

Поверхность. Рентгеновские, синхротронные и нейтронные исследования, 2021, № 12, стр. 18-25
Моделирование поверхностей палладиевых нанокатализаторов из первых принципов
А. Ю. Пневская a, Е. Г. Козырь a, Б. Дж. Ф. Аль-Джаф a, О. А. Усольцев a, *
a Международный исследовательский институт интеллектуальных материалов,
Южный федеральный университет
344090 Ростов-на-Дону, Россия
* E-mail: oleg-usol@yandex.ru
Поступила в редакцию 05.03.2021
После доработки 17.05.2021
Принята к публикации 25.05.2021
Аннотация
Нанокатализаторы палладия широко используются в ряде промышленно значимых реакций гидрирования. Понимание процессов, происходящих на поверхности катализаторов в ходе таких реакций, имеет большую научную и практическую значимость. Настоящая работа посвящена теоретическому исследованию атомной и электронной структуры поверхностей палладия (111) и (100), а также хемосорбированных на них углеводородных молекул и радикалов, являющихся потенциальными промежуточными продуктами реакций гидрирования/дегидрирования ацетилена и этилена. Было обнаружено, что в случае поверхности (111) после релаксации геометрии межплоскостные расстояния вблизи поверхности больше, чем межплоскостные расстояния внутри структуры, в то время как для поверхности (100) наблюдается обратный эффект. Было показано, что данный эффект воспроизводится при варьировании параметра расчетов как в рамках метода присоединенных плоских волн, так и при разложении по орбиталям слейтеровского типа. Показано, что эффект выражен сильнее в обобщенном градиентном приближении, чем в приближении локальной плотности. На полученных поверхностях была оптимизирована геометрия 11 углеводородных молекул и радикалов, а также была оценена вероятность нахождения этих молекул на различных поверхностях. Полученные результаты представляют важную структурную информацию о фундаментальных процессах, которые происходят во время адсорбции углеводородов на наночастицах палладия.
ВВЕДЕНИЕ
Гидрирование ненасыщенных углеводородов – один из важнейших процессов в химической промышленности [1]. Данные реакции также имеют фундаментальное значение для научных исследований и часто используются в качестве модельных. Еще в начале 20 века Сабатье обнаружил, что металлические катализаторы способны активировать гидрирование двойных и тройных связей [2]. С тех пор было проведено большое количество исследований в этой области, нацеленных на понимание каталитической активности различных металлов. Металлические системы на основе палладия находятся в центре внимания многих исследований [3–8]. Особый интерес представляют наночастицы палладия, так как они обладают высокой каталитической активностью и большой удельной площадью поверхности, что, несмотря на высокую стоимость, делает их одним из наиболее широко используемых материалов в промышленном катализе [9, 10].
Именно поэтому изучение процессов, протекающих во время реакций гидрирования и дегидрирования, представляют большой интерес. Так, исследование промежуточных состояний в реакциях гидрирования/дегидрирования ацетилена и этилена на поверхности палладия смогут предоставить информацию о связи между типом углеводорода и адсорбционной способностью выбранной поверхности. В частности, гидрирование ацетилена на палладии и формирование углеводородного слоя были подробно изучены в [11, 12]. В меньшей степени исследована адсорбция других более сложных углеводородов, таких как бутадиен [13, 14] или формальдегид, ацетальдегид и пропаналь [15] на палладиевых поверхностях.
Перспективными способами решения данной проблемы могут служить методы моделирования из первых принципов, которые используются для широкого спектра задач [16–18]. Наиболее широко применяется теория функционала электронной плотности (DFT – density functional theory). В свете обсуждаемой проблемы DFT-расчеты предлагают мощную методологию для оценки структуры, энергетических и спектроскопических свойств углеводородов на кластерах благородных металлов. Однако программные комплексы, реализующие метод молекулярных орбиталей, например ADF (Amsterdam Density Functional) [19], все еще ограничены вычислительными возможностями, и задачи, требующие учета свыше тысячи атомов, оказываются весьма трудоемкими даже для современных суперкомпьютеров. Программные коды, использующие периодические граничные условия, например VASP (Vienna ab initio simulation package) [20–22], больше подходят для бесконечных периодических структур. Поэтому для нахождения свойств, соответствующих наночастицам умеренно малых размеров (3–10 нм), необходимо учитывать как периодические свойства, так и свойства поверхности, что требует использования комбинированного подхода [23–26].
