

Поверхность. Рентгеновские, синхротронные и нейтронные исследования, 2021, № 8, стр. 100-105
Квантово-механическое моделирование взаимодействия углеродных наноструктур с ионами металлов
В. В. Титаренко a, *, Э. Ф. Штапенко a, Е. О. Воронков a, В. А. Заблудовский a, В. Колоджейчик b, **, К. С. Капуста b, В. Н. Кузнецов a
a Днепровский национальный университет железнодорожного транспорта
им. академика В. Лазаряна
49010 Днипро, Украина
b Джексонский государственный университет
39272 Джексон, Миссисипи, США
* E-mail: tytarenko.valentina@gmail.com
** E-mail: dziecial@icnanotox.org
Поступила в редакцию 18.12.2020
После доработки 17.01.2021
Принята к публикации 22.01.2021
Аннотация
Предложены квантово-механические модели формирования металлоуглеродных комплексов ионов Co, Ni, Cu, Zn с молекулами фуллерена С60 и одностенными углеродными нанотрубками (ОУНТ) С48. Результаты расчетов показали, что в водных растворах электролитов возможна адсорбция ионов Co, Ni, Cu, Zn на поверхности фуллерена С60 и ОУНТ С48 с образованием стабильных комплексов углеродный наноматериал–металл (УНМ–М), причем минимальная энергия комплекса С60–М для ионов кобальта и меди соответствуют положению над центром ячейки С6, для иона никеля – над одинарной связью С–С в ячейке С6, а для иона цинка – над атомом углерода С. Оптимизированные состояния комплексов С48–М соответствуют положению ионов металлов над центром ячейки С6.
ВВЕДЕНИЕ
В современном материаловедении широкое применение получили углеродные наноматериалы, особенно в роле наполнителя композиционных материалов [1, 2]. В первую очередь это связано с уникальными свойствами полученных материалов, поскольку добавление углеродного наноматериала (УНМ) в металлическую матрицу приводит к увеличению ее твердости, износостойкости, повышению коррозионной стойкости [3–8]. Однако, учитывая высокую себестоимость УНМ, большой интерес вызывает поверхностное упрочнение, при котором на поверхность деталей наносится металлическая композиционная пленка с углеродными наночастицами в качестве наполнителя. Среди множества существующих способов получения металлических композиционных пленок наиболее широкое распространение получил метод конденсации вещества на подложке из пара или растворов, который дает возможность управлять образованием и ростом металлической пленки в широких пределах. С целью интенсификации и стабилизации процессов соосаждения металлов и УНМ этот процесс может протекать при наложении импульсного тока, ультразвука, магнитного поля и внешнего лазерного излучения.
Уникальные физико-химические свойства электроосаждаемых металлических пленок в значительной степени зависят от концентрации частиц УНМ в металлической матрице. Поэтому особое внимание в последнее время вызывает контроль и управление содержанием частиц УНМ в композиционных металлических пленках. Решение подобной задачи невозможно без изучения механизма формирования структуры углеродсодержащих композиционных металлических пленок. Однако процесс совместного соосаждения на подложке ионов металла и частиц УНМ остается до конца не изученным. Прежде всего, не выяснен механизм транспортировки частиц УНМ из объема раствора на подложку. В ряде работ отмечено, что перемещение частиц УНМ происходит исключительно конвекционным потоком, который создают ионы металла в растворе электролита [9, 10], либо высказывается предположение о том, что частицы УНМ, приобретая заряд в растворе электролита, движутся к катоду под действием электрического поля, созданного разностью потенциалов между анодом и катодом [11, 12]. Механизм образования металлоуглеродных комплексов может быть весьма разнообразным и приобретать заряд частицы УНМ могут различными способами.
