

Прикладная биохимия и микробиология, 2019, T. 55, № 2, стр. 107-123
Связывание эритроцитарного гемоглобина с мембраной как способ осуществления сигнально-регуляторной функции (обзор)
О. В. Космачевская 1, Э. И. Насыбуллина 1, В. Н. Блиндарь 2, А. Ф. Топунов 1, *
1 Институт биохимии им. А.Н. Баха, Федеральный исследовательский центр
“Фундаментальные основы биотехнологии” Российской академии наук
119071 Москва, Россия
2 Национальный медицинский исследовательский центр онкологии им. Н.Н. Блохина Минздрава России
115478 Москва, Россия
* E-mail: aftopunov@yandex.ru
Поступила в редакцию 3.09.2018
После доработки 18.09.2018
Принята к публикации 25.09.2018
Аннотация
Обратимое связывание белков с компонентами мембраны и цитоскелета является одним из механизмов управления клеточным метаболизмом. Этот механизм имеет огромное значение для регуляции метаболизма в безъядерных клетках – эритроцитах млекопитающих, где он реализуется за счет перехода гемоглобина в мембраносвязанное состояние. Взаимодействовать с мембранами Hb может в разных лигандных и окислительно-восстановительных состояниях, что указывает на функционирование этого белка в качестве датчика редокс- и кислородных условий. Через взаимодействие с основным интегральным белком мембраны эритроцита − белком полосы 3 дезоксигенированный Hb в зависимости от кислородных условий изменяет энергетический обмен, морфологию и деформируемость эритроцитов, высвобождение регуляторов сосудистого тонуса − NO и ATP. Сигнальную функцию выполняют также и продукты окислительной денатурации Hb – необратимые гемихромы. Накапливаясь со временем или в результате окислительного стресса, гемихромы несут информацию о редокс-условиях и продолжительности функционирования эритроцита. Высказывается гипотеза о существовании в эритроцитах программы, запускающей внутрисосудистый гемолиз. Обсуждается участие гемоглобина и его мембраносвязанной формы (MBHb) в реализации этой программы. Также обсуждается роль доноров оксида азота в регуляции стабильности эритроцитов. Предложено использовать данные о содержании MBHb в эритроцитах в медицинской диагностике.
За последние два десятилетия представления о функциональных свойствах эритроцитарного гемоглобина (Hb) существенно расширились. Стало ясно, что Hb не только переносчик кислорода, но еще и регулятор его доставки к органам и тканям. Эту функцию Hb осуществляет несколькими способами: 1) через регуляцию механических свойств мембраны эритроцита; 2) через синтез и депонирование NO в эритроците; 3) через высвобождение ATФ из эритроцита. Во всех случаях сигнально-регуляторное действие опосредованно изменениями четвертичной структуры Hb (конформационными R-T-переходами), которые модулируются парциальным давлением кислорода (pO2). Именно благодаря тетрамерной организации Hb и наличию у него аллостерических свойств возможна тонкая настройка как кислородсвязывающих свойств Hb, так и его сигнальных функций. Отметим, что гемоглобин также регулирует энергетический обмен, объем, деформируемость и продолжительность жизни эритроцита.
Поскольку эритроцит млекопитающих представляет собой высокоспециализированную и редуцированную клетку, где отсутствуют ядро, митохондрии и белоксинтезирующий аппарат, то его основной информационной платформой является плазматическая мембрана и связанный с ней примембранный цитоскелет. Связывание Hb с компонентами мембраны формирует основу его сигнально-регуляторной функции.
Обзор посвящен обобщению современных представлениям о путях взаимодействия Hb с мембраной и о физиологическом значении этого процесса для функционирования эритроцита и его участия в поддержании гомеостаза на уровне целого организма.