Расчеты в рамках DFT позволяют проводить теоретические исследования достаточно реалистичных систем. Так, в [27, 28] в рамках DFT была рассчитана адсорбция углерода на поверхностях Pd(111) и Pd(211), а также в приповерхностных октаэдрических междоузлиях. Было показано, что углерод активнее адсорбируется на ступенчатой поверхности Pd(211). Поглощение водорода и способ связывания молекул бензола, а также роль носителя изучали в [29]. Однако самим палладиевым поверхностям уделяют гораздо меньше внимания, хотя выбор расчетных параметров, в частности, обменно-корреляционного потенциала, базиса и метода оптимизации является нетривиальной задачей. В работах по моделированию нанокластеров палладия [30, 31] показано, что в расчетах с использованием обобщенного градиентного приближения (GGA – generalized gradient approximation) межатомные расстояния Pd–Pd больше, чем в металлическом палладии, в то время как приближение локальной плотности (LDA – local-density approximation) даже без учета релятивистских поправок позволяет получить наиболее близкие к эксперименту значения.
В настоящей работе на основе многочисленных расчетов из первых принципов с использованием современных квантово-химических подходов и суперкомпьютерных мощностей показано, как выбор базиса и обменно-корреляционного потенциала влияет на структуру палладиевых поверхностей (111) и (100). Также проведена оптимизация различных углеводородных интермедиатов, которые могут встречаться в реакциях гидрирования/дегидрирования углеводородов, и оценено, на каких поверхностях они адсорбируются с большей вероятностью.
МЕТОДИКА
Оптимизация геометрии поверхностей палладия и молекул углеводорода и радикалов была выполнена в программном пакете VASP, который реализует методы теории функционала электронной плотности. Так как в данном программном комплексе используются только периодические граничные условия, для создания поверхности был добавлен слой вакуума толщиной 10 Å вдоль вектора трансляции z. Расчеты проводили в рамках обменно-корреляционного потенциала PBE (Perdew–Burke–Ernzerhof exchange-correlation functional) [32, 33] и приближения GGA [34]. Для каждой структуры подбирали оптимальное значение энергии отсечки для базиса плоских волн. Для определения минимального количества точек в обратной ячейке использовали метод разбиения Монкхорста–Пака [35, 36], который позволяет пользователю указывать количество точек k1, k2, k3 вдоль каждого из векторов обратного пространства bi. В зависимости от структуры поверхности количество k-точек вдоль векторов обратной ячейки b1 и b2 менялось, а вдоль вектора b3 всегда было равно единице.
Для сравнения полученных результатов был выполнен ряд расчетов в программном пакете ADF Band. В этом программном комплексе была определена зависимость межплоскостного расстояния от типа приближения и размеров базиса, который представлен набором орбиталей слейтеровского типа. Расчеты проводили как в приближении LDA, так и в GGA с обменно-корреляционным потенциалом PBE.
Выбор сетки разбиения в обратном пространстве
Оптимальную сетку разбиения в обратном пространстве определяли путем варьирования чисел k1, k2, k3 в рамках метода разбиения Монкхорста–Пака, позволяющего автоматически создавать набор значений ki с центром в Г-точке. Число k3 было зафиксировано и равно единице во всех тестах. Так как расчетные ячейки для поверхностей (111) и (100) были выбраны симметричными относительно векторов прямой решетки x и y (и, соответственно, векторов обратной решетки b1 и b2), то числа k1, k2 варьировали совместно, принимая k1 = k2.