В работах [13, 14] была теоретически рассмотрена возможность адсорбции атомов металлов на частицах УНМ. Методом теории функционала плотности (ТФП) рассчитаны энергии связи нейтральных атомов переходных металлов c частицами УНМ – фуллереном С60 и ОУНТ С48. Показано, что значения энергий связи атомов, адсорбированных на поверхностях С60 и С48 изменяются в диапазоне 0.05–2 эВ (в зависимости от металла). Однако данные расчеты проведены для адсорбции нейтральных атомов в вакууме, а не положительно заряженных ионов металла в растворе электролита, и не могут быть использованы для моделирования механизмов приобретения заряда частицами УНМ и образования металлоуглеродных комплексов.
Для установления механизма совместного соосаждения частиц УНМ и ионов металла предложены модели образования металлоуглеродных комплексов и проведены расчеты энергий связи ионов Co, Ni, Cu, Zn с фуллереном С60 и ОУНТ С48.
МЕТОДЫ И МОДЕЛИ
Для расчета энергии связи ионов металлов с частицами УНМ необходимо учитывать следующие модельные приближения.
Во-первых, адсорбция происходит в растворах электролитов, в частности, в водных растворах.
Во-вторых, обеспечивая выполнение закона сохранения заряда, в расчетах необходимо учитывать, что адсорбироваться на поверхности частиц УНМ должны ионы металлов, и (как следствие) полученный комплекс УНМ–М должен иметь заряд исходного иона металла.
В-третьих, присоединение комплекса ионов металла к частице УНМ происходит поочередно, т.е. каждый последующий ион металла адсорбируется на частице УНМ, уже имеющей заряд, полученный при предыдущей адсорбции.
Расчеты энергии связи ионов металлов с частицами УНМ проводились в рамках теории функционала плотности (ТФП), которая за последние время прочно заняла место одного из самых популярных методов расчета электронной структуры атомов, молекул, кластеров, твердых тел и т.п. [15, 16]. Растущая популярность ТФП обусловлена, прежде всего, сочетанием достаточно высокой точности получаемых результатов, конкурирующей в ряде случаев с точностью ab initio методов учета электронной корреляции, и с весьма умеренными требованиями к вычислительным ресурсам, позволяющими проводить расчёты систем, состоящих из сотен атомов и представляющих интерес для современной нанотехнологии. Кроме того, метод ТФП используется для исследования адсорбционных характеристик переходных металлов [17, 18], кинетика электрокристаллизации которых исследуется в данной работе.
Многочисленные исследования характеристик молекул и кластеров методом ТФП показали хорошие результаты при правильном выборе обменно-корреляционного функционала. Как отмечено в [19, 20], наиболее подходящим для расчетов структурных и термохимических характеристик комплексов металлов является трехпараметрический гибридный функционал B3LYP. В работе [21] методом ТФП были изучены структура и энергетика образования макроциклических комплексов. Показано, что метод ТФП с использованием гибридного функционала B3LYP дает точную информацию о структурных, энергетических и кинетических характеристиках. Известно также, что теория функционала плотности с использованием гибридных обменно-корреляционных функционалов позволяет успешно рассчитывать структурные и электронные характеристики комплексов переходных и тяжелых металлов с приемлемыми затратами машинного времени [22]. Показано, что при использовании функционала B3LYP для расчета характеристик структуры переходных металлов, точность обеспечивается на уровне ab initio методов. При оптимизации геометрии комплексов в водном растворе нами были учтены также и дисперсионные поправки [23, 24]. Влияние водной среды учитывалось методом самосогласованного реакционного поля в рамках модели поляризационного континуума Томаси и др. [25].
Выбор используемого для расчетов базисного набора атомных орбиталей основывался на том, что расчет энергетических и термодинамических величин производился для тех металлов, для которых наибольшую роль играет взаимодействие валентных электронов. Для описания таких взаимодействий используются валентно-расщепленные наборы базисных орбиталей, в частности базис 6-31g или расширенный 6-31-g(d), содержащий атомные орбитали d-типа для учета поляризации электронной плотности металлов.
Для проведения расчетов энергий молекул, их структур и частот колебаний в конденсированном состоянии, был использован пакет программ GAUSSIAN 09 [26 ] . Температура при расчетах выбиралась 295 К, давление 105 Па.
