

Поверхность. Рентгеновские, синхротронные и нейтронные исследования, 2022, № 9, стр. 3-12
Эволюция молекулярной структуры частично дегидрофторированных пленок поливинилиденфторида при хранении в атмосфере воздуха
В. Е. Живулин a, b, С. Е. Евсюков c, Д. А. Чалов b, В. М. Морилова d, *, В. П. Андрейчук a, Р. Х. Хайранов a, И. Г. Маргамов e, Л. А. Песин a, **
a Южно-Уральский государственный гуманитарно-педагогический университет
454080 Челябинск, Россия
b Южно-Уральский государственный университет (национальный исследовательский университет)
454080 Челябинск, Россия
c Evonik Operations GmbH
69221 Dossenheim, Germany
d Снежинский физико-технический институт НИЯУ МИФИ
456776 Снежинск, Россия
e Институт машиноведения имени Э.С. Горкунова УрО РАН
620049 Екатеринбург, Россия
* E-mail: vika_m@list.ru
** E-mail: pesinla@mail.ru
Поступила в редакцию 12.01.2022
После доработки 02.02.2022
Принята к публикации 17.02.2022
- EDN: AMQSQF
- DOI: 10.31857/S1028096022090217
Аннотация
Методами ИК-спектроскопии, растровой электронной микроскопии и рентгеновской дифракции исследованы изменения молекулярного состава пленки поливинилиденфторида Ф-2МЭ при ее химическом дегидрофторировании и последующем старении, а также идентифицированы продукты побочных химических реакций. Увеличение продолжительности реакции от 10 до 40 мин приводит к увеличению толщины образцов, концентрации двойных углерод–углеродных связей и этоксигрупп, а также пика поглощения с центром при 3100 см–1, предположительно связанного с валентными колебаниями =С–Н-связей при двойной углерод-углеродной связи. Через семь суток после синтеза в спектрах образцов появляются и начинают расти новые пики при 1840 и 2040 см–1, интенсивности которых пропорциональны друг другу и тем больше, чем продолжительнее синтез. Обе особенности характерны для ИК-спектра гидрофторида калия. Проанализированы изменения ИК‑спектров поглощения спустя год хранения образцов в атмосфере воздуха и после дополнительной промывки образца с продолжительностью синтеза 40 мин в смеси ацетона и дистиллированной воды через 467 сут хранения.
ВВЕДЕНИЕ
К настоящему времени большой объем научной литературы связан как с разработками методов синтеза, так и теоретическим моделированием свойств нано- и низкоразмерных углеродных структур, в том числе и карбина [1]. Модель идеального кристалла карбина предполагает наличие линейных углеродных цепей, в которых соседние атомы соединены между собой либо двойными, либо чередованием тройных и одинарных связей. Цепи должны быть упакованы в кристаллы за счет ван-дер-ваальсовских сил [2]. Кристаллический карбин обнаружен в природных минералах [3]. Активно разрабатывают методы его синтеза в лабораторных условиях, проводят расчеты его структуры и свойств [4–9].
Одним из способов синтеза карбиноподобных объектов является карбонизация галогенсодержащих цепочечных полимеров [10–16]. Поливинилиденфторид (ПВДФ) – полимерный материал, обладающий рядом полезных свойств, благодаря которым ему находят широкое применение в мембранных технологиях, электронике, медицине, акустике и так далее [17]. Равное количество фтора и водорода в цепях создает потенциальную возможность использования ПВДФ в качестве исходного материала для создания одномерных углеродных наноструктур путем химического и/или радиационного дегидрофторирования. Согласно известной модели химической карбонизации, под влиянием жидкой дегидрофторирующей среды фтор и водород в равных количествах отщепляются от углеродного каркаса и удаляются в виде молекул фтористого водорода [2, 10, 12].
Ранее, в работах [10, 12–16], изучены процессы, происходящие при химической карбонизации ПВДФ. Методами ИК- и ЯМР-спектроскопии проведены оценки глубины и скорости проникновения дегидрофторирующей смеси в частично кристаллическую пленку Kynar [13, 16]. Проанализированы изменения интенсивности пиков поглощения с максимумами при 532, 765 и 796, 976 см–1, возникающие вследствие колебаний, соответственно, CF2- и СН2-групп. Сделан вывод об одинаковом уменьшении концентрации этих групп на различных стадиях процесса дегидрофторирования.