Для каждого типа поверхности были использованы расчетные ячейки с одним атомом вдоль векторов x и y, которые будем в дальнейшем называть малой расчетной ячейкой, и с большим количеством атомов палладия (девятью для (111) и восемью для (100) соответственно), которые будем в дальнейшем называть большой расчетной ячейкой. Для малой ячейки поверхности (100) значения 14 × 14 × 1 (рис. 1) были выбраны как оптимальный набор k-точек, а для большой ячейки – 12 × 12 × 1. Для малой ячейки поверхности (111) в качестве оптимального набора k-точек был выбран 12 × 12 × 1, а для большой ячейки – 9 × 9 × 1.
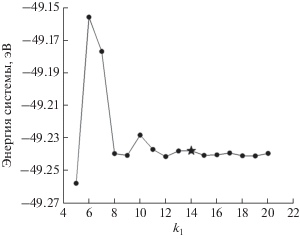
Выбор базиса
Размер базиса плоских волн в VASP, как правило, определяется по минимальному значению энергии отсечки (ENCUT – energy cut-off), выше которого полная энергия всей системы не меняется или меняется незначительно. На рис. 2 представлена зависимость энергии системы от параметра ENCUT в диапазоне от 200 до 800 эВ для большой ячейки поверхности палладия (111). Стоит отметить, что для других типов ячеек и поверхности (100) данная зависимость сохраняется. Поэтому было выбрано значение ENCUT, равное 500 эВ, оптимальное как для поверхностей (111), так и (100). Это значение использовалось для всех структур в дальнейших расчетах. Для расчетов в ADF Band набор слейтеровских орбиталей варьировался от SZ до TZP.
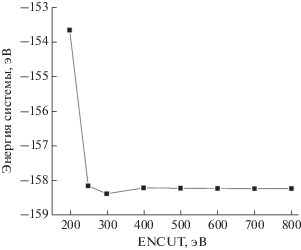
Алгоритм оптимизации геометрии
Для расчетов была выбрана структура из пяти атомных слоев палладия. Было показано, что при изменении числа слоев энергия системы, приходящаяся на один атом, экспоненциально убывает и стремится к значению, соответствующему периодической структуре (рис. 3). В структуре из пяти слоев присутствуют межплоскостные расстояния двух типов: между поверхностным и приповерхностным слоями и между внутренними слоями. Далее будем называть их межплоскостными расстояниями вблизи поверхности (для удобства сравнения поверхностей двух типов приводятся расстояния между атомами палладия, один из которых находится в поверхностном слое, а второй – в приповерхностном (рис. 4а)) и межплоскостными расстояниями внутри структуры (численные значения соответствуют расстояниям между атомами Pd из разных соседних слоев вблизи центра структуры (рис. 4б)).
Рис. 3.
Зависимость полной энергии структуры, приходящейся на один атом палладия, для разного количества слоев поверхности (111) с малой (квадраты) и большой (треугольники) расчетной ячейкой в сравнении с межплоскостным расстоянием идеального кристалла (звездочка).


Периодические граничные условия вдоль всех трансляционных векторов накладывают определенные сложности при оптимизации поверхностей, для которых одно из направлений должно быть не периодическим. Добавление слоя вакуума вдоль вектора z делает процесс оптимизации геометрии многоэтапным. Программный комплекс VASP с помощью метода сопряженных градиентов [37, 38] позволяет оптимизировать: координаты атомов с фиксированной формой и объемом расчетной ячейки (Xopt); только объем ячейки (Vopt) или объем, форму ячейки и координаты атомов одновременно (XVopt). Однако данный метод не позволяет исключить структурную оптимизацию ячейки вдоль одного из векторов.