РАСЧЕТ ЭНЕРГИИ СВЯЗИ
Для расчета энергии связи ионов металлов с частицами УНМ использовался метод функционала плотности, в котором энергия определяется выражением:
(1)
$W = U + {{T}_{S}}\{ {{\varphi }_{i}}(r)\} _{{i{\kern 1pt} = {\kern 1pt} 1}}^{{{{N}_{{{\text{заполн}}}}}}} + {{V}_{{ne}}}[\rho ] + J[\rho ] + {{E}_{{xc}}}[\rho ],$Энергию связи ($\Delta W$) адсорбированного иона металла с частицей УНМ определяли как разность между полной энергией комплекса УНМ с адсорбированным ионом металла (WC + M) и суммой энергий его составных частей (WC, WM):
(2)
$\Delta W = \left| {{{W}_{{{\text{C}} + {\text{M}}}}} - ({{W}_{{\text{C}}}} + {{W}_{{\text{M}}}})} \right|,$В случае поочередной адсорбции ионов металла на поверхность УНМ энергия связи рассчитывалась как разница между полной энергией, которой обладает комплекс УНМ, соответственно, с одним (${{W}_{{{\text{C}} + {\text{М}}}}}$), двумя (${{W}_{{{\text{C}} + 2{\text{М}}}}}$) и тремя (${{W}_{{{\text{C}} + 3{\text{М}}}}}$) адсорбированными ионами металла и энергией составных частей: энергией частицы УНМ (${{W}_{{\text{C}}}}$) и энергией одного иона металла (${{W}_{{\text{М}}}}$)
(3)
$\Delta {{W}_{1}} = \left| {{{W}_{{{\text{C}} + {\text{M}}}}} - ({{W}_{{\text{C}}}} + {{W}_{{\text{M}}}})} \right|,$(4)
$\Delta {{W}_{2}} = \left| {{{W}_{{{\text{C + 2M}}}}} - ({{W}_{{{\text{C + M}}}}} + {{W}_{{\text{M}}}})} \right|,$(5)
$\Delta {{W}_{3}} = \left| {{{W}_{{{\text{C + 3M}}}}} - ({{W}_{{{\text{C + 2M}}}}} + {{W}_{{\text{M}}}})} \right|.$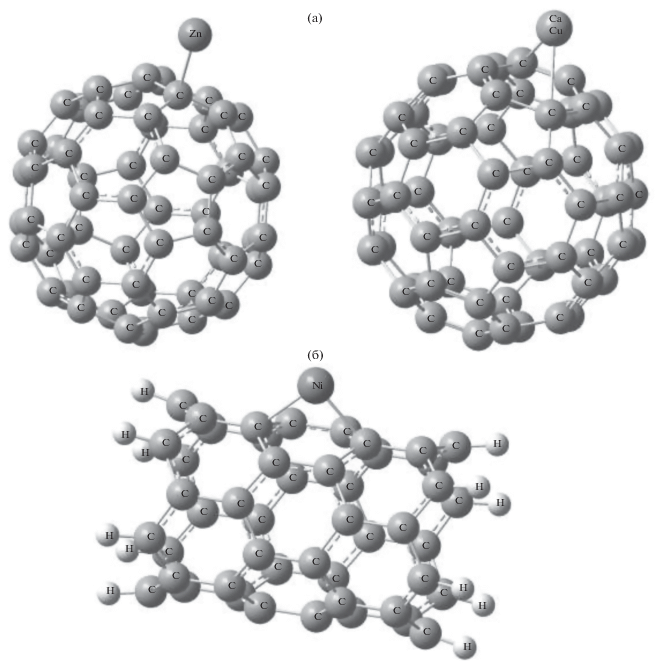
В табл. 1 приведены полученные нами результаты оптимизации расстояния от адсорбированных ионов метала до поверхности частицы УНМ ($d$) для водной среды и результаты расчетов работ [13, 14] для вакуума. В наших расчетах величина $d$ определяет длину перпендикуляра, опущенного из иона на плоскость структурного кольца С6 или С5, в случае адсорбции иона на поверхности фуллерена С60, или длину перпендикуляра, опущенного из иона на плоскость нижних четырех атомов углерода в ячейке С6, в случае адсорбции иона на поверхности ОУНТ С48.
