использовали область 2250–1800 см–1, в которой расположены полосы поглощения, связанные с частотами колебаний линейных (L) и мостиковых (B) карбонилов (~2150–2000 и 2000–1800 см–1 соответственно) для палладия, а также молекул СО, адсорбированных на центрах церия (~2200–2100 см–1) и золота (~2150–2050 см–1). Перед адсорбцией все образцы подвергались обработке в восстановительной водородной атмосфере в течение 30 мин при 200°C. В случае восстановленного CeO2 три пика наблюдались в области карбонилов (рис. 6а) с максимумами при 2172, 2159 и 2104 см–1 (при самом высоком покрытии CO). Все эти пики были смещены в высокочастотную область во время снижения давления СО из-за уменьшения латерального взаимодействия между молекулами СО и полностью исчезали после достижения давления 5 × 10–3 мбар (при –140°С).
Рис. 6.
ИК-спектры, измеренные в ходе десорбции CO при –140°C, предварительно восстановленных при 200°C в 5% смеси H2/Ar: а – CeO2; б – Au/CeO2; в – Pd/CeO2; г – PdAu/CeO2. Спектр, измеренный при наибольшем покрытии CO (PCO = 4–8 мбар), выделен сплошной черной полужирной линией; серия спектров, полученных при уменьшении покрытия СО, показана серым цветом; спектр после непрерывного вакуумирования в течение 10 мин (PCO = 5 × 10–3 мбар) выделен пунктиром. Штриховой линией обозначены результаты аппроксимации спектров с помощью функции Гаусса.
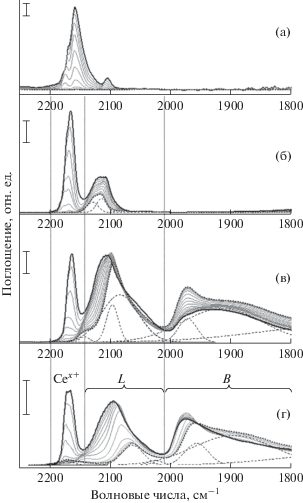
Пик при 2172 см–1 был отнесен к адсорбции CO на центрах Се4+ с координационной ненасыщенностью, что хорошо согласуется с литературными данными для восстановленного оксида церия: 2169 [37], 2170 [38] и 2165 см–1 [39]. Пик около 2159 см−1 можно отнести к адсорбции на Ce4+ с другой координационной ненасыщенностью (Ce4+ cus), который наблюдал ряд авторов в случае невосстановленного диоксида церия при 2150 [39], 2156 [40] и 2151 см–1 [36]. Однако поглощение на аналогичной длине волны при адсорбции СО на Ce3+ наблюдалось также для восстановленного в водороде оксида церия при 2161 [38], 2162 [41] и 2156 см−1 [42]. Последний пик, расположенный при 2104 см–1, вел себя по-другому при снижении покрытия СО в ходе вакуумирования. Он смещался в низкочастотную область, в отличие от ранее упомянутых пиков, и величина сдвига была существенно больше. Этот пик обычно не упоминается в литературе, и его отнесение остается за рамками настоящей работы. Поскольку он полностью исчез во время десорбции при низкой температуре, его возможный вклад в спектры поглощения после снижения покрытия СО при вакуумировании образца был исключен.
В случае остальных трех образцов (PdAu/CeO2, Pd/CeO2 и Au/CeO2) также наблюдались пики, связанные с адсорбцией СО на материале подложки (рис. 6), что выражалось в интенсивных пиках в области 2170–2150 см–1 (при высоких покрытиях СО). Эти пики полностью исчезали после вакуумирования при –140°С. В случае Au/CeO2 (рис. 6б) помимо пиков, связанных с адсорбцией на материале подложки, наблюдались два четко выраженных пика, связанных с адсорбций СО на наночастицах золота. Они заметно сдвигались в высокочастотную область при вакуумировании образца, вплоть до 2114 и 2129 см–1 соответственно при минимальном покрытии СО. Пик при меньшей частоте был отнесен к адсорбции на центрах металлического золота (Au0), высокочастотный пик мог быть связан с адсорбцией на малых золотых нанокластерах с положительным зарядом (Auδ+) [43, 44].