В качестве начального приближения была выбрана идеальная поверхность с одинаковыми межатомными расстояниями Pd–Pd = 2.75 Å и разным количеством слоев (от трех до десяти) с добавлением слоя вакуума толщиной 10 Å над верхней плоскостью вдоль вектора z. Затем было использовано последовательное чередование методов геометрической оптимизации Vopt и Xopt, так как использование метода XVopt невозможно, поскольку алгоритм стремится сократить слой вакуума и привести систему из поверхности в пространственную объемную геометрию. На первом этапе проводилась оптимизация только объема расчетной ячейки, что дало возможность варьировать межатомные расстояния вдоль поверхности. Затем выполнялся метод Xopt для изменения расстояний между плоскостями поверхности. Последовательное чередование методов геометрической оптимизации проводилось до тех пор, пока изменение межатомных расстояний не стало меньше 0.002 Å. В результате остановились на следующем алгоритме: первый шаг – Vopt; второй шаг – Xopt; третий шаг – Vopt и четвертый шаг – Xopt.
Считая, что полученная структура близка к равновесной, провели тест, в ходе которого изменяли длину вектора расчетной ячейки z в интервале [–4%; 4%]. Для каждой точки по z были построены карты зависимости энергии от изменения векторов x и y и найдено минимальное значение. Затем зависимость энергии ячейки (в минимуме для каждой пары x и y) от изменения z была подогнана полиномом второго порядка. В результате было установлено, что межатомные расстояния отличаются менее чем на 1%.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
В рамках работы были теоретически изучены палладиевые поверхности (111) и (100) в конфигурациях большой и малой суперячеек. На рис. 5а представлен один слой поверхности (111), а на рис. 5б – суперячейка в геометрии (100). В качестве начального приближения все межатомные расстояния на поверхности палладия были выбраны равными 2.75 Å вне зависимости от типа поверхности, что соответствует параметру равновесной элементарной ячейки объемной структуры палладия. Затем структуры были оптимизированы с помощью метода сопряженных градиентов.
Межплоскостные расстояния на поверхности и в объеме
Для всех структур после релаксации геометрии были оценены расстояния между ближайшими атомными слоями палладия на поверхности и в объеме рассматриваемых структур. Так, в случае (100) (рис. 6б) межплоскостное расстояние вблизи поверхности оказалось на 1.3% меньше, чем в объеме. Такое поведение ожидаемо и объясняется нескомпенсированными связями поверхностных атомов палладия, в результате происходит перераспределение электронной плотности, приводящее к “сжатию” структуры приповерхностных слоев.
Рис. 6.
Межплоскостные расстояния Pd–Pd вблизи поверхности (кружки) и внутри структуры (квадраты): а – (111); б – (100). Данные для малых расчетных ячеек обозначены открытыми символами, а для больших ячеек – заполненными.
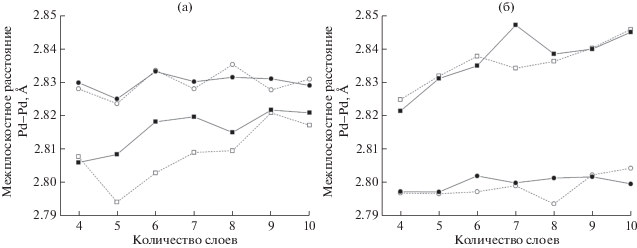
Однако для поверхности (111) (рис. 6а) наблюдается обратный эффект. Поверхностные слои расположены на большем расстоянии друг от друга, чем внутренние. Поскольку данный эффект не является ожидаемым, был проведен ряд дополнительных вычислений с различными расчетными и геометрическими параметрами. Эффект в случае поверхности (111) сохраняется при любом размере ячейки и не зависит от количества слоев в структуре. Как было показано в [30, 31], в приближении GGA межплоскостные расстояния завышены для нанокластеров, что также наблюдается и для моделируемых поверхностей.