Обратимое связывание белков на мембране как способ регуляции метаболизма. Одним из механизмов регуляции метаболизма клетки является обратимый переход белков из растворимого в мембраносвязанное состояние. Это так называемый адсорбционный механизм регуляции [1, 2] или, согласно терминологии, предложенной А.С. Капрельянцем, − топодинамическая регуляция [3]. Обратимая адсорбция ферментов позволяет управлять их каталитической активностью и стабильностью. Идея о том, что обратимое связывание ферментов c субклеточными структурами имеет регуляторное значение, была впервые высказана А.И. Опариным в 1933 г. [4]. Как было показано в его работах, подобный способ регуляции активности ферментов работает и в гелеподобных структурах (коацерватах), что указывает на его эволюционную древность.
Как правило, связывание ферментов с мембранами приводит к снижению их активности и находится в прямой зависимости от энергетического заряда клетки: высокий уровень при низкой концентрации ATP и наоборот. Возможно, это позволяет клетке снизить расход энергии на постоянную каталитическую работу ферментов. Процесс сорбции-десорбции белков обладает высокой чувствительностью к низкоинтенсивным воздействиям. Благодаря этому процессу клетка может в течение нескольких секунд изменить метаболизм за счет ранее связанных ферментов, в то время как для реализации ответа через изменение экспрессии генов требуется гораздо больше времени.
Естественно, возникает вопрос о природе фактора, который является спусковым механизмом для перехода белков из связанного в свободное состояние. Согласно существующим представлениям, таким триггером являются конформационные изменения мембран и белков. Доказано существование взаимосвязи между конформацией белка и редокс-состоянием его SH-групп [5]. Тиоловые группы являются наиболее активными группами белков, способными в мягких физиологических условиях вступать в разнообразные химические реакции (окисление, нитрозилирование, гликирование, алкилирование, тиолирование и др.) [5, 6]. Многие из этих реакций обратимы и поэтому имеют важное значение для регуляции метаболизма клетки. Кроме того, тиол-дисульфидное равновесие чрезвычайно чувствительно к действию редокс-активных соединений [7].
Конформационные переходы в молекуле Hb могут быть вызваны изменением состояния гемовой группы при присоединении лигандов, а также изменением состояния поверхностных остатков цистеина (Cysβ93). Связывание (блокирование) активных тиольных групп меняет распределение электронной плотности на геме, в результате чего происходит ускорение автоокисления Hb, если оно не сопровождается значительными конформационными изменениями [8]. Например, образование супероксидного анион-радикала $\left( {{\text{O}}_{2}^{{ \bullet - }}} \right)$ происходит при переходе низкоспинового оксигенированного гемоглобина (oxyHb) в низкоспиновый окисленный met-hydroxyHb (FeIII–OH) или гемихром (His–FeIII–His). Окисленный гемоглобин, особенно его низкоспиновые формы, имеют повышенную склонность к связыванию с мембраной [9]. Таким образом, различные редокс-активные соединения, образующиеся экзогенно или эндогенно при нарушении метаболизма, могут влиять на перераспределение белка из растворимого в мембраносвязанное состояние.
Известно, что эритроциты обладают многоуровневой способностью к адаптации [10–12]. В отличие от “ядерных” клеток, адаптационные возможности эритроцитов млекопитающих ограничены цитоплазматическими механизмами, которые включают обратимые кооперативные фазовые переходы белков, взаимодействие их с мембраной и цитоскелетом, изменения в мембранных белках и белках цитоскелета, а также нарушение проницаемости мембраны. Поэтому эритроциты являются удобной моделью для изучения формирования адаптационного ответа на уровне цитоплазмы. В следующей главе будут рассмотрены основные пути формирования регуляторного сигнала в этих клетках благодаря связыванию Hb с мембраной.
Центры связывания гемоглобина с мембраной. Способность эритроцитарного гемоглобина к связыванию с мембраной была установлена достаточно давно, при попытке получить тени эритроцитов гипотоническим лизисом. Первое упоминание о содержании Hb в эритроцитарных мембранах можно найти в работе Хоффмана [13], в которой он был назван “структурно фиксированным гемоглобином”. В научной литературе устоялся термин мембраносвязанный гемоглобин (Membrane-Bound Hemoglobin – MBHb). Также к числу ранних исследований MBHb относятся работы Андерсона и Тернера [14, 15], в которых было показано, что отмытые мембраны эритроцитов содержат около 3% от всего гемоглобина клетки. Затем Фунг и Эйзингер [16, 17] подтвердили факт взаимодействия Hb с мембранами эритроцитов в физиологических условиях.