Картина адсорбции СО на наночастицах палладия Pd/CeO2 была более сложной в интервале 2150–1800 см−1 (рис. 6в). Две группы полос были отнесены к молекулам CO, линейно связанным с центрами палладия (обозначенными L), или так называемым линейным карбонилам и к нелинейным (мостиковым) карбонилам (обозначенным B). Первая группа включала интенсивный пик с максимумом примерно при 2108 см–1, испытавший красное смещение до 2096 см–1 при вакуумировании, который также имел плечо в области низких частот. Согласно результатам деконволюции это плечо связано с низкочастотным пиком при 2056 см–1. Высокочастотный пик можно отнести к случаю линейной адсорбции СО на высокодисперсных, но не кристаллических частицах палладия [45, 46]. Снижение интенсивности низкочастотного пика происходило значительно медленнее, чем высокочастотной компоненты, что позволило отнести пик при 2056 см–1 к линейной адсорбции СО на атомах палладия с низким координационным числом, также известных как дефекты (углы, ступени, ребра) кристаллических частиц Pd(111). Такое поведение при вакуумировании хорошо согласуется с тем, что CO сильнее связывается с дефектами палладия [47]. Адсорбция СО на ионных участках Pd2+ и Pd+ могла бы способствовать поглощению при 2170 и 2120 см–1 соответственно [48, 49], но эти пики не наблюдались в ходе эксперимента. Однако при уменьшении покрытия наблюдался пик низкой интенсивности при 2140 см–1, который был скрыт большим пиком на частоте 2108 см–1. Можно предположить, что этот пик связан с одиночными атомами палладия, координированными атомами кислорода, что подтверждается результатами моделирования методом теории функционала плотности, выполненного в [50] для моделей PdO2/CeO2(111), что привело к частоте 2137 см–1. Другие возможные конфигурации координированных кислородом одиночных атомов на гранях диоксида церия, такие как PdO/CeO2(111) или Pd/CeO2(111), должны были располагаться при 2098 и 2047 см–1 согласно результатам [50], и в этом случае они перекрывались с другими пиками.
Вторая группа пиков поглощения была представлена двумя основными широкими пиками поглощения: один из них располагался около 1960 см–1, другой был намного шире и простирался от 1950 до 1850 см–1. Их можно отнести к молекулам СО, образующим мостиковые карбонилы с двумя или тремя атомами палладия соответственно. Интенсивность этих пиков увеличивалась при вакуумировании и температуре –140°С (что более заметно для пика, приписываемого к мостиковым карбонилам, связанным с двумя атомами металла) за счет перехода молекул CO из энергетически менее выгодных положений линейных карбонилов в мостиковую координацию [23].
Для биметаллического образца PdAu/CeO2 в процессе десорбции наблюдалось практически идентичное Pd/CeO2 поведение, однако скорость, с которой уменьшалась интенсивность, была значительно выше (рис. 6г). Отдельных полос поглощения, соответствующих адсорбции СО на золотых центрах, обнаружено не было, что может быть объяснено, в частности, меньшей высотой данного пика, скрытого более интенсивными и перекрывающимися с ним полосами поглощения палладия в области линейных карбонилов (L). Другим объяснением может служить больший размер наночастиц золота в биметаллическом образце по сравнению с высокодисперсными частицами палладия, и, как следствие, меньшая площадь поверхности, доступной для адсорбции молекул-проб CO, что согласуется с результатами РПЭМ, где для золотых частиц наблюдались крупные агломераты.
На основе деконволюции пиков (рис. 6в) были оценены размеры частиц для Pd/CeO2 в соответствии с методикой, описанной в [51]. Для этого рассчитывали отношение общей площади под пиками CO, адсорбированного на мостиковых участках палладия (В) и линейно адсорбированных молекул СО на палладии (L), для различных давлений СО (давление СО варьировалось от 7.5 до 5 × 10–3 мбар) при фиксированной температуре –140°С. Согласно полученным спектрам этот коэффициент увеличивается вместе с уменьшением покрытия СО, и этот факт находится в хорошем согласии с данными [23]. Количество молекул, адсорбированных на мостиковых участках, было пропорционально площади поверхности, что соответствует более высоким соотношениям B/L для наночастиц большего размера. Поэтому можно оценить размер частицы и сравнить этот коэффициент для различных структур. По сравнению с [52] для наночастиц палладия, нанесенных на TiO2, соотношение B/L соответствует размеру меньше 1.5–2 нм (т.е. это свидетельствует о высокой дисперсности наночастиц палладия в исследуемом образце).