В программном комплексе ADF Band [19], реализующем разложение по базису орбиталей слейтеровского типа, был проведен ряд расчетов по релаксации атомной структуры малых пятислойных геометрических моделей (111) и (100) в приближении LDA и GGA (табл. 1). Действительно, в приближении LDA межплоскостные расстояния значительно меньше, чем при GGA-расчетах обеих поверхностей. В то же время разница между межплоскостными расстояниями вблизи поверхности и внутри структуры практически исчезает, что может означать слабую чувствительность метода для описания поверхностных свойств, а, следовательно, и для адсорбированных молекул. Стоит отметить, что во всех расчетах поверхности (111) межплоскостные расстояния вблизи поверхности после релаксации были больше межплоскостных расстояний внутри структуры, хотя в приближении LDA разница незначительна. В случае приближения GGA с увеличением набора слейтеровских орбиталей от SZ до TZP межплоскостные расстояния Pd–Pd внутри структуры становятся меньше и оказываются близкими для поверхностей (111) и (100): 2.779 и 2.778 Å. Поверхностные межплоскостные расстояния для (100) принимают меньшие значение 2.770 Å, а для поверхности (111), наоборот, большие – 2.796 Å.
Таблица 1.
Межплоскостные расстояния Pd–Pd (Å) для поверхностей (111) и (100), рассчитанные в приближении LDA и GGA для базисных наборов SZ, DZ и TZP
| (111) | (100) | ||||||
|---|---|---|---|---|---|---|---|
| SZ | DZ | TZP | SZ | DZ | TZP | ||
| GGA | Вблизи поверхности | 2.882 | 2.795 | 2.796 | 2.858 | 2.776 | 2.770 |
| Внутри структуры | 2.849 | 2.789 | 2.779 | 2.833 | 2.790 | 2.778 | |
| LDA | Вблизи поверхности | 2.767 | 2.700 | 2.703 | 2.746 | 2.684 | 2.682 |
| Внутри структуры | 2.724 | 2.688 | 2.680 | 2.720 | 2.679 | 2.679 | |
Интересно отметить, что при выборе базисного набора SZ для поверхности (100) и приближения GGA межплоскостное расстояние вблизи поверхности становится существенно больше, чем расстояние внутри структуры. Это свидетельствует о том, что базисного набора SZ недостаточно для расчетов таких поверхностей, а подбор базиса для поверхности (111) может скорректировать эффект увеличенных межплоскостных расстояний вблизи поверхности.
Таким образом, при расчетах был обнаружен нетривиальный эффект, который необходимо учитывать при квантово-химическом моделировании металлических поверхностей. Несмотря на то, что этот результат воспроизводится в рамках различных подходов, для широкого спектра геометрических структур (размеров ячейки и количества слоев), а также с различными расчетными параметрами, в литературе данный эффект не обсуждается.
Адсорбция углеводородов
Известно, что молекулы типа этила и метина адсорбируются на палладии в положении “hollow site”, т.е. располагаются в центре треугольника и образуют связь с тремя атомами, поэтому могут быть представлены только на поверхности (111). Однако другие углеводороды (рис. 7) могут присутствовать как на поверхности (111), так и на (100). Поэтому на следующем шаге были получены равновесные структуры 11 углеводородных конфигураций на обеих поверхностях и посчитаны значения свободной энергии для каждой из систем. Затем энергия соответствующей поверхности без углеводородов была вычтена, и на рис. 8 представлена разность значений энергии E(100) – E(111) для каждого углеводорода на поверхностях (111) и (100) соответственно.
Рис. 7.
Молекулы различных оптимизированных углеводородов на поверхности палладия: этил (а); ди-σ-(б); π-адсорбированный этилен (в); этилиден (г); µ-винил (д); винилиден (е); ди-σ (ж); π-адсорбированный ацетилен (з); этинил (и); метил (к); метилен (л).
