

Поверхность. Рентгеновские, синхротронные и нейтронные исследования, 2020, № 8, стр. 42-49
Некоторые особенности электронного обмена ионов с поверхностью металлов, обусловленные ее атомной структурой
А. Ф. Александров a, И. К. Гайнуллин a, *, М. А. Сонькин b
a Московский государственный университет им. М.В. Ломоносова
119992 Москва, Россия
b Томский политехнический университет, Инженерная школа
информационных технологий и робототехники
634034 Томск, Россия
* E-mail: ivan.gainullin@physics.msu.ru
Поступила в редакцию 16.12.2019
После доработки 04.01.2020
Принята к публикации 17.01.2020
Аннотация
Изучены некоторые особенности электронного обмена ионов с поверхностью металлов, обусловленные ее атомной структурой. Моделирование основано на трехмерной реализации метода распространения волновых пакетов с применением псевдопотенциалов, описывающих металл на атомном уровне. С помощью теории функционала плотности были построены трехмерные псевдопотенциалы для поверхностей Cu(100), Cu(110) и Cu(111), которые хорошо воспроизводят известные закономерности электронного обмена. При рассмотрении модельной “статической” задачи было показано, что латеральное положение иона слабо влияет на основные характеристики электронного обмена и на движение иона вдоль одного из кристаллических направлений. Однако, трехмерные псевдопотенциалы, учитывающие атомную структуру металла, позволяют получить более реалистичную картину электронного перехода по сравнению с широко используемыми одномерными модельными псевдопотенциалами. Например, при моделировании скользящего рассеяния с применением одномерных псевдопотенциалов электрон после перехода в металл сохраняет параллельную компоненту скорости, что не соответствует действительности. Если же используются трехмерные потенциалы, то параллельная компонента скорости движения электрона в металле уменьшается, что является более правильным.
ВВЕДЕНИЕ
Изучение зарядового (электронного) обмена при взаимодействии атомных частиц с поверхностью твердых тел представляет фундаментальный и прикладной интерес для нескольких областей физики и химии. Для фундаментальной науки зарядовый обмен интересен при изучении рассеяния, распыления, адсорбции и диссоциации молекул [1–3]. Практическая важность электронного обмена обусловлена различными приложениями, включая миниатюризацию полупроводниковой электроники за счет напыления тонких пленок, реактивное ионное травление, катализ, модификацию и анализ поверхности (ВИМС) [2, 4–7].
При изучении зарядового обмена атомной частицы с металлической поверхностью важную роль играет резонансное электронное туннелирование [8–19]. Если энергетические ограничения отсутствуют, то именно этот процесс доминирует в обмене зарядом, так как его вероятность велика по сравнению с нерезонансными переходами и оже-процессами.
В настоящей работе численно изучается резонансный зарядовый обмен ионов с металлическими поверхностями. Показывается, что учет атомной структуры поверхности в ряде случаев оказывает существенное влияние на электронный обмен.
В работе используется атомная система единиц: ${{m}_{e}} = e = \hbar = 1,$ 1 а. е. расстояния = 5.292 × 10–11 м, 1 а. е. времени = 2.419 × 10–17 c, 1 а. е. скорости = = 2.188 × 106 м/с.
ОПИСАНИЕ ЧИСЛЕННОГО МЕТОДА И АЛГОРИТМА ПОСТРОЕНИЯ ТРЕХМЕРНЫХ ПСЕВДОПОТЕНЦИАЛОВ
Постановка задачи и физическая модель
В общем случае резонансный электронный обмен между ионом и поверхностью твердого тела описывается многоэлектронным нестационарным уравнением Шредингера:
(1)
$i\frac{{\partial \Psi ({{{\mathbf{r}}}_{1}},...,{{{\mathbf{r}}}_{N}},t)}}{{\partial t}} = \overset{\lower0.5em\hbox{$\smash{\scriptscriptstyle\frown}$}}{H} ({{{\mathbf{r}}}_{1}},...,{{{\mathbf{r}}}_{N}},t)\Psi ({{{\mathbf{r}}}_{1}},...,{{{\mathbf{r}}}_{N}},t).$При рассмотрении резонансного электронного обмена ионов и атомов с металлами часто применяется так называемая концепция замороженного атомного ядра и одного туннелирующего (переходящего) электрона. То есть рассматривается водородно-подобный атом, состоящий из экранированного атомного ядра и одного электрона, участвующего в зарядовом обмене [5, 20]. Наш подход к моделированию резонансного электронного обмена базируется на методе распространения волновых пакетов [21], который не опирается на адиабатическое приближение, что позволяет моделировать относительно сложные системы и неадиабатические эффекты электронного обмена. Суть метода распространения волновых пакетов заключается в нахождении эволюции волнового пакета туннелирующего электрона ψ(r, t) путем численного решения нестационарного трехмерного уравнения Шредингера:
(2)
$i\frac{{d\psi ({\mathbf{r}},t)}}{{dt}} = \left( { - \frac{\Delta }{2} + {{V}_{{{\text{atom}}}}}({\mathbf{r}},t) + {{V}_{{{\text{surf}}}}}({\mathbf{r}})} \right)d\psi {\kern 1pt} ({\mathbf{r}},t).$Уравнение (2) описывает туннелирование электрона через потенциальный барьер, разделяющий ион/атом и нанокластер. Более подробное описание физической модели и численного метода приведено в работах [25–30].
