

Поверхность. Рентгеновские, синхротронные и нейтронные исследования, 2022, № 3, стр. 101-106
Моделирование механических свойств поверхности методом молекулярной динамики с потенциалом ReaxFF
Ю. В. Русалев a, b, *, А. А. Гуда a, b, Д. М. Пашков a, b, О. А. Беляк b, В. И. Колесников b, А. В. Солдатов a
a Международный исследовательский институт интеллектуальных материалов,
Южный федеральный университет
344090 Ростов-на-Дону, Россия
b Ростовский государственный университет путей сообщения
344038 Ростов-на-Дону, Россия
* E-mail: yuri.rusalev@gmail.com
Поступила в редакцию 21.07.2021
После доработки 25.08.2021
Принята к публикации 30.08.2021
- EDN: NWMNBK
- DOI: 10.31857/S1028096022030189
Аннотация
Морфология, дефектность, степень кристалличности и механические свойства поверхности определяют ее трибологические и износостойкие характеристики. На разных пространственных масштабах ключевую роль играют шероховатость поверхности, размеры кристаллитов, дислокации, точечные дефекты. Благодаря развитию новых методов суперкомпьютерного моделирования появляется возможность проведения численного эксперимента на атомном, нано- и микромасштабах. Рассмотрен процесс наноиндентирования для системы с числом частиц ~1000, где точные подходы теории функционала плотности уже не работают, а эмпирические потенциалы являются слишком грубыми. На масштабе 2.5 нм показаны процессы образования дефектов, влияние аморфизации на механические свойства, необратимые процессы деформации материала индентором. Полученные результаты открывают новые возможности для исследования механических и трибологических свойств материалов с помощью численного эксперимента наноиндентирования.
ВВЕДЕНИЕ
Метод молекулярной динамики (МД) является мощным инструментом для исследования материалов в реалистичных технологических условиях, например для исследования каталитических реакций [1], роста нанотрубок [2], диффузии в протонообменных мембранах [3], процессов трения и износа [4]. Благодаря прогрессу в вычислительных методах и компьютерной технике появилась возможность моделирования систем с большим числом частиц на масштабах объектов от нанометрового до субмикронного. Учет дислокаций, границ раздела зерен и неоднородности поверхности на таких масштабах позволяет рассчитывать механические и трибологические свойства реальных материалов. Среди примеров можно отметить работы по исследованию коэффициента трения [5], тензора упругих постоянных [6], модуля Юнга [7], процесса развития трещин и смещения дислокаций на атомарном уровне [8]. Таким образом, расчеты МД позволяют установить зависимость между микроструктурой пленок и поверхностей и их макроскопическими свойствами.
Одним из наиболее часто используемых экспериментальных методов исследования механических свойств поверхности является наноиндентирование [9]. Индентирование – это процесс, при котором индентор давит или царапает поверхность материала. Измеряют силу, действующую на индентор, в зависимости от глубины погружения или расстояния смещения вдоль поверхности. Численное моделирование процесса наноиндентирования позволяет правильно интерпретировать результаты эксперимента и предсказывать структурные свойства материала на основе кривой нагрузки–разгрузки. Например, в [5] с помощью численного наноиндентирования были исследованы трибологические свойства металлических стекол Cu64.5Zr35.5. Было изучено поведение коэффициента трения при царапании индентором поверхности в зависимости от температуры. Проводилось моделирование многослойных тонких пленок с помощью наноиндентирования методом МД. В ходе индентирования наблюдался рост температуры около кончика индентора, что приводило к аморфизации поверхности и снижению нагрузки на кривой наноиндентирования.
Результаты численного моделирования зависят от выбора потенциала межатомного взаимодействия. Самым точным методом является квантовая молекулярная динамика в подходе AI-PIMD [10], но такое приближение подходит для систем с малым числом частиц на временны́х масштабах менее 1 пс. Подход на основе теории функционала электронной плотности и классической ньютоновской динамики для атомов позволяет моделировать системы, содержащие сотни атомов на временны́х масштабах до наносекунд. Для большего числа частиц и длительных временны́х интервалов существуют различные приближения. Среди наиболее часто используемых – DFTB [11], MOPAC [12], EAM [13]. Выбор межатомного потенциала сильно влияет на результаты численного наноиндентирования поверхности [14]. Рекомендуется тренировать модельные потенциалы на выборке искомых свойств материала, например, механических, на основе расчетов ab initio.