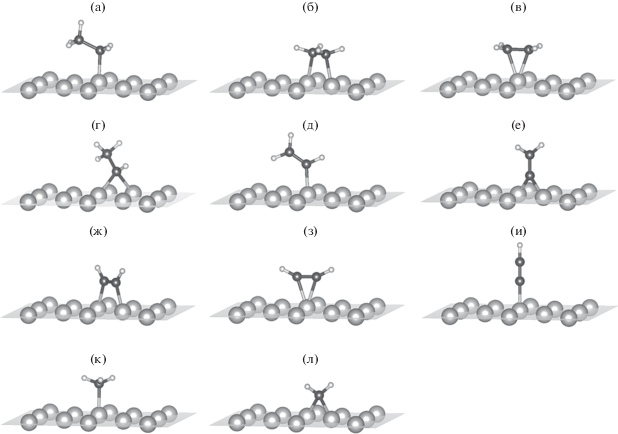
Рис. 8.
Разность значений энергии E(100)– E(111) адсорбированных молекул на поверхностях (100) и (111), соответственно, для: этила (а); ди-σ-(б); π-адсорбированного этилена (в); этилидена (г); µ-винила (д); винилидена (е); ди-σ (ж); π-адсорбированного ацетилена (з); этинила (и); метила (к); метилена (л).
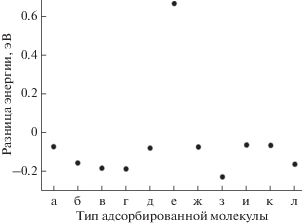
Значения для этила (рис. 8а), μ-винила (рис. 8д) ди-σ-адсорбированного этилена (рис. 8ж), этинила (рис. 8и) и метила (рис. 8к) близки к нулю, что говорит об одинаковых значениях энергии связи на обеих поверхностях. В то же время π-адсорбированный ацетилен (рис. 8к) обладает большей (порядка 0.22 эВ) энергией связи на поверхности (111). Несколько меньшая разность (порядка 0.19 эВ) соответствует π-адсорбированному этилену (рис. 8в), этилидену (рис. 8г) и метину (рис. 8л), и значение 0.17 эВ соответствует ди-σ-адсорбированному ацетилену, что говорит о большей вероятности обнаружить данные молекулы на поверхности (100).
Винилиден при релаксации на поверхности (111) имеет конфигурацию μ3–η2, образуя связи с тремя атомами палладия, что не происходит на поверхности (100), где сохраняется структура μ‑винилидена. Образование дополнительной связи с поверхностным атомом палладия объясняет большую вероятность нахождения данной структуры на поверхности (111) (рис. 8е).
ВЫВОДЫ
В рамках работы были получены модели равновесной геометрии палладиевых поверхностей (111) и (100) для различного количества атомов в ячейке. Обнаружено, что в случае поверхности (111) межплоскостные расстояния вблизи поверхности больше межплоскостных расстояний в объеме структуры. Количественно этот эффект зависит от выбора базисного набора и приближения (LDA или GGA), однако качественно воспроизводится во всех рассмотренных расчетах. В приближении GGA межатомные расстояния увеличены в отличие от расчетов с помощью LDA, однако приближение локальной плотности позволяет получить очень близкие межатомные расстояния на поверхности и внутри структуры, что может говорить о нечувствительности данного подхода к расчету свойств поверхностей.
Было проведено моделирование 11 углеводородных молекул и радикалов, адсорбированных на поверхностях (111) и (100) палладия. Молекула винилидена с наибольшей вероятностью может адсорбироваться на поверхности (111), а молекулы π‑адсорбированного ацетилена, π‑адсорбированного этилена, этилидена, метина и ди-σ-адсорбированного ацетилена – на поверхности (100). Молекулы этила, μ-винила, ди‑σ‑адсорбированного этилена, этинила и метила обладают одинаковой энергией связи и могут быть обнаружены на поверхностях обоих типов.
Список литературы
Handbook of Heterogeneous Catalysis / Eds. Arnold H., Dobert F. et al. Weinheim: Wiley–VCH, 1997.
Sabatier P. The Method of Direct Hydrogenation by Catalysis. Nobel lecture, December 11, 1912. Nobel Lectures in Chemistry 1901–1921.