Построение трехмерных псевдопотенциалов
В данном разделе рассмотрены псевдопотенциалы поверхностей Cu(100), Cu(110) и Cu(111), полученные при помощи теории функционала плотности (DFT). Монокристалл меди часто используется в экспериментах по электронному обмену. Кроме того, поверхности Cu(110) с одной стороны и Cu(100)/Cu(111) с другой, являются примерами поверхностей со свободным и ограниченным движением электрона.
Теоретическим базисом для построения одноэлектронного потенциала кристалла, содержащего большое количество электронов, являются теоремы Кона–Шема [31, 32]. При квантово-механическом рассмотрении твердых тел и молекул принято разделять атомную и электронные подсистемы. При этом часто применяется приближение Борна–Оппенгеймера, в котором движение атомной подсистемы считается заданным (например, тепловые колебания атомов) и (практически) не зависящим от электронной подсистемы, так как масса атомов много больше массы электронов. В этом случае стационарное состояние электронной подсистемы, состоящей из N электронов, описывается стационарным уравнением Шредингера:
(3)
$\begin{gathered} \overset{\lower0.5em\hbox{$\smash{\scriptscriptstyle\frown}$}}{H} \Psi = \left[ {\sum\limits_i^N {\left( { - \frac{{{{\hbar }^{2}}}}{{2{{m}_{i}}}}\nabla _{i}^{2}} \right)} + \sum\limits_i^N {{{V}_{{{\text{e}} - {\text{I}}}}}({{{\mathbf{r}}}_{i}})} + } \right. \\ \left. { + \,\,\sum\limits_{i < j}^N {{{V}_{{{\text{e}} - {\text{e}}}}}({{{\mathbf{r}}}_{i}},{{{\mathbf{r}}}_{j}})} + {{U}_{{{\text{external}}}}}} \right]\Psi = E\Psi , \\ \end{gathered} $Аналитически вышеуказанная система уравнений решается только для очень простых физических систем. Сложность численного решения многоэлектронного уравнения Шредингера обусловлена двумя факторами. Во-первых, волновая функция зависит от координат всех электронов. Если для записи волновой функции одного электрона в трехмерном пространстве требуется задать порядка 109 значений (по 103 на каждую координату), то для записи волновой функции системы из 10 электронов требуется задать 1090 значений, что больше числа атомов в наблюдаемой Вселенной и существенно превышает суммарные возможности существующих на сегодняшний день компьютерных ресурсов. Во-вторых, потенциал отталкивания электронов зависит от их положения (т.е. от волновой функции электронной подсистемы). Таким образом, требуется искать самосогласованное решение уравнения (3). Существуют различные приближенные (итеративные) подходы к решению вышеуказанного уравнения (например, метод Хартри–Фока), но все они применимы к системам с малым количеством электронов (до нескольких штук).