Аналогичная оценка размеров была проведена для биметаллического образца PdAu/CeO2. С помощью деконволюции спектра (рис. 6г) при давлении PCO = 5 × 10–3 мбар (после 10 мин вакуумирования) установлено, что отношение площадей под пиками B/L составило 6.52, что значительно больше полученного аналогичным образом (и в полностью идентичных условиях) значения B/L для Pd/CeO2 (1.42). Кроме того, если сравнивать размер исключительно наночастиц палладия, для более точной оценки необходимо также вычесть вклад адсорбции СО на золотых центрах в суммарную площадь под пиками линейных карбонилов (L). Однако учет данного вклада приведет только к уменьшению площади под пиками поглощения линейных карбонилов на центрах палладия, а, значит B/L только возрастет. Это приводит к выводу, что для данного образца размер наночастиц палладия больше, чем для образца Pd/CeO2, и согласно данным [52] находится в диапазоне 3–4 нм или даже больше. После каталитических испытаний образца Pd/CeO2 отношение площадей под пиками нелинейных и линейных карбонилов оставалось неизменным с точностью 2.8% (спектры не показаны, поскольку полностью воспроизводят исходный спектр). Наблюдались небольшие изменения относительных интенсивностей для нелинейных карбонилов на двух- и трехатомных центрах палладия. Последнее свидетельствует о реорганизации поверхности наночастиц, в то время как средний размер, определяемый с помощью соотношения B/L, остался практически неизменным.
Каталитическая активность
Образец Pd/CeO2 продемонстрировал высокую каталитическую активность: 10%-конверсия достигалась уже при комнатной температуре и увеличивалась до 100% при ~50°С (рис. 7). Для сравнения, образец Au/CeO2 достигал 82%-конверсии только при 70°C. Активность биметаллического образца была ниже, и сопоставимые значения конверсии достигались при еще более высокой температуре. Такая высокая каталитическая активность синтезированных материалов может быть объяснена сильным взаимодействием палладия или золота с поверхностью CeO2, а также большой емкостью хранения кислорода за счет образования окислительно-восстановительной пары Ce4+/Ce3+, которая позволяет CeO2 эффективно высвобождать каталитически активные атомы кислорода [21, 53]. Известно, что наночастицы золота и палладия при их одинаковом размере, а также концентрации должны проявлять близкую каталитическую активность [2]. Различия в активности синтезированных материалов вызваны эффектом границы периметра наночастицы [54, 55]. Размеры кластеров палладия в среднем менее 2 нм, а золотые наночастицы значительно больше. Более мелкие палладиевые кластеры позволяют максимизировать площадь контакта между частицами металла и подложкой из оксида переходного металла, одновременно минимизируя площадь “голых” наночастиц палладия. Также уменьшение размера палладиевых наночастиц приводит к увеличению числа активных центров на их поверхности. В результате образец Pd/CeO2 проявляет более высокую каталитическую активность по сравнению с Au/CeO2 и PdAu/CeO2.
Рис. 7.
Зависимость конверсии CO от температуры, при которой протекает реакция окисления в смеси 16.9% O2, 62.4% N2 и 20.7% CO при расходе 120 000 ч−1: первый (1) и третий (2) циклы для образца Pd/CeO2; один цикл для образца Au/CeO2 (3); один цикл для образца PdAu/CeO2 (4). Молярное отношение газов в смеси O2/CO = 0.82.

Анализ литературных данных показал, что некоторые ранее описанные аналогичные системы обладают заметно меньшей каталитической активностью. Так, авторы [2, 3, 7] сообщают о полном окислении монооксида углерода наночастицами палладия на различных подложках только при температурах от 110 до 250°C. Как показано в [3], золотые наночастицы проявляют сходную каталитическую активность, но несколько меньшую, чем палладиевого катализатора. Аналогичные значения конверсии CO характерны для температурного интервала 150–250°C. Также в [56] сообщается о конверсии CO на биметаллическом (PdAu) катализаторе. Температура полной конверсии наночастиц на поверхности SiO2 составила 140°C, на поверхности TiO2 – 60°C. Такая значительная разница объясняется тем, что подложка из SiO2 не способна быть донором активированного кислорода за счет высокой степени инертности оксида кремния (IV) к восстановительным процессам. Активация происходит исключительно на поверхности металлических наночастиц. Это приводит к конкуренции параллельно протекающих процессов активации CO и O2, что снижает каталитическую активность. Подложка из TiO2 способна поставлять активированный кислород благодаря легкости протекания окислительно-восстановительных процессов Ti4+/Ti3+ и образования нестехиометрического оксида TiO2 – x. Пониженная каталитическая активность биметаллического образца, изучаемого в настоящем исследовании, может быть объяснена более крупными размерами наночастиц.