Studt F., Abild-Pedersen F., Bligaard T., Sørensen R. Z., Christensen C. H., Nørskov J. K. // Ang. Chem. 2008. V. 120. № 48. P. 9439. https://doi.org/10.1002/ange.200802844
Skorynina A.A., Tereshchenko A.A., Usoltsev O.A., Bugaev A.L., Lomachenko K.A., Guda A.A., Groppo E., Pellegrini R., Lamberti C., Soldatov A.V. // Rad. Phys. Chem. 2020. V. 175. P. 108079. https://doi.org/10.1016/j.radphyschem.2018.11.033
Bugaev A.L., Usoltsev O.A., Guda A.A., Lomachenko K.A., Brunelli M., Groppo E., Pellegrini R., Soldatov A.V., van Bokhoven J. // Faraday Discuss. 2021. V. 229. P. 197. https://doi.org/10.1039/c9fd00139e
Kamyshova E.G., Skorynina A.A., Bugaev A.L., Lamberti C., Soldatov A.V. // Rad. Phys. Chem. 2020. V. 175. P. 108 144. https://doi.org/10.1016/j.radphyschem.2019.02.003
Kirichkov M.V., Bugaev A.L., Skorynina A.A., Butova V.V., Budnyk A.P., Guda A.A., Trigub A.L., Soldatov A.V. // Metals. 2020. V. 10. № 6. P. 810. https://doi.org/10.3390/met10060810
Bugaev A.L., Guda A.A., Pankin I.A., Groppo E., Pellegrini R., Longo A., Soldatov A.V., Lamberti C. // Catalysis Today. 2019. V. 336. P. 40. https://doi.org/10.1016/j.cattod.2019.02.068
Ruban A., Hammer B., Stoltze P., Skriver H.L., Nørskov J.K. // J. Mol. Catal. A. 1997. V. 115. P. 421. https://doi.org/10.1016/S1381-1169(96)00348-2
Gruber-Woelfler H., Radaschitz P.F., Feenstra P.W., Haas W., Khinast J.G. // J. Catal. 2012. V. 286. P. 30. https://doi.org/10.1016/j.jcat.2011.10.013
Sárkány A., Horváth A., Beck A. // Appl. Catal. 2002. V. 229. № 1–2. P. 117. https://doi.org/10.1016/S0926-860X(02)00020-0
Studt F., Abild-Pedersen F., Bligaard T., Sorensen R.Z., Christensen C.H., Norskov J.K. // Science. 2008. V. 320. № 5881. P. 1320. https://doi.org/10.1126/science.1156660
Mittendorfer F., Thomazeau C., Raybaud P., Toulhoat H. // J. Phys. Chem. B. 2003. V. 107. № 44. P. 12287. https://doi.org/10.1021/jp035660f
Sautet P., Paul J.-F. // Catal. Lett. 1991. V. 9. № 3–4. P. 245.
Davis J.L., Barteau M.A. // J. Am. Chem. Soc. 1989. V. 111. № 5. P. 1782. https://doi.org/10.1021/ja00187a035
Alkorta I., Elguero J. // Int. J. Mol. Sci. 2003. V. 4. P. 64. https://doi.org/10.3390/i4030064
Laurent A.D., Jacquemin D. // Int. J. Quantum Chem. 2013. V. 113. № 17. P. 2019. https://doi.org/10.1002/qua.24438
Schleder G.R., Padilha A.C.M., Acosta C.M., Costa M., Fazzio A. // J. Phys.: Mater. 2019. V. 2. № 3. P. 032001. https://doi.org/10.1088/2515-7639/ab084b
De Velde G., Bickelhaupt F.M., Baerends E.J., Fonseca Guerra C., van Gisbergen S.J., Snijders J.G., Ziegler T. // J. Comput. Chem. 2001. V. 22. № 9. P. 931. https://doi.org/10.1002/jcc.1056
Kresse G., Furthmüller J. // Phys. Rev. B. 1996. V. 54. № 16. P. 11169. https://doi.org/10.1103/PhysRevB.54.11169