В 1964 г. Хоэнберг (P. Hohenberg) и Кон (W. Kohn) показали [28, 29], что электронная плотность многоэлектронной системы n(r) в основном состоянии (т.е. с минимальной энергией) может быть найдена путем решения одноэлектронных уравнений Кона–Шэма для вспомогательной системы, в которой электрон-электронное взаимодействие учтено в так называемом эффективном потенциале:
(4)
$\left[ {\left( { - \frac{{{{\hbar }^{2}}}}{{2{{m}_{i}}}}\nabla _{i}^{2}} \right) + {{V}_{{{\text{eff}}}}}({\mathbf{r}})} \right]{{\varphi }_{i}}({\mathbf{r}}) = {{\varepsilon }_{i}}{{\varphi }_{i}}({\mathbf{r}}),$(5)
$\begin{gathered} {{V}_{{{\text{eff}}}}}({\mathbf{r}}) = {{U}_{{{\text{external}}}}}({\mathbf{r}}) + {{V}_{{{\text{e}} - {\text{I}}}}}({\mathbf{r}}) + \\ + \,\,\int {d{{{\mathbf{r}}}^{{'3}}}\frac{{{{e}^{2}}n{\kern 1pt} ({\mathbf{r}}{\kern 1pt} ')}}{{\left| {{\mathbf{r}} - {\mathbf{r}}{\kern 1pt} '} \right|}}} + {{V}_{{{\text{X}}C}}}(n{\kern 1pt} ({\mathbf{r}})), \\ \end{gathered} $Для задания обменно-корреляционного потенциала (функционалов обменной и корреляционной энергии) используется ряд приближений, такие как приближение локальной плотности (local density approximation) LDA и метод обобщенного градиентного приближения (generalized gradient approximation) GGA. После решения системы уравнений (4) электронная плотность находится как
(6)
$n({\mathbf{r}}) = \sum\limits_i^N {{{{\left| {{{\varphi }_{i}}({\mathbf{r}})} \right|}}^{2}}} .$Таким образом, для расчета эффективного одноэлектронного потенциала в кристалле необходимо определить распределение электронной плотности. Для этого мы применяем стандартные программные реализации (SIESTA [33], VASP [34], QuantumEspresso [35]) техники суперячеек (элементарных ячеек) в сочетании с периодическими граничными условиями. Для вычисления псевдопотенциала каждой из поверхностей проводилось две серии расчетов: для приповерхностной области и для неограниченного кристалла. Для расчета приповерхностных областей задается размер ячейки: 14 монослоев (в направлении кристалла, перпендикулярном поверхности), причем одна половина ячейки (7 монослоев) заполнена атомами меди согласно рассматриваемой кристаллической структуре, а вторая соответствует вакууму. Для расчета электронной структуры неограниченного кристалла используется ячейка толщиной в один монослой. Размеры и атомная структура ячейки в плоскости поверхности задаются в соответствии с ячейкой для расчета приповерхностной области. Благодаря такому соответствию упрощается “сшивка” и репликация полученных потенциалов на весь объем кристалла, моделируемый с помощью уравнения Шредингера.
DFT-расчеты проводились в базисе плоских волн с использованием приближения локальной плотности (local-density approximation) для задания обменно-корреляционного потенциала. Интегрирование по зоне Бриллюэна проводилось методом тетраэдров (tetrahedron method) с учетом поправок Блохла (Blochl corrections). Зона Бриллюэна аппроксимировалась сеткой из 11 × 11 × × 11 точек в k-пространстве по алгоритму Монхорст–Пака (Mankhorst–Pack grid). При расчетах использовалась энергия отсечения (cutoff energy), равная 1000 эВ. Комбинация вышеприведенных параметров расчета обеспечивает сходимость численной схемы при расчете электронной структуры кристалла меди. Далее одноэлектронный псевдопотенциал вычислялся как сумма потенциалов ионных остовов, потенциала отталкивания электронов и обменно-корреляционного потенциала (5).
Для расчета псевдопотенциалов использовалась заданная кристаллическая структура меди (гранецентрированная решетка с длиной ребра куба 3.61 Å) без учета релаксации и реконструкции поверхности. Расчеты электронной структуры выполнялись для изолированной поверхности, т.е. без находящейся рядом атомной частицы. Можно заметить, что для проведения полностью первопринципных DFT-расчетов необходимо рассчитывать совместный псевдопотенциал поверхности и находящейся рядом атомной частицы. Но такой подход не жизнеспособен для моделирования задачи электронного обмена, так как расчет псевдопотенциала придется выполнять в каждой точке траектории движения атомной частицы. Поэтому в данной работе используется общепринятое приближение, в котором совместный псевдопотенциал рассчитывается как сумма псевдопотенциалов поверхности и атомной частицы. Для корректного учета индуцированного электроном заряда к нулевому псевдопотенциалу поверхности в области вакуума добавляется потенциал взаимодействия с зарядом изображения электрона, подробно описанный в работах [5, 17]. Описанное приближение является корректным и широко используется для моделирования электронного обмена, если расстояние от атомной частицы до поверхности превышает 3 ат. ед. [17]. Необходимо отметить, что с практической точки зрения интерес представляет диапазон расстояний 5–15 ат. ед., так как именно в нем происходит формирование зарядового состояния атомной частицы [17].