В литературе также упоминаются материалы с низкотемпературной каталитической активностью (например, данные, упомянутые в [5, 6, 8], близке к данным, полученным в настоящей работе для образца Pd/CeO2). Такая разница температур полного превращения может быть связана с расположением металлических наночастиц на различных поверхностях CeO2 – (100) или (111). Авторы [7] показали, что преобразование при низких температурах требует, чтобы металлические наночастицы находились на поверхности (111) подложки, на поверхности (100) преобразование происходит при более высоких температурах. Ими также было обнаружено, что палладий на поверхности (111) диоксида церия проявляет более высокую активность из-за бимолекулярного механизма Ленгмюра–Хиншельвуда. Приведенные в работе этих авторов микрокинетические симуляции методом теории функционала плотности каталитических циклов окисления СО и рассчитанные скорости реакции показывают, что для поверхности (100) CeO2 окисление CO происходит в соответствии с механизмом Марса–Ван Кревелена. Этот эффект можно объяснить более низкой энергией связи Ce–O по сравнению с поверхностью (111). В целом, каталитический механизм окисления СО на Pd/CeO2(100) включает более высокий барьер свободной энергии, чем на Pd/CeO2(111), в соответствии с экспериментально наблюдаемой разницей в активности. Таким образом, высокая каталитическая активность материалов, приведенных в настоящей работе, может быть объяснена большим количеством наночастиц металлов, расположенных на поверхности (111) CeO2, и косвенно указывает на каталитический механизм Ленгмюра–Хиншельвуда.
ЗАКЛЮЧЕНИЕ
В настоящем исследовании представлены результаты синтеза монометаллических наночастиц палладия и золота, и биметаллической системы палладий–золото на модифицированной подложке диоксида церия. Образец Pd/CeO2 показал наиболее высокую каталитическую активность в реакции окисления CO, продемонстрировав термическую стабильность в ходе циклических испытаний, – полная конверсия была достигнута уже при 50°С. Каталитическая активность образцов Au/CeO2 и PdAu/CeO2 была ниже, синергетический эффект в случае биметаллического образца не наблюдался.
Из-за низкого Z-контраста и небольших размеров наночастиц благородных металлов для характеризации полученных образцов была применена ИК-фурье-спектроскопия с молекулами-пробами CO. Благодаря присутствию в спектрах частот колебаний, характерных для нелинейных карбонилов, образованных при адсорбции СО на двух и трех атомах палладия, доказано наличие ультрамалых наночастиц, а не одиночных атомов палладия на поверхности образцов Pd/CeO2 и PdAu/CeO2. При использовании соотношения площадей под пиками, соответствующими нелинейным и линейным карбонилам, была проведена оценка среднего размера наночастиц палладия, который составил менее 1.5–2 нм в случае монометаллического и более 3–4 нм в случае биметаллического образца, что согласуется с измерениями методом РПЭМ и XANES-спектрами, а также с результатами каталитических испытаний. Возможное доминирование поверхностных граней CeO2(111) также может служить объяснением высокой каталитической активности.
Список литературы
Tang Z., Edwards J., Bartley J. et al. // J. Catalysis. 2007. V. 249. № 2. P. 208. https://doi.org/10.1016/j.jcat.2007.04.016
Yang Y., Saoud K.M., Abdelsayed V. et al. // Catalysis Commun. 2006. V. 7. № 5. P. 281. https://doi.org/10.1016/j.catcom.2005.11.014