Kresse G., Furthmüller J. // Comput. Mater. Sci. 1996. V. 6. P. 15. https://doi.org/10.1016/0927-0256(96)00008-0
Kresse G., Joubert D. // Phys. Rev. B. 1999. V. 59. № 3. P. 1758. https://doi.org/10.1103/PhysRevB.59.1758
Bugaev A.L., Zabilskiy M., Skorynina A.A., Usoltsev O.A., Soldatov A.V., van Bokhoven J.A. // Chem. Commun. 2020. V. 56. № 86. P. 13097. https://doi.org/10.1039/D0CC05050D
Bugaev A.L., Usoltsev O.A., Guda A.A., Lomachenko K.A., Pankin I.A., Rusalev Y.V., Emerich H., Groppo E., Pellegrini R., Soldatov A.V., van Bokhoven J.A., Lamberti C. // J. Phys. Chem. C. 2018. V. 122. № 22. P. 12029. Doi: https://doi.org/10.1021/acs.jpcc.7b11473
Usoltsev O.A., Pnevskaya A.Y., Kamyshova E.G., Tereshchenko A.A., Skorynina A.A., Zhang W., Yao T., Bugaev A.L., Soldatov A.V. // Nanomaterials (Basel). 2020. V. 10. № 9. P. 1643. https://doi.org/10.3390/nano10091643
Usoltsev O.A., Bugaev A.L., Guda A.A., Guda S.A., Soldatov A.V. // Topics in Catalysis. 2020. V. 63. № 1–2. P. 58. https://doi.org/10.1007/s11244-020-01221-2
Studt F., Abild-Pedersen F., Bligaard T., Sorensen R.Z., Christensen C.H., Norskov J.K. // Ang. Chem. Int. Ed. Engl. 2008. V. 47. № 48. P. 9299. https://doi.org/10.1002/anie.200802844
Teschner D., Revay Z., Borsodi J., Havecker M., Knop-Gericke A., Schlogl R., Milroy D., Jackson S.D., Torres D., Sautet P. // Ang. Chem. Int. Ed. Engl. 2008. V. 47. № 48. P. 9274. https://doi.org/10.1002/anie.200802134
Kubicka H. // J. Catal. 1968. V. 12. № 3. P. 223. https://doi.org/10.1016/0021-9517(68)90102-4
Krüger S., Vent S., Nörtemann F., Staufer M., Rösch N. // J. Chem. Phys. 2001. V. 115. № 5. P. 2082. https://doi.org/10.1063/1.1383985
Yudanov I.V., Neyman K.M., Rösch N. // Phys. Chem. Chem. Phys. 2004. V. 6. № 1. P. 116. https://doi.org/10.1039/b311054k
Paier J., Hirschl R., Marsman M., Kresse G. // J. Chem. Phys. 2005. V. 122. № 23. P. 234102. https://doi.org/10.1063/1.1926272
Paier J., Marsman M., Hummer K., Kresse G., Gerber I.C., Angyan J.G. // J. Chem. Phys. 2006. V. 124. № 15. P. 154 709. https://doi.org/10.1063/1.2187006
Perdew J.P. // Phys. Rev. Lett. 1985. V. 55. № 16. P. 1665. https://doi.org/10.1103/PhysRevLett.55.1665
Evarestov R.A., Smirnov V.P. // Phys. Rev. B. 2004. V. 70. № 23. P. 233101.https://doi.org/10.1103/PhysRevB.70.233101
Pack J.D., Monkhorst H.J. // Phys. Rev. B. 1977. V. 16. № 4. P. 1748. https://doi.org/10.1103/PhysRevB.13.5188
Shewchuk J.R. // An Introduction to the Conjugate Gradient Method Without the Agonizing Pain / Carnegie–Mellon University, Department of Computer Science, 1994.
Steihaug T. // SIAM J. Numer. Anal. 1983. V. 20. № 3. P. 626. https://doi.org/10.1137/0720042
Дополнительные материалы отсутствуют.
Инструменты
Поверхность. Рентгеновские, синхротронные и нейтронные исследования