Метод ReaxFF [15] является одним из наиболее удачных способов задания межатомного потенциала для систем с большим числом частиц. Благодаря применению формализма порядка связи вместе с учетом поляризации зарядов в системе удается учитывать электростатические, ковалентные и слабые взаимодействия, что позволяет применять данный вид потенциала к любым соединениям: от металлов до органических молекул. Целью настоящей работы была разработка методики численного моделирования механических свойств металлических пленок с большой концентрацией дефектов методом наноиндентирования с потенциалом ReaxFF.
МЕТОДЫ
Расчеты методом МД были проведены с помощью программного обеспечения LAMMPS [16] (Large-scale Atomic/Molecular Massively Parallel Simulator) с пакетом reax/c [17]. Визуализацию результатов осуществляли с помощью программ OVITO [18] [18]и VESTA [19] [19]. Для проведения расчетов была построена поверхность ГЦК-пленки кобальта размером 50 × 50 × 24 Å (6300 атомов) с параметром решетки 3.42 Å. Вдоль горизонтальных осей x, y были наложены периодические граничные условия. В ходе моделирования частицы находились в микроканоническом ансамбле, а для контроля температуры использовали термостат Берендсена [20] с параметром демпинга 100 фс.
В качестве индентора был выбран абсолютно твердый шар. Индентор двигается вдоль оси z (рис. 1). Во время расчета индентор равномерно двигается к нижней границе пленки, а затем возвращается вдоль того же направления на исходную высоту. На каждом шаге по времени измеряется сила, действующая на поверхность индентора. Сила F описывается выражением:
где K – это константа взаимодействия, r – расстояние от атома до центра индентора, R – его радиус. Сила F полностью отталкивающая и равна нулю при r > R. Прочие параметры расчета представлены в табл. 1.
Таблица 1.
Параметры индентора в численном эксперименте
| Радиус индентора, Å | 10.5 |
| Максимальная глубина погружения, Å | 7 |
| Скорость погружения, м/с; Å/пс | 10; 0.1 |
| Константа взаимодействия материала индентора с атомами пленки K, эВ/Å2 | 10 |
| Температура образца в процессе наноиндентирования, К | 10 |
Для моделирования был выбран потенциал типа ReaxFF. В данном потенциале энергия системы E – непрерывная дифференцируемая функция, описываемая выражением:
(2)
$\begin{gathered} E = {{E}_{{{\text{bond}}}}} + {{E}_{{{\text{over}}}}} + {{E}_{{{\text{angle}}}}} + {{E}_{{{\text{torsion}}}}} + \\ + \,\,{{E}_{{{\text{disp}}}}} + {{E}_{{{\text{electro}}}}} + {{E}_{{{\text{specific}}}}}, \\ \end{gathered} $В настоящей работе использован потенциал для металлического кобальта, разработанный в [21] для описания кристаллической структуры, поверхностей, кластеров, а также различных дефектов в решетке металла. Для параметризации данного потенциала брали не только значения энергии различных кристаллических фаз и поверхностей, но и компоненты тензора упругих постоянных, а также значения энергии дефектов упаковки, вакансий и дополнительных атомов на поверхности кобальта. Свойства поверхности кобальта лучше описываются данным потенциалом, чем аналогичным потенциалом EAM.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Моделирование взаимодействия наноиндентора с поверхностью осуществляли для четырех пленок с различной степенью кристалличности. Для создания дефектов исходную пленку со структурой ГЦК нагрели до температур 300, 2000, 2250 и 2500 К в течение 30 пс (табл. 2). Затем ее резко охладили и полученную структуру оптимизировали методом градиентного спуска. Рассчитанные функции радиального распределения атомов Co для четырех пленок показаны на рис. 2, где приведены также изображения структур вдоль оси Z.
Таблица 2.
Соответствие номера образца и температуры, до которой нагревалась пленка в течение 30 пс в расчетах методом молекулярной динамики для создания требуемой концентрации дефектов
| Образец | Температура, К |
|---|---|
| 1 | 300 |
| 2 | 2000 |
| 3 | 2250 |
| 4 | 2500 |
Рис. 2.
Радиальные функции распределения и структуры образцов пленок кобальта № 1–4 (а–г) вдоль оси Z для различных температур нагрева.