С другой стороны, существует вероятность присоединения гидроксильных групп к освободившимся от фтора и водорода позициям в углеродной цепи, а также увеличения концентрации С–Н-связей в образцах за счет протекания побочных реакций с водородсодержащими ингредиентами дегидрофторирующей смеси [18]. Описанные в [14, 15, 18–21] вариации ЭПР-поглощения и формы ИК-спектров при старении дегидрофторированных пленок ПВДФ, демонстрируют существенную и долговременную нестабильность их свойств. Поэтому интересной научной и практически важной задачей представляется изучение эволюции молекулярной структуры как по мере протекания реакции дегидрофторирования, так и при последующем старении продуктов химической карбонизации ПВДФ. Такую возможность предоставляет метод ИК-спектроскопии, обладающий уникальной чувствительностью к изменениям молекулярной структуры.
В настоящей статье исследованы образцы химически дегидрофторированных пленок ПВДФ. Первичную промывку образцов после их извлечения из дегидрофторирующей смеси осуществляли в этаноле. В более ранних исследованиях [13–16, 18–21] для этой цели использовали ацетон, что приводило к разбуханию пленки и эффективному удалению из нее непрореагировавших компонентов смеси и побочных продуктов процесса ДГФ. Однако для получения более полных сведений о протекании реакции дегидрофторирования необходима идентификация этих продуктов. Использование метода рентгеновской дифракции позволило идентифицировать некоторые кристаллические фазы, образующиеся в результате побочных химических реакций процесса ДГФ полимера.
СИНТЕЗ ОБРАЗЦОВ И МЕТОДИКА ИЗМЕРЕНИЙ
Исследуемые образцы изготовлены химическим дегидрофторированием пленки ПВДФ Ф-2МЭ (ООО “Пластполимер”, Санкт-Петербург) при комнатной температуре в течение шести различных промежутков времени: 10, 15, 20, 25, 30 и 40 мин (соответственно, образцы 1–6). Дегидрофторирующая смесь состояла из насыщенного раствора КОН в этаноле и химически чистого ацетона в объемном соотношении 1 : 9 [10]. Для ее приготовления в стеклянную колбу наливали 50 мл этанола и добавляли 10 г едкого калия. Перемешивание проводили с помощью магнитной мешалки в течение 30 мин, при этом КОН полностью растворялся. Далее к получившемуся раствору добавляли 450 мл ацетона. Полученную смесь вновь перемешивали. Общий объем приготовленной смеси, таким образом, составил 500 мл.
Из исходной пленки были вырезаны шесть квадратных заготовок (20 × 20 мм). Каждую заготовку помещали в отдельную стеклянную посуду, содержащую 50 мл готовой дегидрофторирующей смеси. При увеличении продолжительности химического воздействия первоначально прозрачные заготовки темнели, меняя цвет от светло-коричневого до матово-черного. Сразу после изъятия из смеси образцы промывали в этаноле с постоянным перемешиванием на вибрационном стенде в течение 30 мин, а затем помещали между двумя листами фильтровальной бумаги, что предотвращало их деформацию при просушивании. Высохшие образцы прочно закрепляли в отдельных одинаковых держателях с прямоугольными отверстиями и выдерживали в форвакуумной камере при комнатной температуре и давлении остаточных газов не выше 5 × 10–2 мм рт. ст. в течение двух суток, после чего были получены первые спектры. В этой же камере и при тех же условиях образцы хранились в промежутках времени между измерениями в течение первых 36 сут после их синтеза. Далее образцы хранились в атмосфере воздуха в течение года.
Регистрацию ИК-спектров проводили с помощью ИК-Фурье спектрометра Shimadzu IRAffinity-1 в геометрии на пропускание в интервале волновых чисел 4000–400 см–1. При всех измерениях держатели образцов помещали в кюветное отделение спектрометра идентичным образом. Экспериментальные значения пропускания Т измеряли в процентах и пересчитывали на величины оптической плотности операцией логарифмирования как ln(100/T).
Торцевые срезы образцов изучены с помощью сканирующего электронного микроскопа Jeol JSM-7001F. Для подготовки торцевой поверхности часть образца зажимали металлической струбциной, после чего при помощи специального ножа выполнен срез строго перпендикулярно лицевой поверхности пленки. ПВДФ и его химически дегидрофторированные производные обладают высоким электросопротивлением, поэтому для уменьшения эффекта электростатической зарядки образцов на них методом магнетронного напыления наносили тонкий слой золота.