Показано, что комплексы С60–М с минимальной энергией в случае ионов меди и кобальта соответствуют структуре, в которой ион находится над центром ячейки С6. Положение иона никеля над серединой связи C–C оказалось минимальным для комплекса C60–Ni, в то время как для комплекса C60–Zn наиболее стабильной является структура, в которой ион находится над атомом углерода С. Аналогично, оптимизированные состояния комплексов С48–М соответствуют положению ионов металлов над центром ячейки С6. Тенденция увеличения оптимизированного расстояния в ряду Ni < Cu <Co < Zn наблюдалась как для C60, так и для С48.
Поскольку теоретически является возможной адсорбция более одного иона металла на поверхности частицы УНМ, нами были учтены модели, в которых одна наночастица связывает до трех ионов металла. Учитывая то, что с добавлением каждого нового иона металла заряд системы должен увеличиваться на 2e (e – элементарный электрический заряд), были предложены и оптимизированы модели комплексов с одним, двумя или тремя связанными ионами металлов, которые имеют соответственно заряды +2e, +4e и +6e.
В табл. 2 приведены результаты оптимизации расстояния между адсорбированным ионом никеля и поверхностью фуллерена С60 для разных зарядов комплекса, рассчитанные при взаимодействии в вакууме и воде. Как показали расчеты (табл. 2), с увеличением заряда расстояние между адсорбированным ионом метала и поверхностью частицы УНМ увеличивается.
Таблица 2.
Расстояние между адсорбированным ионом никеля и поверхностью фуллерена С60 для разных зарядов комплекса
| q | +2e | +4e | +6e | |
|---|---|---|---|---|
| d, Å | Вакуум | 1.869 | 1.881 | 1.999 |
| Вода | 0.987 | 1.392 | 1.410 | |
В табл. 3 приведены результаты расчета энергии связи ($\Delta W$) адсорбированного иона металла с частицей УНМ. Из табл. 3 видно, что все рассматриваемые ионы металлов прочно связываются с частицами С48, в отличие от соединений с С60, так как их энергия связи значительно превышает энергию теплового движения 0.025 эВ.
Таблица 3.
Энергия связи ионов Co, Ni, Cu, Zn с частицами УНМ
| $\Delta W$, эВ | ||||
|---|---|---|---|---|
| Co | Ni | Cu | Zn | |
| Фуллерена С60 | 1.406 | 1.907 | 2.933 | 0.026 |
| ОУНТ С48 | 2.005 | 2.234 | 3.158 | 0.533 |
При последовательном присоединении нескольких ионов металла к одной частице УНМ энергия их связи уменьшается, как видно из приведенных в табл. 4 результатов расчетов для фуллерена С60 и иона никеля. Оптимизация геометрии комплексов С60 с несколькими ионами никеля привела к следующим результатам: четыре иона располагаются в виде квадрата с фуллереном в центре, три иона принимают форму прямоугольного равнобедренного треугольника, два иона расположены по диаметру фуллерена. Во всех случаях ионы располагаются над одинарной связью С–С. Взаимодействие частиц УНМ с двумя и более ионами металлов не приводит к образованию стабильных комплексов (табл. 4), и все дальнейшие расчеты проводились для заряда +2е, что соответствует валентности одного иона металла.
Таблица 4.
Энергия связи комплекса С60–Ni в зависимости от количества адсорбированных ионов никеля
| Количество адсорбированных ионов | |||
|---|---|---|---|
| 1 | 2 | 3 | |
| $\Delta W$, эВ | 1.907 | 1.582 | 0.240 |
Сравнения полученных значений энергии связи комплекса УНМ–М с энергией теплового движения показывают, что в водных растворах электролитов возможна адсорбция ионов Co, Ni, Cu, Zn на поверхности фуллерена С60 и ОУНТ С48 с образованием стабильных комплексов УНМ–М. Следовательно, перенос частиц УНМ из объема водного раствора электролита к поверхности катода возможен в следствии приобретения комплексом УНМ–М положительного заряда.