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Общий вид трехмерных потенциалов и сравнение результатов расчетов с использованием одномерных и трехмерных потенциалов
На рис. 1, 2 показана изоповерхность трехмерного псевдопотенциала Cu(100). Экстремумы псевдопотенциала соответствуют положению ионных остовов меди, поэтому изоповерхность воспроизводит кубическую гранецентрированную решетку. Радиус изосфер, соответствующих поверхностному слою атомов, несколько меньше в силу уменьшения модуля псевдопотенциала при переходе в область вакуума.

Рис. 2.
Изоповерхность пространственного распределения трехмерного псевдопотенциала Cu(100), вид сверху.

На рис. 3 показана сравнительная зависимость эффективности электронного перехода от расстояния ион–поверхность. Видно, что результаты расчетов с помощью одномерных и трехмерных псевдопотенциалов с хорошей точностью совпадают для различных поверхностей.

Влияние латерального положения атомной частицы
На рис. 4, 5 показаны кристаллические структуры поверхностей Cu(100) и Cu(110). Для каждой из поверхностей выбрано несколько реперных точек, соответствующих различным латеральным позициям атомной частицы.
Рис. 4.
Кристаллическая структура поверхности Cu(100). Круглыми маркерами обозначен верхний слой атомов поверхности Cu(100), квадратными маркерами следующий за ним. Выноски A, B, C, D обозначают точки, используемые для расчета зависимости электронного обмена от латерального положения иона. Координаты точек и значения по осям даны в Ангстремах.
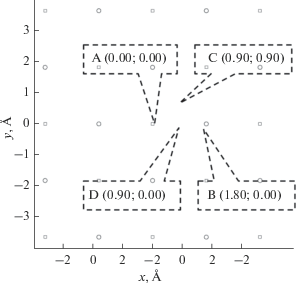
Рис. 5.
Кристаллическая структура поверхности Cu(110). Круглыми маркерами обозначен верхний слой атомов поверхности Cu(110), квадратными маркерами следующий за ним. Выноски A, B, C, D обозначают точки, используемые для расчета зависимости электронного обмена от латерального положения иона.

На рис. 6–8 показана заселенность иона H–, находящегося вблизи поверхностей Cu(100) и Cu(110), как функция времени взаимодействия. Можно заметить, что изменение латеральной позиции иона не приводит к значительным изменениям характера электронного перехода. При малых расстояниях ион–поверхность влияние латерального положения иона проявляется несколько сильнее (рис. 8).
Рис. 6.
Иллюстрация влияния латерального положения иона на электронный обмен. Показана зависимость заселенности уровня от времени для различных латеральных положений иона H– (A, B, C, D). Расстояние между ионом H– и поверхностью Cu(100) равно 12 ат. ед.

Рис. 7.
Иллюстрация влияния латерального положения иона на электронный обмен. Показана зависимость заселенности уровня от времени для различных латеральных положений иона H– (A, B, C, D). Расстояние между ионом H– и поверхностью Cu(110) равно 12 ат. ед.

Рис. 8.
Иллюстрация влияния латерального положения иона на электронный обмен. Показана зависимость заселенности уровня от времени для различных латеральных положений иона H– (A, B, C, D). Расстояние между ионом H– и поверхностью Cu(110) равно 5 ат. ед.

Следует отметить, что при малых расстояниях до поверхности Cu(110) заселенность атомной частицы испытывает небольшие осцилляции, характерные для электронного обмена с поверхностями с ограниченным движением электрона. Такое поведение является логичным следствием применения трехмерного псевдопотенциала. В отличие от “плоского” одномерного псевдопотенциала Cu(110), трехмерный псевдопотенциал, описывает металлическую поверхность на атомном уровне и не является постоянным внутри металла. При малых расстояниях до поверхности электронный обмен осуществляется преимущественно через один из атомов поверхности, поэтому проявляется дискретная структура и осцилляции заселенности атомного уровня (рис. 7). При больших расстояниях в электронном обмене задействуется несколько поверхностных атомов, поэтому дискретная структура поверхности усредняется, а заселенность атомного уровня экспоненциально убывает (рис. 8).
В динамическом случае, когда ион движется относительно поверхности, влияние латерального положения иона в точке наименьшего сближения практически не заметно (за исключением движения иона по нормали к поверхности). Это происходит в силу того, что ион одновременно взаимодействует с несколькими ближайшими атомами поверхности, а благодаря движению иона в плоскости поверхности данное взаимодействие “усредняется”.