Среди способов связывания Hb с мембраной выявлены следующие: связывание deoxyHb по белку полосы 3 за счет электростатических взаимодействий [17–21], ковалентная пришивка к мембранным компонентам дисульфидными связями [22, 23], адсорбция к мембранным липидам с помощью гидрофобных взаимодействий. Также Hb может связываться с белками цитоскелета: спектрином [24, 25], гликофорином [26–28], актином и тубулином [29, 30].
Белок полосы 3 (Band3) – представитель семейства (SLC4) переносчиков (транспортеров) растворенных веществ [31], он является основным интегральным белком мембран (25% от всех мембранных белков). Мономерный белок полосы 3 состоит из трех доменов: домен, пронизывающий мембрану, который осуществляет обмен Cl−/${\text{CO}}_{2}^{ - };$ короткий C-концевой цитоплазматический домен и длинный N-концевой цитоплазматический домен. С-концевой цитоплазматический домен связывает карбоангидразу II, N-концевой цитоплазматический домен (Cytoplasmic Domain of Band 3 protein, CDB3) связывает гликолитические ферменты, Hb и гемихромы, образуя комплекс белка полосы 3. Одна из основных функций N-концевого домена заключается в связывании цитоскелетного каркаса с мембраной, что определяет морфологию и стабильность эритроцита. Комплекс белка полосы 3 осуществляет координацию мембранно-цитоскелетной структуры с энергетическим состоянием и кислородными условиями внутри клетки. Структура этого белка определяет антигенные детерминанты эритроцитарной мембраны; полиморфные варианты Band3 определяют группу крови Диего [32].
Band3 является центром организации мембраны эритроцитов и регулятором ионного гомеостаза, поэтому даже незначительные модификации в его регуляторных сайтах приводят к изменению структуры и функции этих клеток. Фосфорилирование тирозинов Band3 нарушает связывание Hb и белков цитоскелета [33]. Можно предположить, что и иные модификации в связывающем участке, например, нитрование тирозинов, могут приводить к нарушению кислородзависимой регуляции метаболических потоков в эритроците. CDB3 содержит реакционноспособные цистеины, участвующие в детекции редокс-состояния внутри клетки [34].
Связывание Hb с мембраной может быть обратимым и необратимым. С применением флуоресцентных зондов было показано, что deoxyHb с высоким сродством (в 8 раз превышающим сродство для oxyHb) обратимо связывается с CDB3 [18, 35–37]. Необратимым является ковалентное связывание Hb с компонентами мембраны вследствие действия окислительных агентов, вызывающих образование свободных радикалов, феррильных и оксоферрильных форм гемоглобина (гем–Fe(IV)=O) [38–41]. Чем выше уровень окисления гемоглобина (от FeIII до FeV), тем выше доля связанного Hb, при этом примерно 50% Hb связывается с помощью SH-групп [41]. Помимо Band3, гемоглобин также способен образовывать комплекс с цитоскелетным узлом, образованным спектрином, анкирином и белком полосы 4.2. Связанный таким образом окисленный metHb сохраняет окислительно-восстановительные свойства и способность к взаимодействию с лигандами [41].
С мембраной взаимодействует частично окисленный Hb [42], что, вероятно, имеет физиологическое значение, поскольку в примембранной области происходит его восстановление мембраносвязанной НАДФН-зависимой metHb-редуктазой [43].
Известна также способность гемоглобина образовывать комплексы с липидами, например, с холестерином [44], причем содержание аддуктов Hb с холестерином в зимний период выше, чем в летний (30% и 19% от общего Hb соответственно), что отражает уровень холестерина в липопротеидов высокой плотности (ЛПВП) в плазме.