Дифрактограммы получены при помощи дифрактометра Ultima IV (Rigaku) в геометрии Брэгга–Брентано с использованием излучения CuKα1 (λ = 1.5405 Å).
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
В результате дегидрофторирования в ИК-спектрах всех исследуемых образцов появляются и растут с увеличением продолжительности реакции новые полосы поглощения, отсутствующие в спектре исходного полимера. Форма и интенсивность этих пиков изменяется при хранении. На рис. 1 представлены наиболее информативные участки спектров исходной пленки ПВДФ, а также образцов 2, 4, 6, измеренных через двое суток после синтеза. Спектры образцов 1, 3 и 5 не приведены, чтобы не усложнять рисунок.
Рис. 1.
ИК-спектры поливинилиденфторида (ПВДФ) и дегидрофторированных образцов 2, 4 и 6 через двое суток после синтеза в интервалах частот валентных колебаний С=С, С=О (а) и С–Н, О–Н (б) связей.
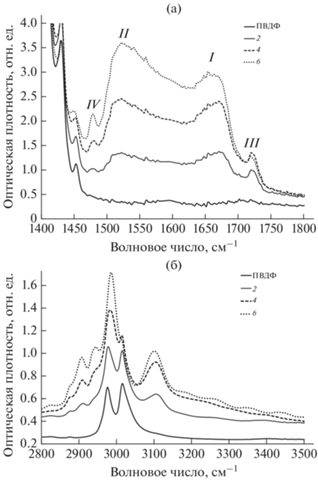
Из рис. 1 видно, что увеличение продолжительности дегидрофторирования приводит к увеличению площадей полос поглощения за счет валентных колебаний С=С, С=О и С–Н, О–Н связей в интервалах, соответственно, 1400–1800 (a) и 2800– 3500 см–1 (б). Полоса поглощения в первом интервале (рис. 1а), отсутствующая в спектре ПВДФ, имеет две узкие и две широкие визуально различимые особенности. Широкая линия I с центром вблизи 1670 см–1, предположительно, обусловлена валентными колебаниями двойных углерод–углеродных связей в коротких кумуленовых фрагментах углеродных цепей [16] и с увеличением продолжительности дегидрофторирования растет немонотонно. В спектре образца 3 она несколько выше, чем в спектре образца 4, а в спектре образца 5 она несколько выше таковой в спектре образца 6. Широкая особенность II с центром ~1520 см–1, вероятно, связана с валентными колебаниями двойных углерод–углеродных связей во фторзамещенных полиеновых участках цепи. С увеличением продолжительности дегидрофторирования эта особенность растет монотонно, обусловливая общий рост площади обсуждаемой полосы поглощения. Можно полностью исключить даже слабое влияние на форму и интенсивность спектра в области волновых чисел 1400–1800 см–1 гидрофторида калия (KHF2) [22], поскольку его другие характерные пики спустя двое суток после синтеза еще не были обнаружены. Узкий пик III небольшой интенсивности с центром около 1720 см–1 соответствует колебаниям карбонильных связей; с увеличением продолжительности ДГФ от 10 до 20 мин он заметно возрастает, затем стабилизируется. Представляет интерес узкая слабая, но монотонно возрастающая при дегидрофторировании линия IV при ~1480 см–1, отсутствующая в спектре ПВДФ. Можно предположить, что ее обусловливают валентные колебания двойных углерод–углеродных связей, к одному из атомов которой прикреплена некоторая массивная молекулярная группа, например, этоксигруппа.
Появление этоксигрупп в молекулярной структуре образцов и рост их концентрации при увеличении продолжительности дегидрофторирования происходит по следующей схеме:
Наличие этоксигрупп проявляется в ИК-спектрах в виде четырех характерных особенностей, соответствующих симметричным sCH2, sCH3 и асимметричным aCH2, aCH3 валентным колебаниям [18] (рис. 1б). Из этого же рис. заметно, что с ростом продолжительности дегидрофторирования ослабляется вклад в спектр С–Н-дублета исходного ПВДФ и увеличивается особенность с центром около 3100 см–1, которая, скорее всего, связана с валентными колебаниями =С–Н-связи при двойной углерод-углеродной связи [23] во фторзамещенных полиеновых фрагментах углеродной цепи. Таким образом, форма С–Н-полосы поглощения определяется вкладом, по меньшей мере, трех перечисленных компонент.