На рис. 9 сравнивается распределение электронной плотности в верхнем слое Cu(100) в начале электронного перехода для разных латеральных положений иона и расстояний ион–поверхность. Общая закономерность такова, что при больших расстояниях до поверхности ион взаимодействует с большим количеством соседних атомов. Например, для положения (B) взаимодействие происходит с пятью атомами при расстоянии 12 ат. ед. и только одним при расстоянии 5 ат. ед. Аналогичная картина наблюдается для положений иона C и D. Если ион равноудален от нескольких ближайших к нему атомов (A), то в независимости от расстояния до поверхности, ион будет взаимодействовать с ними одновременно.
Рис. 9.
Распределение электронной плотности в верхнем слое Cu(100) на начальном этапе электронного обмена (10 ат. ед. времени) для различных латеральных положений иона H– (A, B, C, D см. рис. 4). Расстояние ион–поверхность равно 12 ат. ед. в левой части и 5 ат. ед. в правой.

Распространение электрона в металле и эффект торможения при скользящем рассеянии
Применение трехмерных псевдопотенциалов позволяет получить реалистичную картину распространения электрона внутри металла. Например, в случае использования одномерных псевдопотенциалов (постоянное значение в плоскости X–Y), электрон после туннелирования в металл сохраняет латеральную компоненту скорости, которую он имел, двигаясь вместе с ионом. Но в случае использования трехмерных псевдопотенциалов, распространение электрона в плоскости, параллельной поверхности, ограничено в силу периодичности псевдопотенциала. Следовательно, в металле происходит торможение электрона по сравнению с движущимся ионом. На рис. 10 показана изоповерхность распределения электронной плотности в момент времени 150 ат. ед. с начала взаимодействия. Ион H– отлетает от поверхности Cu(111). Начальное расстояние ион-поверхность выбрано 5 ат. ед., скорость вдоль нормали 0.02 ат. ед., параллельная компонента скорости равна 0.5 ат. ед.

ЗАКЛЮЧЕНИЕ
В работе были изучены особенности электронного обмена ионов с поверхностью металлов, обусловленные ее атомной структурой. С помощью теории функционала плотности были построены трехмерные псевдопотенциалы для поверхностей Cu(100), Cu(110) и Cu(111). Сравнение с результатами расчетов с применением широко используемых одномерных пседопотенциалов показало, что построенные трехмерные псевдопотенциалы достаточно хорошо воспроизводят базовые закономерности электронного обмена.
Для выявления особенностей электронного обмена иона с поверхностью, связанных с атомной структурой поверхности металла, была проведена серия расчетов в которых варьировалось латеральное положение иона. Было показано, что латеральное положение иона существенно не влияет на основные характеристики электронного обмена, также не оказывает влияния движение иона вдоль одного из кристаллических направлений.
Была рассмотрена эволюция электронной плотности при скользящем рассеянии. При описании поверхности металла с помощью одномерных псевдопотенциалов, не учитывающих атомную структуру металла, электрон после перехода в металл сохраняет параллельную компоненту скорости, что не соответствует действительности. Если же используются трехмерные потенциалы, то параллельная компонента скорости движения электрона в металле уменьшается.
Таким образом, предложенные трехмерные псевдопотенциалы позволяют получить более правдивую картину электронного перехода по сравнению с широко используемыми одномерными модельными псевдопотенциалами.
Список литературы
Brako R., Newns D.M. // Rep. Prog. Phys. 1989. V. 52. P. 655.
Shao H., Langreth D.C., Nordlander P. // Low Energy Ion-Surface Interactions / Ed. Rabalais J.W. N.Y.: Wiley, 1994.
Hecht T., Winter H., Borisov A.G., Gauyacq J.P., Kazansky A.K. // Phys. Rev. Lett. 2000. V. 84. P. 2517.
Gauyacq J.P., Borisov A.G., Teillet-Billy D. // Formation/Destruction of Negative Ions in Heavy Particle-Surface Collisions / Ed. Esaulov V.A. Cambridge: Cambridge University Press, 1996.
Geerlings J.J.C., Los J. // Phys. Rep. 1990. V. 190. P. 133.
Chakraborty H., Niederhausen T., Thumm U. // Phys. Rev. A. 2004. V. 70. P. 052903.