Способы определения мембраносвязанного гемоглобина. В литературе содержатся противоречивые сведения о содержании MBHb в эритроцитах, что объясняется различным физиологическим состоянием эритроцитов и способом приготовления теней (рН, иона сила, количество отмывок).
В настоящее время концентрацию MBHb в эритроцитах чаще всего определяют спектрофотометрически, оценивая гемоглобин, ассоциированный с тенями эритроцитов [14, 45–51]. Нами был разработан простой спектрофотометрический метод определения MBHb в пробах крови, позволяющий проводить оценку концентрации Hb в рассеивающих средах [51]. Используются также ДДС-электрофорез белков теней в ПААГ с последующей денситометрией полосы Hb [53, 54]; измерение тушения гемоглобином флуоресценции зондов, встроенных в мембрану и специфически взаимодействующих с белком полосы 3 [17, 21]; люминол-активированная хемилюминесценция [20, 52]. Среди достоинств последнего метода следует отметить высокую чувствительность (до 0.01 пмоля MBHb в образце) и возможность проводить измерения в цельных эритроцитах. В табл. 1 суммирована имеющаяся по этому вопросу информация.
Таблица 1.
Содержание MBHb в тенях эритроцитов здоровых доноров по оценкам различных авторов
| Содержание MBHb, %* |
Метод определения | Источник |
|---|---|---|
| ≤2 | Спектрофотометрический цианметгемоглобиновый метод (метод Драбкина) | [45] |
| ~3 | Спектрофотометрический цианметгемоглобиновый метод (метод Драбкина) | [14] |
| 7−10 | Спектрофотометрический при длине волны 536 нм | [46] |
| 3.6 | » | [47] |
| 4.04 ± 0.54 | » | [48] |
| 4.43 ± 2.2 | » | [49] |
| 5.54 ± 0.32 | » | [50] |
| 4.53 ± 1.05 | Спектрофотометрическая оценка комплексов Hb с пиридином (метод Риггса) | [51] |
| 7−8 | Активированная хемилюминесценция | [52] |
По данным двумерного электрофореза содержание Hb в мембранах эритроцитов составило 1.740 ± 0.074% от общего белка мембран и было наименьшим, по сравнению с содержанием остальных мембранных белков [54]. Близкие значения (1.3 ± 0.3% от общего белка мембран) были получены в работе [41].
Быстрый и точный метод количественного определения связанного deoxyHb с CDB3 основан на измерении тушения гемоглобином флуоресценции зеленого флуоресцентного белка (eGFP), связанного с COOH-концом CDB3 [21]. Необычный способ анализа MBHb с помощью спектроскопии гигантского комбинационного рассеивания был предложен в работе [55]. Для этого эритроциты предварительно иммобилизовывали на покрытиях в виде кольцевых наноструктур серебра. Такой подход позволяет оценить изменения конформации и кислородсвязывающих свойств MBHb в неповрежденных эритроцитах.
МЕМБРАНОСВЯЗАННЫЙ ГЕМОГЛОБИН КАК НОСИТЕЛЬ БИОЛОГИЧЕСКОГО СИГНАЛА
Обратимый характер связывания Hb с мембраной указывает на физиологическую роль этого процесса. Рассмотрим физиологические и патофизиологические эффекты, обусловленные переходом Hb в мембраносвязанное состояние.
Аллостерическая регуляция CDB3. Гемоглобин связывается с белком полосы 3 за счет встраивания анионного сегмента CDB3 в катионную центральную полость, образованную β-цепями, которая одновременно является сайтом связывания 2.3-бифосфоглицерата (2.3-БФГ, 2.3-DPG) – аллостерического регулятора Hb. Такое взаимодействие позволяет рассматривать белок полосы 3 по отношению к deoxyHb как гетеротропного аллостерического эффектора, индуцирующего переход в Т-конформацию или стабилизирующего Т-конформацию [56]. Со своей стороны, Hb регулирует анион-транспортные свойства белка полосы 3: Na+/K+ – транспорт, K+–Cl− и Na+–K+–2Cl− – котранспорт [56].