Для проведения компонентного анализа использовали программу Fityk [24]. Предварительно из спектров образцов и исходного ПВДФ в интервале 2850–3170 см–1 вычитали линейный фон. Спектр ПВДФ без фона использовали далее в качестве специальной функции, не входящей в пакет стандартных функций Fityk, для моделирования вклада участков полимера, не прореагировавших со смесью. Вклады четырех мод колебаний С–Н-связей этоксигрупп и вклад колебания С–Н-связи при двойной углерод-углеродной связи симулировали комбинацией пяти функций Гаусса. Результаты компонентного анализа представлены в табл. 1. Совпадение результатов такого моделирования с измеренным спектром существенно улучшилось при включении небольшого вклада еще одной кривой Гаусса с центром ~3014 см–1. Хотя происхождение этой компоненты (столбец “Пик Х”) остается неясным, ее площадь настолько мала, что не может существенно повлиять на площади остальных компонент.
Таблица 1.
Площади компонент CH-полосы поглощения и СF2-групп через двое суток после синтеза, а также толщина образцов, измеренная спустя один год методом РЭМ
| Образец | Время синтеза, мин | sCH2 | sCH3 | aCH2 | aCH3 | ПВДФ | Пик Х | –HC= | EtO | CF2 | C=C | Толщина, мкм |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 10 | 0.65 | 1.15 | 6.35 | 4.32 | 28.9 | 0.94 | 7.21 | 12.5 | 112 | 77.4 | 28.26 |
| 2 | 15 | 0.95 | 1.72 | 8.50 | 4.91 | 31.2 | 1.05 | 7.87 | 16.1 | 123 | 96.5 | 31.31 |
| 3 | 20 | 2.60 | 3.76 | 12.1 | 15.3 | 24.3 | 1.49 | 15.2 | 33.8 | 107 | 235 | 42.84 |
| 4 | 25 | 3.39 | 4.11 | 16.6 | 15.9 | 28.4 | 2.47 | 17.5 | 40.0 | 114 | 250 | 44.91 |
| 5 | 30 | 4.38 | 8.50 | 13.3 | 34.2 | 12.6 | 2.57 | 23.6 | 60.3 | 77.0 | 379 | 48.66 |
| 6 | 40 | 4.16 | 10.1 | 17.6 | 35.0 | 13.8 | 3.74 | 20.0 | 66.9 | 80.2 | 380 | 50.63 |
Из данных рис. 1 и табл. 1 следует, что увеличение продолжительности дегидрофторирования приводит к росту поглощения за счет увеличения вклада двойных углерод–углеродных связей, С–Н-связей этоксигрупп и колебаний с частотой ~3014 см–1 (столбцы “С=С”, “EtO” и “Пик Х”). Отметим, что в столбце “EtO” указана сумма возрастающих вкладов четырех отдельных колебательных мод этоксигрупп. Это возрастание происходит непропорционально друг другу. Подобную зависимость наблюдали и ранее [21], но ее причина до сих пор остается неясной. Самая высокочастотная компонента С–Н-полосы спектра (~3100 см–1) также в целом возрастает с увеличением продолжительности дегидрофторирования, но в образце 5 она проявляется сильнее, чем в образце 6. На рис. 2–4 на примере спектров образцов 2, 3 и 6 показано их разложение на компоненты.
Рис. 2.
Разложение на компоненты полосы поглощения С–Н связей в ИК-спектре образца 2 (15 мин дегидрофторирования). Сплошная, пунктирные и штриховая линии обозначают, соответственно, экспериментальный спектр, компоненты разложения и их сумму.
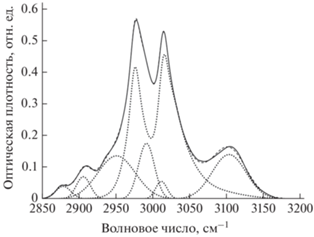
Рис. 3.
Разложение на компоненты полосы поглощения С–Н связей в ИК-спектре образца 3 (20 мин дегидрофторирования). Сплошная, пунктирные и штриховая линии обозначают, соответственно, экспериментальный спектр, компоненты разложения и их сумму.

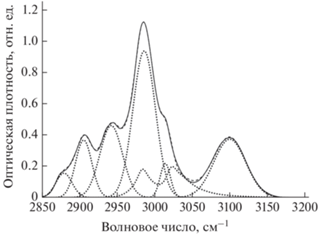
Рис. 4.