Glaspell G., Fuoco L., El-Shall M.S. // J. Phys. Chem. B. 2005. V. 109. P. 17350. https://doi.org/10.1021/jp0526849
Zou J., Si Z., Cao Y. et al. // J. Phys. Chem. C. 2016. V. 120. № 51. P. 29116. https://doi.org/10.1021/acs.jpcc.6b08630
Jeong H., Bae J., Han J.W. et al. // ACS Catalysis. 2017. V. 7. № 10. P. 7097. https://doi.org/10.1021/acscatal.7b01810
Slavinskaya E.M., Gulyaev R.V., Zadesenets A.V. et al. // Appl. Catalysis. B. 2015. V. 166–167. P. 91. https://doi.org/10.1016/j.apcatb.2014.11.015
Spezzati G., Benavidez A.D., DeLaRiva A.T. et al. // Appl. Catalysis. B. 2019. V. 243. P. 36. https://doi.org/10.1016/j.apcatb.2018.10.015
Zhu H., Qin Z., Shan W. et al. // J. Catal. 2005. V. 233. P. 41. https://doi.org/10.1016/j.jcat.2005.04.033
Kinoshita K.J. // J. Electrochem. Soc. 1990. V. 137. № 3. P. 845.https://doi.org/10.1149/1.2086566
Rojluechai S., Chavadej S., Schwank J.W. et al. // Catalysis Commun. 2007. V. 8. № 1. P. 57. https://doi.org/10.1016/j.catcom.2006.05.029
Buil M.L., Esteruelas M.A., Niembro S. et al. // Organometallics. 2010. V. 29. № 19. P. 4375. https://doi.org/10.1021/om1003072
Shim J., Yoo D.-Y., Lee J.-S. // Electrochim. Acta. 2000. V. 45. № 12. P. 1943. https://doi.org/10.1016/S0013-4686(99)00414-4
Nie L., Mei D., Xiong H. et al. // Science. 2017. V. 358. № 6369. P. 1419. https://doi.org/10.1126/science.aao2109
Guo M., Li C., Yang Q. // Catalysis Sci. Technol. 2017. V. 7. № 11. P. 2221. https://doi.org/10.1039/C7CY00394C
Li Z., Liu J., Xia C. et al. // ACS Catalysis. 2013. V. 3. № 11. P. 2440. https://doi.org/10.1021/cs400506q
Wong A., Liu Q., Griffin S. et al. // Science. 2017. V. 358. № 6369. P. 1427. https://doi.org/10.1126/science.aao6538
Wei J., Zou L., Li Y. // J. Porous Mater. 2018. V. 26. P. 157. https://doi.org/10.1007/s10934-018-0632-1
Chaudhary S., Sharma P., Singh D. et al. // ACS Sustainable Chem. Eng. 2017. V. 5. № 8. P. 6803. https://doi.org/10.1021/acssuschemeng.7b01041
Xu J., Li L., Li G. // J. Dispersion Sci. Technol. 2008. V. 29. № 8. P. 1072. https://doi.org/10.1080/01932690701815879
Jorge A.B., Sakatani Y., Boissière C. et al. // J. Mater. Chem. 2012. V. 22. № 7. P. 3220. https://doi.org/10.1039/c2jm15230d
Fu Q., Weber A., Flytzani-Stephanopoulos M. // Catalysis Lett. 2001. V. 77. № 1. P. 87. https://doi.org/10.1023/A:1012666128812
Safonova O.V., Guda A.A., Paun C. et al. // J. Phys. Chem. C. 2014. V. 118. № 4. P. 1974. https://doi.org/10.1021/jp409571b
Groppo E., Bertarione S., Rotunno F. et al. // J. Phys. Chem. C. 2007. V. 111. № 19. P. 7021. https://doi.org/10.1021/jp0666434
Lamberti C., Zecchina A., Groppo E. et al. // Chem. Soc. Rev. 2010. V. 39. № 12. P. 4951. https://doi.org/10.1039/C0CS00117A
Abbott H.L., Aumer A., Lei Y. et al. // J. Phys. Chem. C. 2010. V. 114. № 40. P. 17099. https://doi.org/10.1021/jp1038333
Benmouhoub C., Kadri A., Benbrahim N. et al. // Mater. Sci. Forum. 2009. V. 609. P. 189. doi 10.4028/www.scientific.net/MSF.609.189
Petříček V., Dušek M., Palatinus L. // Z. Kristallogr. 2014. V. 229. № 5. P. 345. https://doi.org/10.1515/zkri-2014-1737
Schneider C.A., Rasband W.S., Eliceiri K.W.// Nature Methods. 2012. V. 9. P. 671. https://doi.org/10.1038/nmeth.2089
Chernyshov A.A., Veligzhanin A.A., Zubavichus Ya.V. // Nucl. Instrum. Methods Phys. Res. A. 2009. V. 603. P. 95.