Регуляция газообмена. Тесный контакт Hb и карбоангидразы с участием CDB3 позволяет скоординировать поглощение углекислого газа и выделение кислорода в капиллярах [57]: высвобождаемые при образовании бикарбоната протоны локализуются в области комплекса полосы 3, где они благодаря эффекту Бора способствуют дезоксигенации Hb.
Регулирующее воздействие на газообмен Hb может оказывать еще одним способом, изменяя структуру примембранного цитоскелета (субмембранного барьера) [58]. Усиление контакта цитоскелета с мембраной приводит к сокращению ячеек цитоскелетного каркаса, снижая диффузию газов. Этот механизм представляется вполне вероятным при регуляции поступления NO в эритроциты [42, 59, 60].
Регуляция энергетического метаболизма. Известно, что CDB3 связывает гликолитические ферменты: альдолазу, фосфофруктокиназу и глицеральдегид-3-фосфатдегидрогеназу [61]. Эти ферменты, а также пируваткиназа и лактатдегидрогеназа, образуют мультиферментный комплекс (метаболон) [62, 63]. Организация ферментов в метаболон обеспечивает более эффективное регулирование гликолитического пути. В связанном состоянии гликолитические ферменты, как правило, имеют низкую каталитическую активность [64]. Конформационные изменения в молекуле Hb при дезоксигенации приводят к повышению его сродства к CDB3, вытеснению ферментов гликолиза с сайта связывания и переходу их в растворимое активное состояние [19, 57, 65]. Таким образом, связывание deoxyHb с CDB3 переключает метаболизм с пентозофосфатного пути на гликолитический в ответ на изменение кислородного статуса клетки [57, 64, 66, 67]. С одной стороны, это способствует возрастанию уровня внутриклеточного ATP и, одновременно, 2,3-БФГ, а, с другой, уменьшает поток глюкозы, поступающей в пентозофосфатный путь, в котором образуется НАДФН, необходимый для восстановления metHb и антиоксидантных ферментов.
Органические фосфаты (ATP и 2.3-БФГ) являются звеном, связывающим энергетику клетки с функциональной активностью содержащихся в них молекул Hb. Специфически присоединяясь к Hb, они снижают его сродство к кислороду [68]. Это один из способов регуляции доставки кислорода в условиях гипоксии: чем ниже рО2, тем выше доля deoxyHb, тем выше концентрация ATP и 2.3‑DPG, и тем выше отдача О2 гемоглобином. При этом регуляция кислородсвязывающих свойств Hb за счет аллостерического механизма может быть очень быстрой, поскольку часть 2.3-БФГ (примерно 30%) находится в связанном с мембранами состоянии [68]. Таким образом, deoxyHb и 2.3-ДФГ могут конкурировать за сайты связывания с мембраной.
Переключение энергетического метаболизма на преимущественный синтез органических фосфатов имеет большое значение для стабилизации мембраны, для создания физиологически оптимальной формы эритроцита [69], а также для регуляции локального кровотока в условиях гипоксии [70–72]. Снижение содержания ATФ на 15% и более изменяет характер взаимодействия спектрина, актина и других интегральных белков эритроцитарной мембраны. Другой метаболит гликолиза – 2.3-БФГ, обратимо связываясь со спектрином, также оказывает влияние на свойства мембранного цитоскелета. Увеличение его концентрации повышает деформируемость мембраны.
Регуляция капиллярного кровотока. Описаны два взаимодополняющих механизма вовлечения эритроцитов в регуляцию капиллярного кровотока (локального внутрисосудистого давления): быстрое снижение вязкости крови за счет увеличения деформируемости эритроцитов и медленное, но устойчивое увеличение диаметра сосуда за счет выделения регуляторов сосудистого тонуса (NO и ATP) в его просвет. Последний механизм имеет три пути реализации. Это аллостерически регулируемое высвобождение NO с участием SH-групп β-субъединиц Hb (SNO-Hb) [73, 74], образование NO в реакции восстановления нитрит-ионов deoxyHb [75–77] и локальная пуринэргическая сигнализация с помощью ATP, высвободившегося из эритроцитов [72, 78, 79]. Все перечисленные механизмы реализуются в условиях пониженного рO2 и модулируются взаимодействием Hb с компонентами мембраны и цитоскелета.