Разложение на компоненты полосы поглощения С–Н связей в ИК-спектре образца 6 (40 мин дегидрофторирования). Сплошная, пунктирные и штриховая линии обозначают, соответственно, экспериментальный спектр, компоненты разложения и их сумму.
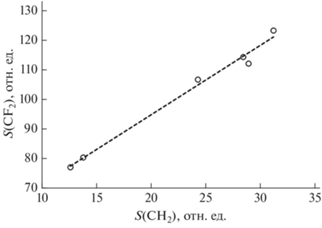
Кроме обсуждаемых выше спектральных особенностей, относящихся к химически модифицированной части объема образцов, в области спектра, соответствующей связям С–Н, проявляется дублет sCH2 и aCH2, характерный для исходного полимера. Ожидалось обнаружить монотонное убывание этого вклада в спектр с увеличением продолжительности дегидрофторирования. Однако, как видно из табл. 1 (столбец “ПВДФ”), это уменьшение происходит немонотонно. Скорее всего, такое поведение связано с несколько отличающимися толщинами пленок, из которых вырезали заготовки образцов. В результате объем не прореагировавшей со смесью части образца зависел не только от продолжительности реакции, но и от толщины заготовки. Поскольку, согласно модели химического дигидрофторирования [2] и экспериментальным данным [13, 16], в процессе реакции атомы фтора и водорода удаляются в равных количествах, между площадями СН2 дублета, характерного для ПВДФ, и CF2 полосы должна быть корреляция (соответственно, столбцы “ПВДФ” и “CF2” табл. 1). На рис. 5 графически представлена взаимосвязь обсуждаемых параметров. Корреляция, как видно из рисунка, действительно имеет место, что косвенно подтверждает предположение о влиянии на них толщины исходной заготовки.
Рис. 5.
Корреляция между интегральными величинами полос поглощения S, соответствующих СН2-дублету, характерному для поливинилиденфторидa (ПВДФ), и CF2, для всех исследованных образцов.
Перейдем к рассмотрению результатов старения образцов. Первые 36 сут после синтеза образцы большую часть времени хранились в вакуумной камере, их извлекали только на время измерений ИК-спектров. Затем образцы хранились в атмосфере воздуха. Спустя семь суток после синтеза в ИК-спектрах всех образцов были обнаружены новые особенности. Из них наиболее заметны линии поглощения при 1840 и 2040 см–1, причем и на этом, и на дальнейших этапах старения они тем интенсивнее, чем больше продолжительность дегидрофторирования. Измерения с вычитанием наклонных базовых линий показали пропорциональность площадей этих линий друг другу в спектрах всех образцов на всех этапах старения. Обе особенности характерны для ИК-спектра гидрофторида калия (KHF2) [22].
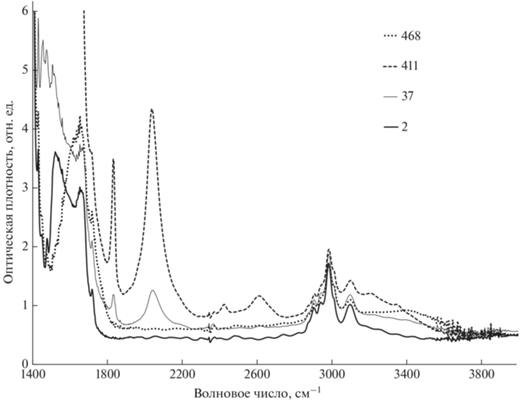
Спустя 411 сут после синтеза эти линии становятся еще интенсивнее и в спектрах образцов 3–6 доминируют в интервале 1800–4000 см–1. Отчетливо наблюдается еще один удвоенный пик существенно меньшей интенсивности, смещенный относительно первого приблизительно на 600 см–1 в высокочастотную область, также характерный для KHF2 [22]. В то же время в спектрах образцов 3–6 появляются слабые линии при 650 и 680 см–1, характерные для внеплоскостных либрационных колебаний тетрамеров воды. О появлении молекулярных кластеров воды также свидетельствует специфическая форма полосы поглощения гидроксильных групп, в которой присутствуют максимумы при 3200 и 3350 см–1. Кластеризация воды, скорее всего, связана с формированием мелкопористой структуры в объеме образца на данном этапе старения [18]. Заметно некоторое уменьшение поглощения этоксигруппами и =С–Н-связями при двойной углерод-углеродной связи. Мощный рост поглощения происходит в области ниже 1600 см–1, наиболее вероятной причиной которого является увеличение количества гидрофторида калия в исследуемых образцах.