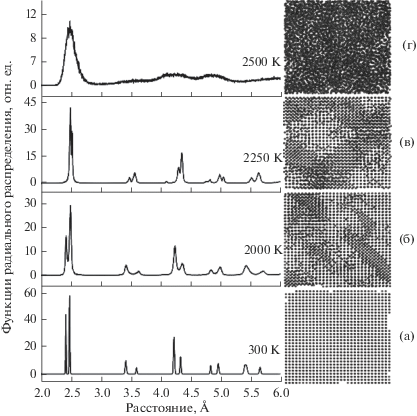
Пленка № 1 представляла собой почти идеальный кристалл с ГЦК-решеткой кобальта. При нагреве до 2000 К в образце № 2 появлялись дефекты междоузлий, в том числе дефекты упаковки в объеме материала. Интенсивность пиков функции радиального распределения уменьшилась в два раза, а их ширина на половине высоты увеличилась. Такое поведение объясняется увеличением степени беспорядка в первой координационной сфере кобальта, вызванного смещениями атомов из их идеальных позиций в кристаллической решетке. В образце № 3 наблюдался переход, который выражался в смещении максимумов функции радиального распределения и перераспределении их интенсивности, в то время как ширина отдельных пиков практически не изменялась. Образец № 4 представлял собой аморфную фазу кобальта, о чем свидетельствовали широкий первый пик функции радиального распределения и практически полное отсутствие других узких пиков.
Полученные таким образом структуры пленок стали стартовыми для проведения численного эксперимента по наноиндентированию. Кривая нагрузки–разгрузки для кристаллического образца № 1 представлена на рис. 3. Кривая нагрузки монотонно возрастает до точки 1, где достигается максимальное значение нагрузки 180 нН. Затем кривая претерпевает разрыв, и значение нагрузки падает до точки 2. Для того чтобы объяснить такое поведение кривой, в точках 1 и 2 для всех атомов был рассчитан параметр центральной симметрии [22] согласно формуле Кельхнера. В твердых телах этот параметр является удобным способом описания локального беспорядка решетки вокруг атома и может использоваться для классификации принадлежности атома к идеальной решетке, локальным дефектам, дислокации или поверхности. Для атома в упорядоченном кристалле значение этого параметра близко к нулю.
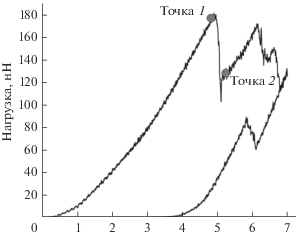
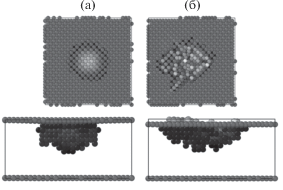
На рис. 4 показаны атомы образца № 1 в точках 1 и 2 положения индентора, для которых параметр центральной симметрии больше 0.1. На верхней части рисунка приведено изображение поверхности вдоль оси Z, на нижней части рисунка – вдоль оси Y. До разрыва кривой нагрузки пятно контакта симметрично и повторяет поверхность сферы индентора. Однако в точке 2 после разрыва на поверхности образуется трещина, и часть материала пленки выдавливается на поверхность. Резкое снижение величины нагрузки на кривой индентирования свидетельствует об образовании и распространении новых дефектов, в результате материал становится мягче.
Рис. 4.
Изображения атомов пленки № 1 в точках 1 (а) и 2 (б) на кривые нагрузки–разгрузки (рис. 3). Верхняя часть рисунка – проекция перпендикулярно оси Z, нижняя часть рисунка – проекция перпендикулярно оси Y. На рисунке изображены атомы с параметром центральной симметрии более 0.1.
На рис. 5 изображены кривые нагрузки–разгрузки образцов № 2–4. Пленки № 2 и № 3 выдерживают максимальную нагрузку порядка 110 нН, а пленка № 4 – только 17 нН при максимальном погружении индентора в материал. Заметим, что для образцов № 1–3 значительный рост нагрузки начинается с глубины внедрения hi ~ 0.2–0.5 Å, в то время как пленка № 4 на такой глубине практически не оказывает сопротивления индентору, а величина нагрузки заметно растет начиная с глубины порядка 3.9 Å. Данный факт можно объяснить тем, что плотность поверхности образца № 4 существенно ниже ввиду сильного разупорядочения, и первые несколько ангстрем погружения индентора соответствуют уплотнению верхних слоев, не вызывающих заметного отклика.