Обсуждается участие гемоглобина в экспорте NO из эритроцитов. Акцептором NO-группы от SNO-Hb в мембране является CDB3 [80]. Взаимодействие Hb с CDB3 благоприятствует переходу белка в T-конформацию, что сопровождается переносом NO-группы в реакции транснитрозилирования от цистеинов β-цепей Hb на вицинальные остатки цистеина (Cys201, Cys317) CDB3 [80, 81], поэтому окисление тиолов CDB3 приводит к нарушению молекулярного механизма внутриклеточного переноса NO [80]. Связанный с CDB3 deoxyHb может также восстанавливать нитрит-ионы и, таким образом, генерировать NO на внутренней поверхности мембраны эритроцитов [82].
В 1992 г. появилась работа, в которой было показано участие Hb в гипоксическом выбросе ATФ из эритроцитов [78]. Предполагаемый механизм этого процесса включал связывание desoxyHb с CDB3, что приводило к активации G-белка и запуску каскада реакций сигнального пути, на заключительном этапе которого происходит высвобождение ATP через определенные каналы в плазматической мембране [79]. Этот эритроцитарный ATФ связывается с P2Y-пуринэргическими рецепторами на поверхности эндотелиальных клеток, активирует синтез NO и других сосудорасширяющих факторов, тем самым способствуя увеличению скорости капиллярного кровотока и эффективности доставки O2 к тканям. Вопрос о том, какие именно каналы участвуют в селективном транспорте АТФ, до сих пор не решен. Предположительно, это паннексиновые каналы (Panx1) и механочувствительные неселективные катионные каналы (Piezo 1) [83]. Экспериментальное подтверждение получила гипотеза о высвобождении АТФ из лизированных эритроцитов [70–72].
Наряду с гипоксией, выброс ATФ из эритроцитов может быть вызван их механической деформацией при прохождении через капилляры, закислением среды и избыточным количеством CO2 в крови [84]. Показано, что высвобождение ATФ из эритроцитов человека увеличивается в присутствии нитритов, проникающих аналогов цAMФ, активаторов цAMФ-опосредованной сигнальной системы (адреналин, изопротеренол, простациклин PGI2, форсколин, папаверин) и диметилсульфоксида [84].
В экспериментах in vitro и in vivo было показано, что увеличение синтеза и высвобождения ATФ в присутствии нитритов сопровождается снижением артериального давления [20], причем этот эффект наблюдался и в нормоксических, и в гипоксических эритроцитах, что указывает на его независимость от нитритредуктазной реакции. Также было показано, что в эритроцитах, обработанных физиологическими концентрациями NaNO2, возрастало содержание MBHb (9.9 ± 0.2 мкМ по сравнению с 2.0 ± 0.6 мкМ в контроле). Совокупность этих фактов позволила авторам работы [20] предположить участие гемоглобина в нитрит-зависимом высвобождении ATФ. В какой именно форме Hb связывается с мембраной в данном случае, пока не известно. Предположительно, это окисленные низкоспиновые формы белка, которые образуются в реакции deoxy- и oxyHb с ${\text{NO}}_{{\text{2}}}^{ - }{\text{:}}$ metHb–NO2 и бис-гистидин–metHb [9]. Обсуждается возможность формирования при участии CDB3 внутриэритроцитарного пула ATФ, связанного с мембраной, который высвобождается в условиях гипоксии, когда активируется путь передачи сигнала для выхода ATФ.