Спустя 467 сут хранения образцов после синтеза часть образца 6 подверглась дополнительной промывке для удаления гидрофторида калия. Для этого пленку помещали в смесь, состоящую из ацетона и воды в соотношении объемов 1 : 4. После суточной выдержки в смеси все обсуждавшиеся выше особенности, появившиеся при старении, исчезают, толщина пленки уменьшается на 40%. Также полностью исчезает широкий пик в области ~1520 см–1. Форма полосы поглощения в частотном интервале 3100–3800 см–1 радикально изменяется в результате отсутствия вклада валентных колебаний О–Н-связей в кластерах воды. С другой стороны, спектры образца 6, зарегистрированные спустя двое суток после синтеза и после дополнительной промывки, как видно из рис. 6, существенно отличны друг от друга по форме. Это свидетельствует о том, что процесс старения заключается не только в накоплении внутри карбонизированного образца гидрофторида калия, но и в увеличении количества кумуленовых звеньев углеродных цепей. Эволюция формы ИК-спектров образца 6 при старении и после дополнительной промывки представлена на рис. 6.
Рис. 6.
Эволюция формы ИК-спектров образца 6 при старении и после дополнительной промывки в смеси ацетона и воды. Числа в верхней правой части рисунка соответствуют продолжительности старения в сутках.
Метод сканирующей электронной микроскопии показал, что увеличение продолжительности дегидрофторирования увеличивает толщину образцов (рис. 7; табл. 1, колонка “Толщина”). В ранних исследованиях при использовании для промывки ацетоном такого эффекта не наблюдали. Этот факт свидетельствует о том, что побочные продукты дегидрофторирования при промывке этанолом не удаляются и препятствуют усадке карбонизированного полимера. Дополнительная промывка образца 6, дегидрофторированного в течение 40 мин, проведенная в смеси воды и ацетона через 467 суток хранения после синтеза, привела к уменьшению его толщины с 50.6 до 29.0 мкм.
Рис. 7.
Изображения, полученные методом растровой электронной микроскопии торцевого среза образцов 1–6, соответственно, (а)–(е).

Интересным представляется формирование существенно неоднородной и пористой структуры в срединной части образца 6, что хорошо заметно на рис. 7е. Можно с достаточной уверенностью предположить, что именно увеличение пористости при старении пленок и накопление в мелких порах кластеров воды, а в крупных – гидрофторида калия является основной причиной увеличения их толщины. Из этого же рис. 7 видно существование более однородной “корки” небольшой и приблизительно одинаковой толщины с обеих сторон торцевой поверхности. Поскольку дегидрофторирующая смесь проникает в глубину пленки постепенно, то, скорее всего, в этих поверхностных областях пленки дегидрофторирование прошло наиболее полно. Поэтому остаточное количество калия и фтора в них оказывается недостаточным для формирования гидрофторида калия.
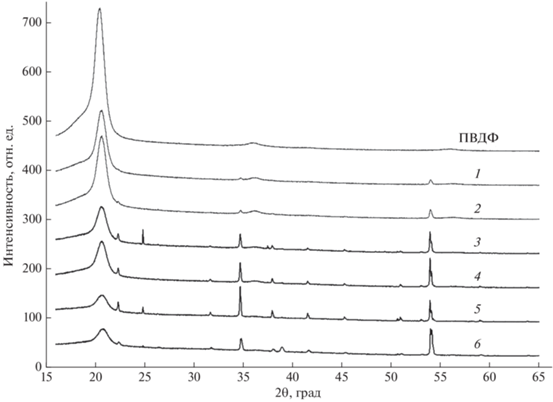
На рис. 8 представлены спектры рентгеновской дифракции исходного ПВДФ и образцов 1–6. Из рис. видно, что в результате дегидрофторирования интенсивность дифракционных максимумов, относящихся к кристаллической фазе ПВДФ [25–27] уменьшается. В карбонизированных образцах обнаруживаются рефлексы от кристаллов KHF2 [28], а также небольшой рефлекс при 2θ = = 39°, соответствующий кристаллу NaF [29, 30]. Вероятнее всего, Na входит в состав стекла посуды, использованной для синтеза образцов, а молекулы HF, образующиеся в результате реакции дегидрофторирования, разрушают стекло с образованием фтористого натрия. В результате дополнительной промывки образца 6 через 467 сут после синтеза в смеси ацетона и дистиллированной воды практически все рефлексы, не относящиеся к ПВДФ, исчезают, что хорошо согласуется с существенным уменьшением его толщины.