ЗАКЛЮЧЕНИЕ
Предложенный в работе механизм адсорбции ионов металлов на поверхности частиц УНМ с дальнейшим продвижением заряженного комплекса в направлении катода был проверен вычислительным методом. Сравнение результатов расчета энергий связи комплексов УНМ–М с энергиями теплового движения показало, что в водных растворах электролитов возможна адсорбция ионов Co, Ni, Cu, Zn на поверхности фуллеренов C60 и ОУНТ C48 с образованием стабильных комплексов УНМ–М. Для комплексов С60–М и С48–М энергии связи увеличиваются в последовательности Zn < Co < Ni <Cu. Показано, что комплексы, адсорбированные более чем одним ионом металла, не являются стабильными.
Таким образом, перенос частиц УНМ из объема раствора к катоду может протекать через стадию адсорбции на их поверхности катионов осаждаемого металла. Получив заряд, металлоуглеродный комплекс переносится к катоду и там заращивается разряжающимися ионами металла.
Список литературы
Gheorghies C., Rusu D.E., Bund A., Condurache-Bota S., Georgescu L.P. // Appl. Nanosci. 2014. № 4. P. 1021. https://doi.org/10.1007/s13204-013-0285-y
Burkat G.K., Fujimura T., Dolmatov V.Yu., Orlova E.A., Veretennikova M.V. // Diamond & Related Materials. 2005. № 14. P. 1761. https://doi.org/10.1016/j.diamond.2005.08.004
Isakov V.P., Lyamkin A.I., Nikitin D.N., Shalimova A.S., Solntsev A.V. // Protection of Metals and Physical Chemistry of Surfaces. 2010. № 46. P. 578. https://doi.org/10.1134/S2070205110050138
Liping Wang, Yan Gao, Qunji Xue, Huiwen Liu, Tao Xu // Materials Science and Engineering, A. 2005. V. 390. P. 313. https://doi.org/10.1016/j.msea.2004.08.033
Заблудовський В.О., Дудкіна В.В., Штапенко Е.П. // Наука та прогрес транспорту. Вісник Дніпропетровського національного університету імені академіка В. Лазаряна. 2013. Т. 47. № 5. С. 70. https://doi.org/10.15802/stp2013/17968
Дудкина В.В., Заблудовский В.А., Штапенко Э.Ф. // Металлофизика и новейшие технологии. 2015. Т. 37. № 5. С. 713. https://doi.org/10.15407/mfint.37.05.0713
Титаренко В.В., Заблудовский В.А. // Металлофизика и новейшие технологии. 2016. Т. 38. № 4. С. 519. https://doi.org/10.15407/mfint.38.04.0519
Tytarenko V.V., Zabludovsky V.A., Shtapenko E.Ph., Tytarenko I.V. // Galvanotechnik. 2019. № 4. P. 648.
Chiganova G.A., Mordvinova L.E. // Inorganic Materials. 2011. V. 47. № 7. P. 717. https://doi.org/10.1134/S0020168511070089
Hiroshi Matsubara, Yoshihiro Abe, Yoshiyuki Chiba, Hiroshi Nishiyama, Nobuo Saito, Kazunori Hodouchi, Yasunobu Inoue // Electrochimica Acta. 2007. V. 52. P. 3047. https://doi.org/10.1016/j.electacta.2006.09.043
Sam Zhang, Deen Sun, Yongqing Fu, Hejun Du // Surface and Coatings Technology. 2003. V. 167. P. 113.https://doi.org/10.1016/S0257-8972(02)00903-9
Hou J.G., Xiang Li, Haiqian Wang, BingWang // J. Physics and Chemistry of Solids. 2000. V. 61. P. 995. https://doi.org/10.1016/S0022-3697(99)00349-2
Hubert Valencia, Adrià Gil, Gilles Frapper // J. Physical Chemistry C. 2010. V. 114. P. 14141.