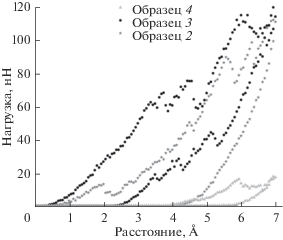
При обратном движении индентора кривая разгрузки всех образцов не возвращается в начало кривой нагрузки, что свидетельствует о необратимой пластической деформации в образце [23]. Значение, при котором разгрузка обращается в ноль, обозначим hf. Тогда разность d между hi и hf будет равна глубине кратера, образовавшегося после извлечения индентора. Рассчитанные значения hi, hf, d приведены в табл. 3. Глубина кратера минимальна в аморфном образце № 4, что свидетельствует о его большей пластичности по сравнению с образцами № 1–3, в которых кристаллическая структура частично сохранена, а наличие точечных дефектов может препятствовать распространению глубоких дислокаций при движении индентора.
Таблица 3.
Результаты эксперимента, численного индентирования образцов № 1–4
| Образец | hi, Å | hf, Å | d, Å | E, ГПа |
|---|---|---|---|---|
| 1 | 0.22 | 3.36 | 3.14 | 343 |
| 2 | 0.56 | 4.13 | 3.57 | 303 |
| 3 | 0.38 | 2.38 | 2 | 235 |
| 4 | 3.88 | 5.44 | 1.56 | 81 |
C помощью метода Оливера–Фара [24] из кривых наноиндентирования был рассчитан модуль Юнга E (табл. 3). Он убывает от 343 до 81 ГПа с ростом концентрации дефектов от пленки № 1 с высокой степенью кристалличности до аморфной пленки № 4. Полученные данные численного моделирования находятся в хорошем согласии с имеющимися экспериментальными данными для некристаллических структур кобальта. В [25] были экспериментально исследованы микроструктуры кобальта с большим количеством дефектов. Полученный авторами модуль Юнга в экспериментах по наноиндентированию уменьшался с ростом количества дефектов и находился в диапазоне 181–218 ГПа в зависимости от способа приготовления образца. Полученные авторами образцы наиболее близко соответствуют пленке № 3 в настоящей работе, для которой модуль Юнга равен 235 ГПа. В [26] исследовали наноструктуры кобальта с высокой пористостью. Модуль Юнга для структуры, состоящей на 82% из пористого ГЦК-кобальта, оказался равен 94–102 ГПа. Этому диапазону соответствует аморфная пленка № 4, полученная в ходе быстрого отжига при высокой температуре 2500 К, для которой модуль Юнга в численном эксперименте равен 81 ГПа.
ЗАКЛЮЧЕНИЕ
В настоящем исследовании была разработана методика численного наноиндентирования металлических пленок с различной степенью кристалличности методом МД с потенциалом ReaxFF. Степень разупорядочения структуры варьировали с помощью кратковременного нагрева образца выше температуры плавления и последующего резкого охлаждения. Для четырех образцов были получены кривые нагрузки–разгрузки в численном эксперименте по наноиндентированию со сферическим индентором радиусом 10.5 Å. Было показано, что скачки нагрузки на кривых соответствуют появлению протяженного дефекта, соответствующего образованию трещины и выдавливанию материала пленки индентором из кратера на поверхность. Из кривых была получена глубина области неупругой деформации поверхности, а также рассчитан модуль Юнга методом Оливера–Фара. С ростом концентрации дефектов модуль Юнга уменьшается от 353 ГПа для идеально кристаллической пленки до 81 ГПа для аморфной пленки. Полученные значения хорошо согласуются с экспериментальными данными по наноиндентированию дефектных микроструктур кобальта, а методика численного моделирования открывает новые перспективы компьютерного дизайна новых материалов нанопокрытий для получения заранее заданных механических и трибологических свойств.