Ravel B., Newville M. // 2005. V. 12. P. 537. https://doi.org/10.1107/S0909049505012719
Baylet A., Marécot P., Duprez D. et al. // Phys. Chem. Chem. Phys. 2011. V. 13. № 10. P. 4607. https://doi.org/10.1039/C0CP01331E
Zhou W., Lee J.Y. // J. Phys. Chem. C. 2008. V. 112. № 10. P. 3789. https://doi.org/10.1021/jp077068m
Xia Y., Ye J., Cheng D.-G. et al. // Catalysis Sci.Technol. 2018. V. 8. № 20. P. 5137. https://doi.org/10.1039/C8CY01590B
Cargnello M., Montini T., Polizzi S. et al. // Dalton Transac. 2010. V. 39. № 8. P. 2122. https://doi.org/10.1039/B916035C
Qi W., Huang B., Wang M. // Nanoscale Res. Lett. 2009. V. 4. № 3. P. 269. https://doi.org/10.1007/s11671-008-9236-z
Binet C., Daturi M., Lavalley J.-C. // Catalysis Today. 1999. V. 50. № 2. P. 207. https://doi.org/10.1016/S0920-5861(98)00504-5
Zaki M.I., Knözlngem H. // Spectrochim. Acta. A. 1987. V. 43. № 12. P. 1455. https://doi.org/10.1016/S0584-8539(87)80030-2
Badri A., Binet C., Lavalley J.-C. // J. Chem. Soc., Faraday Transac. 1996. V. 92. № 9. P. 1603. https://doi.org/10.1039/FT9969201603
Bozon-Verduraz F., Bensalem A. // J. Chem. Soc., Faraday Transac. 1994. V. 90. № 4. P. 653. https://doi.org/10.1039/FT9949000653
Li C., Sakata Y., Arai T. et al. // J. Chem. Soc., Faraday Transac. 1989. V. 85. № 4. P. 929. https://doi.org/10.1039/F19898500929
Mudiyanselage K., Kim H.Y., Senanayake S.D. et al. // Phys. Chem. Chem. Phys. 2013. V. 15. № 38. P. 15856. https://doi.org/10.1039/C3CP52295D
Chen S., Cao T., Gao Y. et al. // J. Phys. Chem. C. 2016. V. 120. № 38. P. 21472. https://doi.org/10.1021/acs.jpcc.6b06158
Manzoli M., Boccuzzi F., Chiorino A. et al. // J. Catal. 2007. V. 245. P. 308. https://doi.org/10.1016/j.jcat.2006.10.021
Tabakova T., Boccuzzi F., Manzoli M. et al. // Appl. Catalysis. A. 2003. V. 252. P. 385. https://doi.org/10.1016/S0926-860X(03)00493-9
Padilla J.M., Del Angel G., Navarrete J. // Catalysis Today. 2008. V. 133–135. P. 541. https://doi.org/10.1016/j.cattod.2007.12.053
Di Gregorio F., Bisson L., Armaroli T. et al. // Appl. Catalysis. A. 2009. V. 352. № 1. P. 50. https://doi.org/10.1016/j.apcata.2008.09.034
Yudanov I.V., Sahnoun R., Neyman K.M. et al. // J. Phys Chem. B. 2003. V. 107. № 1. P. 255. https://doi.org/10.1021/jp022052b
Wang X., Wu G., Guan N. et al. // Appl. Catalysis B. 2012. V. 115–116. P. 7. https://doi.org/10.1016/j.apcatb.2011.12.011
Baidya T., Bera P., Mukri B. и дp. // J. Catal. 2013. V. 303. № P. 117. https://doi.org/10.1016/j.jcat.2013.03.020
Spezzati G., Su Y., Hofmann J.P. et al. // ACS Catalysis. 2017. V. 7. № 10. P. 6887. https://doi.org/10.1021/acscatal.7b02001
Sheu L., Karpinski Z., Sachtler W.M.H. // J. Phys. Chem. (USA). 1989. V. 93. № 12. P. 4890. https://doi.org/10.1021/j100349a042
Fan Q., He S., Hao L. et al. // Sci. Rep. 2017. V. 7. P. 42 172. https://doi.org/10.1038/srep42172
Iglesias-Juez A., Hungría A.B., Martínez-Arias A. et al. // Catalysis Today. 2009. V. 143. № 3. P. 195. https://doi.org/10.1016/j.cattod.2008.12.013
Qi J., Chen J., Li G. et al. // En. Environ. Sci. 2012. V. 5. № 10. P. 8937. https://doi.org/10.1039/c2ee22600f
Cargnello M., Doan-Nguyen V.V., Gordon T.R. et al. // Science. 2013. V. 341. № 6147. P. 771. https://doi.org/10.1126/science.1240148
Guczi L., Beck A., Horváth A. et al. // J. Mol. Catalysis. A. 2003. V. 204–205. P. 545. https://doi.org/10.1016/S1381-1169(03)00337-6
Дополнительные материалы отсутствуют.
Инструменты
Поверхность. Рентгеновские, синхротронные и нейтронные исследования