ЗАКЛЮЧЕНИЕ И ВЫВОДЫ
Синтезирована серия образцов химически карбонизированных производных поливинилиденфторида марки Ф-2МЭ, отличающихся продолжительностью синтеза от 10 до 40 мин. При первичной промывке образцов этой серии не использовали ацетон. Это позволило сохранить в объеме материала, а затем идентифицировать методами ИК-спектроскопии и рентгеновской дифракции гидрофторид калия (KHF2), образующийся и накапливающийся в результате побочных реакций в течение длительного времени после синтеза при хранении образцов в атмосфере воздуха. Проведенные измерения показали, что и форма, и интенсивность спектров поглощения образца 6 (синтезированного в течение 40 мин), зарегистрированных спустя двое суток после синтеза и после дополнительной промывки, в интервале, соответствующем колебаниям CF2-групп, практически идентичны. Следовательно, источником фтора для накопления гидрофторида калия при хранении не могут являться остаточные CF2-группы. Роль компонентов атмосферного воздуха в этом процессе, которые способны влиять на скорость его протекания, также не выяснена, это требует проведения дополнительных исследований.
Проведен компонентный анализ полосы поглощения валентных колебаний С–Н-связей. Установлено, что при увеличении продолжительности дегидрофторирования возрастает вклад этоксигрупп и С–Н-связей при двойных углерод–углеродных связях. Величина интегральной интенсивности С–Н-дублета той части объема образца поливинилиденфторида, которая не прореагировала со смесью, изменяется немонотонно, что связано с несколько отличающимися толщинами пленок-прекурсоров.
Методом растровой электронной микроскопии выявлена монотонная зависимость толщины образцов от продолжительности дегидрофторирования. Это свидетельствует о том, что кристаллы гидрофторида калия препятствуют усадке образцов после синтеза. Дополнительная промывка образца, дегидрофторированного в течение 40 мин, проведенная в смеси воды и ацетона через 467 сут хранения после синтеза, привела к уменьшению его толщины на 40%, исчезновению рентгеновских дифракционных рефлексов и всех полос ИК-поглощения, характерных для гидрофторида калия.
Список литературы
Шахова И.В., Беленков Е.А. // ФТТ. 2011. Т. 53. Вып. 11. С. 2265.
Heimann R.B., Evsyukov S.E., Kavan L. Carbyne and carbynoid structures. Dordrecht: Kluwer Academic Publishers, 1999. 446 p.
Шумилова Т.Г., Данилова Ю.В., Горбунов М.В., Исаенко С.И. // Доклады Академии наук. 2011. Т. 436. Вып. 3. С. 394.
Zatsepin A.F., Buntov E.A., Zatsepin D.A., Boqizoda D.A., Guseva M.B., Vyatkina S.P., Kas’yanova A.V. // Phys. Atomic Nuclei. 2018. V. 81. Iss. 11. P. 1660. https://doi.org/10.1134/S1063778818110224
Bettini L.G., Foglia D.F., Piseri P., Milani P. // Nanotechnology. 2016. V. 27(11). P. 115403. https://doi.org/10.1088/0957-4484/27/11/115403
Ma C.R., Xiao J., Yang G.W. // J. Materials Chem. C. 2016. V. 4. P. 4692. https://doi.org/10.1039/C6TC00648E
Casari C.S., Milani A. // MRS Comm. 2018. V. 8. Iss. 2. P. 207. https://doi.org/10.1557/mrc.2018.48
Krishnamoorthy K., Mariappan V.K., Pazhamalai P., Sahoo S., Kim S.-J. // Nano Energy. 2019. V. 59. P. 453. https://doi.org/10.1016/j.nanoen.2019.02.041
Pan B., Xiao J., Li J., Liu P., Wang Ch., Yang G. // Sci. Adv. 2015. V. 1. Iss. 9. P. 1500857. https://doi.org/10.1126/sciadv.1500857
Кудрявцев Ю.П., Евсюков С.Е., Бабаев В.Г. // Изв. АН СССР: Сер. хим. 1992. Вып. 5. С. 1223.