Gainullin I.K. // Physics Uspekhi. https://doi.org/10.3367/UFNe.2019.11.038691
Gainullin I.K., Urazgildin I.F. // Bulletin of the Russian Academy of Sciences, Physics. 2006. V. 70. P. 1024.
Gainullin I.K., Usman E.Yu., Urazgildin I.F. // Nucl. Instrum. Methods Phys. Res. Sect. B: Beam Interactions with Materials and Atoms. 2005. V. 232. P. 22.https://doi.org/10.1016/j.nimb.2005.03.019
Gainullin I.K., Usman E.Yu., Song Y.W., Urazgil’din I.F. // Vacuum. 2004. V. 72. P. 263. https://doi.org/10.1016/j.vacuum.2003.07.001
Gainullin I.K., Satarin K.K., Usman E.Yu., Urazgildin I.F. // J. Surface Invest.: X-ray, Synchrotron Neutron Techniques. 2005. V. 1. P. 39. (in Russian).
Gainullin I.K., Urazgildin I.F. // Phys. Rev. B. 2006. V. 74. P. 205403.https://doi.org/10.1103/PhysRevB.74.205403
Magunov A.A., Shestakov D.K., Gainullin I.K., Urazgildin I.F. // J. Surface Invest.: X-Ray, Synchrotron Neutron Techniques. 2008. V. 2. P. 764.https://doi.org/10.1134/S1027451008050170
Shestakov D.K., Polivnikova T. Yu., Gainullin I.K., Urazgildin I.F. // Nucl. Instrum. Methods Phys. Res. B. 2009. V. 267. P. 2596.https://doi.org/10.1016/j.nimb.2009.05.043
Усман Е.Ю., Гайнуллин И.К., Уразгильдин И.Ф. // Вестник Московского университета. 2005. Т. 2. С. 23.
Amanbaev E.R., Gainullin I.K., Zykova E.Yu., Urazgildin I.F. // Thin Solid Films. 2011. V. 519. P. 4737. https://doi.org/10.1016/j.tsf.2011.01.026
Gainullin I.K., Sonkin M.A. // Mathematical Models and Computer Simulations. 2019. V. 11. P. 964.https://doi.org/10.1134/S2070048219060048
Gainullin I.K. // Moscow University Physics Bulletin. 2019. V. 74. P. 585.https://doi.org/10.3103/S0027134919060158
Gainullin I.K. // Phys. Rev. A. 2019. V. 100. P. 032712. https://doi.org/10.1103/PhysRevA.100.032712
Winter H. // Phys. Rep. 2002. V. 367. P. 387.
Ermoshin V.A., Kazansky A.K. // Phys. Lett. A. 1996. V. 218. P. 99.
Bardsley J.N. // Case Studies At. Phys. 1974. V. 4. P. 299.
Jennings P.J., Jones R.O., Weinert M. // Phys. Rev. B. 1988. V. 37. P. 6113.
Chulkov E.V., Silkin V.M., Echenique P.M. // Surface Sci. 1999. V. 437. P. 330.
Gainullin I.K., Sonkin M.A. // Comput. Phys. Communicat. 2015. V. 188. P. 68.https://doi.org/10.1016/j.cpc.2014.11.005
Gainullin I.K. // Comput. Phys. Communicat. 2017. V. 210. P. 72. https://doi.org/10.1016/j.cpc.2016.09.021
Gainullin I.K. // Phys. Rev. A. 2017. V. 95. P. 052705. https://doi.org/10.1103/PhysRevA.95.052705
Gainullin I.K., Sonkin M.A. // Phys. Rev. A. 2015. V. 92. P. 022710.https://doi.org/10.1103/PhysRevA.92.022710
Gainullin I.K. // Surf. Sci. 2018. V. 677. P. 324.https://doi.org/10.1016/j.susc.2018.08.007
Gainullin I.K. // Surf. Sci. 2019. V. 681. P. 158. https://doi.org/10.1016/j.susc.2018.11.003
Hohenberg P., Kohn W. // Phys. Rev. B. 1964. V. 136. P. 864.
Kohn W., Sham L.J. // Phys. Rev. A. 1965. V. 140. P. 1133.
Soler J.M. et al. // J. Phys.: Condens. Matter. 2002. V. 14. P. 2745.
Kresse G., Hafner J. // Phys. Rev. B. 1993. V. 47. P. 558.
Giannozzi P. et al. // J. Phys.: Condens. Matter. 2009. V. 21. P. 395 502.
Дополнительные материалы отсутствуют.
Инструменты
Поверхность. Рентгеновские, синхротронные и нейтронные исследования