Список литературы
Chenoweth K., van Duin A.C.T., Goddard W.A. // Ang. Chem. Int. Ed. 2009. V. 48. № 41. P. 7630. https://doi.org/10.1002/anie.200902574
Neyts E.C., Shibuta Y., van Duin A.C.T. et al. // ACS Nano. 2010. V. 4. № 11. P. 6665. https://doi.org/10.1021/nn102095y
Achtyl J.L., Unocic R.R., Xu L.J. et al. // Nature Commun. 2015. V. 6. № P. https://doi.org/10.1038/ncomms7539
Vakis A.I., Yastrebov V.A., Scheibert J. et al. // Tribology Int. 2018. V. 125. № P. 169. https://doi.org/10.1016/j.triboint.2018.02.005
Avila K.E., Kuchemann S., Alhafeez I.A. et al. // Tribology Int. 2019. V. 139. № P. 1. https://doi.org/10.1016/j.triboint.2019.06.017
Krief M., Ashkenazy Y. // Phys. Rev. E. 2021. V. 103. № 6. P. 063307. https://doi.org/10.1103/PhysRevE.103.063307
Radue M.S., Jensen B.D., Gowtham S. et al. // J. Polymer Sci. B. 2018. V. 56. № 3. P. 255. https://doi.org/10.1002/polb.24539
Zhang Y.Q., Jiang S.Y. // Metals. 2017. V. 7. № 10. P. https://doi.org/10.3390/met7100432
Cohen S.R., Kalfon-Cohen E. // Beilstein J. Nanotechnol. 2013. V. 4. P. 815. https://doi.org/10.3762/bjnano.4.93
Geng H.Y. // J. Comput. Phys. 2015. V. 283. Iss. C. P. 299. https://doi.org/10.1016/j.jcp.2014.12.007
Ruger R., van Lenthe E., Lu Y. et al. // J. Chem. Theory Comput. 2015. V. 11. № 1. P. 157. https://doi.org/10.1021/ct500838h
Stewart J.J.P. // J. Mol. Modeling. 2013. V. 19. № 1. P. 1. https://doi.org/10.1007/s00894-012-1667-x
Daw M.S., Baskes M.I. // Phys. Rev. B. 1984. V. 29. № 12. P. 6443. https://doi.org/10.1103/PhysRevB.29.6443
Pratt D.R., Morrissey L.S., Nakhla S. // Mol. Simulation. 2020. V. 46. № 12. P. 923. https://doi.org/10.1080/08927022.2020.1791859
Senftle T.P., Hong S., Islam M.M. et al. // NPJ Comput. Mater. 2016. V. 2. № 1. P. 15011. https://doi.org/10.1038/npjcompumats.2015.11
Plimpton S. // J. Comput. Phys. 1995. V. 117. № 1. P. 1. https://doi.org/10.1006/jcph.1995.1039
Aktulga H.M., Fogarty J.C., Pandit S.A. et al. // Parallel Computing. 2012. V. 38. № 4. P. 245. https://doi.org/10.1016/j.parco.2011.08.005
Stukowski A. // Model. Simulation Mater. Sci. Eng. 2009. V. 18. № 1. P. 015012. https://doi.org/10.1088/0965-0393/18/1/015012
Momma K., Izumi F. // J. Appl. Crystallogr. 2011. V. 44. № 6. P. 1272. https://doi.org/10.1107/S0021889811038970
Berendsen H.J.C., Postma J.P.M., van Gunsteren W.F. et al. // J. Chem. Phys. 1984. V. 81. № 8. P. 3684. https://doi.org/10.1063/1.448118
LaBrosse M.R., Johnson J.K., van Duin A.C.T. // J. Phys. Chem. A. 2010. V. 114. № 18. P. 5855. https://doi.org/10.1021/jp911867r
Kelchner C.L., Plimpton S.J., Hamilton J.C. // Phys. Rev. B. 1998. V. 58. № 17. P. 11085. https://doi.org/10.1103/PhysRevB.58.11085
Golovin Y.I. // Phys. Solid State. 2008. V. 50. № 12. P. 2205. https://doi.org/10.1134/S1063783408120019
Oliver W.C., Pharr G.M. // J. Mater. Res. 2011. V. 19. № 1. P. 3. https://doi.org/10.1557/jmr.2004.19.1.3
Barry A.H., Dirras G., Schoenstein F. et al. // Mater. Charact. 2014. V. 91. P. 26. https://doi.org/10.1016/j.matchar.2014.02.004
Fellah F., Schoenstein F., Dakhlaoui–Omrani A. et al. // Mater. Charact. 2012. V. 69. P. 1. https://doi.org/10.1016/j.matchar.2012.03.017
Дополнительные материалы отсутствуют.
Инструменты
Поверхность. Рентгеновские, синхротронные и нейтронные исследования