Vointseva I.I., Gil’man L.M., Kudryavtsev Yu.P., Evsyukov S.E., Pesin L.A., Gribov I.V., Moskvina N.A., Khvostov V.V. // Europ. Polymer J. 1996. V. 32. № 1. P. 61. https://doi.org/10.1016/0014-3057(95)00104-2
Евсюков С.Е., Кудрявцев Ю.П., Коршак Ю.В. // Успехи химии. 1991. Т. 60. № 4. С. 764.
Волегов А.А., Песин Л.А., Маргамов И.Г., Евсюков С.Е., Корякова О.В., Кочедыков В.А. // Известия Челябинского научного центра. 2006. Т. 34. Вып. 4. С. 26.
Мавринская Н.А., Песин Л.А., Баумгартен М., Байтингер Е.М., Мавринский А.В., Евсюков С.Е. // Вестник Южно-Уральского государственного университета. 2008. Т. 107. № 7. С. 80.
Мавринская Н.А., Мавринский А.В., Баумгартен М., Байтингер Е.М., Евсюков С.Е., Песин Л.А. // Вестник Южно-Уральского государственного университета. 2008. Т. 122. № 22. С. 88.
Zhivulin V.E., Chernov V.M., Osipov A.A., Shtenberg M.V., Evsyukov S.E., Pesin L.A. // Phys. Solid State. 2017. V. 59. № 7. P. 1414. https://doi.org/10.1134/S1063783417070332
Ferroelectric polymers: chemistry, physics and applications / Ed. Nalwa H.S. N.Y.: Marcel Dekker Inc., 1995. 912 p.
Zhivulin V.E., Khairanov R.Kh., Zlobina N.A., Doroshenko O.P., Evsyukov S.E., Doroshenko I.Yu., Pesin L.A. // Results Mater. 2021. V. 9. P. 100163. https://doi.org/10.1016/j.rinma.2020.100163
Zhivulin V.E., Pesin L.A., Belenkov E.A., Greshnyakov V.A., Zlobina N., Brzhezinskaya M. // Polym. Degrad. Stab. 2020. V. 172. P. 109059. https://doi.org/10.1016/j.polymdegradstab.2019.109059
Zhivulin V.E., Khairanov R.Kh., Zlobina N.A., Pesin L.A. // J. Surf. Invest.: X-Ray, Synchrotron Neutron Tech. 2020. V. 14. № 6. P. 1144. https://doi.org/10.1134/S1027451020060178
Zhivulin V.E., Zlobina N.A., Evsyukov S.E., Pesin L.A. // J. Surf. Invest.: X-Ray, Synchrotron Neutron Tech. 2021. V. 15. № 1. P. 12. https://doi.org/10.1134/S1027451021010171
Spectra Base (2021), John Wiley & Sons, Inc. https://spectrabase.com/spectrum/7ZyNYK5kxKE. Cited December 2021.
Socrates G. Infrared and Raman Characteristic Group Frequencies: Tables and Charts. 3rd Edition. Wiley, 2004. 364 p.
Wojdyr M. // J. Appl. Cryst. 2010. V. 43. P. 1126. https://doi.org/10.1107/S0021889810030499
Кочервинский В.В. // Успехи химии. 1996. Т. 65. С. 936.
Weinhold S., Litt M., Lando J., Takahashi Y., Tadokoro H. // Macromolecules. 1980. V. 13. P. 1317.
Hasegawa R., Takahashi, Y., Chatani Y., Tadokoro H. // Polymer J. 1972. V. 3. № 5. P. 600. https://doi.org/10.1295/POLYMJ.3.600
Pierrard A., Gredin P., de Kozak A. // Powder Diffraction. 1996. V. 11. P. 121.
Sueyoshi H., Yamada K., Miyazaki M., Okada T., Ashie N., Kousaka Y. // International J. Nonferrous Metallurgy. 2018. V. 7. № 1. P. 1. https://doi.org/10.4236/ijnm.2018.71001
McMurdie H.F., Morris M.C., Evans E.H., Paretzkin B., Wong-Ng W., Ettlinger L., Hubbard C. // Powder Diffraction. 1986. V. 1. P. 64.
Дополнительные материалы отсутствуют.
Инструменты
Поверхность. Рентгеновские, синхротронные и нейтронные исследования