J. Andreas Larsson, Simon D. Elliott, James C. Greer, Jascha Repp, Gerhard Meyer, Rolf Allenspach // Physical Review B 77. 2008. P. 115434-1. https://doi.org/10.1103/Phys
Штапенко Э.Ф., Заблудовский В.А., Воронков Е.О. // Поверхность. Рентген., синхротр. и нейтрон. исслед. 2010. № 12. С. 95.
Koch W., Holthausen M.C. Chemists Guide to Density Functional Theory, 2nd Edition. N.Y.: Wiley – VCH, 2001. 293 p.
Lopez N., Almora-Barrios N., Carchini G. // Catalysis Science & Technology. 2012. № 2. P. 2405. https://doi.org/10.1039/C2CY20384G
Allison T.C., J Tong Y.Y. // Physical Chemistry Chemical Physics. 2011. V. 13. P. 12858. https://doi.org/10.1039/C1CP20376B
Schreckenbach G., Hay P.J., Martin R.L. // Inorganic Chemistry. 1998. V. 37. P. 4442. https://doi.org/10.1002/(SICI)1096-987X(19990115)20: 1<70::AID-JCC9>3.0.CO;2-F
Miehlich B., Savin A., Stoll H., Preuss H. // Chemical Physics Letters. 1989. V. 157. P. 200. https://doi.org/10.1016/0009-2614(89)87234-3
Keire David A., Hee Jans Yun, Lin Li at al. // Inorganic Chemistry. 2001. V. 40, JNM7. P. 4310. https://doi.org/10.1021/ic0010297
Andzelm J., Labanowski J. Density Functional Methods in Chemistry. Heidelberg: Springer Verlag, 1991. 443 p. https://doi.org/10.1007/978-1-4612-3136-3
Grimme S. Grimme, Antony J., Ehrlich S., Krieg H. // J. Physical Chemistry. 2010. M. 132. P. 154104. https://doi.org/10.1063/1.3382344
Grimme S. Grimme, Ehrlich S., Goerigk L. // J. Computational Chemistry. 2011. V. 32. P. 1456. https://doi.org/10.1002/jcc.21759
Tomasi J., Mennucci B., Cammi R. // Chem. Rev. 2005. V. 105. P. 2999. https://doi.org/10.1021/cr9904009
Gaussian 03, Revision C.02, Frisch M.J., Trucks G.W., Schlegel H.B., Scuseria G.E., Robb M.A., Cheeseman J.R., Montgomery Jr., J.A., Vreven T., Kudin K.N., Burant J.C., Millam J.M., Iyengar S.S., Tomasi J., Barone V., Mennucci B., Cossi M., Scalmani G., Rega N., Petersson G.A., Nakatsuji H., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Klene M., Li X., Knox J.E., Hratchian H.P., Cross J.B., Bakken V., Adamo C., Jaramillo J., Gomperts R., Stratmann R.E., Yazyev O., Austin A.J., Caimni R., Pomelli C., Ochterski J.W., Ayala P.Y., Morokuma K., Voth G.A., Salvador P., Dannenberg J.J., Zakrzewski V.G., Dapprich S., Daniels A.D., Strain M.C., Farkas O., Malick D.K., Rabuck A.D., Raghavachari K., Foresman J.B., Ortiz J.V., Cui Q., Baboul A.G., Clifford S., Cioslowski J., Stefanov B.B., Liu G., Liashenko A., Piskorz P., Komaromi I., Martin R.L., Fox D.J., Keith T., Al-Laham M., Peng C.Y., Nanayakkara A., Challacombe M., Gill P.M.W., Johnson B., Chen W., Wong M.W., Gonzalez C., Pople J.A. Gaussian, Inc., Wallingford CT, 2004.
Дополнительные материалы отсутствуют.
Инструменты
Поверхность. Рентгеновские, синхротронные и нейтронные исследования