Индукция перераспределения Hb из растворимого в мембраносвязанное состояние нитритом натрия наблюдалась и в наших экспериментах с изолированными эритроцитами [51], причем нитрозотиолы, в концентрациях на порядок превышающих концентрацию нитрит-ионов, не приводили к образованию дополнительного MBHb. Это согласуется с результатами работы [41], в которой было показано, что инкубация эритроцитов с нитритом натрия приводила к 10-кратному увеличению MBHb.
Регуляция деформируемости эритроцитов. Снабжение тканей кислородом зависит не только от свойств Hb, но и от реологических характеристик мембраны эритроцитов. Высокая деформируемость этих клеток играет важную роль в обеспечении микроциркуляции, поскольку позволяет клеткам проходить через сосуды с диаметром меньше их собственного размера [85]. Было показано, что комплекс Hb-CDB3 принимает участие в перестройке цитоскелета в микроциркуляторном русле в зависимости от кислородных условий [21, 86, 87]. Связывание deoxyHb с CDB3 в непосредственной близости от сайтов связывания анкиринов, соединяющих Band3 и цитоскелет, приводит к вытеснению анкирина из комплекса с Band3 [21, 86]. Благодаря этому в течение периода дезоксигенации происходит ослабление мембранно-цитоскелетных взаимодействий, что значительно повышает деформируемость эритроцитов. Наоборот, повышение pO2 способствует усилению взаимодействия анкиринов с Band3, что стабилизирует мембрану эритроцитов во время их движения в турбулентном потоке из легких в капилляры.
Лабилизация цитоскелета в периоды длительной дезоксигенации (ишемия, гипоксия) может приводить к внутрисосудистому образованию везикул и гемолизу. Существует точка зрения, что этот неблагоприятный на первый взгляд процесс может быть физиологически оправданным. Данный вопрос будет рассмотрен ниже.
Индукция внутрикапиллярного окислительного стресса. Различные формы окисленного Hb (metHb, ferrylHb, oxoferrylHb) и продукты его окислительной денатурации (необратимые гемихромы) и деградации (гем и двухвалентное железо) могут прочно связываться с компонентами мембраны и быть источником активных форм кислорода (АФК), инициирующих перекисное окисление липидов (ПОЛ) [39–41, 52, 88–93]. Поскольку примембранная область практически недоступна для цитозольных антиоксидантных ферментов, преципитация Hb к мембране эритроцитов может только усилить процесс свободнорадикального окисления липидов и спровоцировать гемолиз.
Источником АФК может также быть и oxyHb в условиях гипоксии. Если в обычных условиях Hb отдает тканям около одной трети связанного О2, то при пониженном pO2 гемоглобин достигает состояния полунасыщения, при котором он легко окисляется и теряет стабильность. Интенсифицировать процесс автоокисления также может вызванный гипоксией глубокий ацидоз. В гипоксических эритроцитах благодаря высокому уровню deoxyHb возрастает содержание Hb в примембранной области, что уменьшает вклад гексозомонофосфатного шунта в энергетику клетки и в синтез НАДФН. Это может усиливать негативные эффекты АФК.
Описанными выше процессами генерации АФК в эритроцитах объясняется индукция воспалительного ответа в условиях гипоксии [94]. Образуемый эритроцитами H2O2 выходит в просвет капилляра, диффундирует до микрососудистого эндотелия, где запускает Ca2+-зависимый рекрутинг лейкоцитов. Возможность осуществления такого сценария in vivo была продемонстрирована на легких крыс [94]. Таким образом, Hb является триггером развития воспалительных нарушений, таких, как острое повреждение легких или сердечно-сосудистые заболевания с низким сердечным выбросом.
Редокс сигнализация. Увеличение доли связанных с мембраной окисленных форм Hb не всегда имеет негативные последствия для клетки и в некоторых случаях может быть физиологически детерминированным процессом, направленным на удаление старых, поврежденных и инфицированных эритроцитов из кровотока [95, 96]. Функционально неполноценные эритроциты имеют измененные антигенные детерминанты, благодаря чему они узнаются фагоцитами и макрофагами и уничтожаются.
Предложено несколько гипотез, объясняющих роль Hb в формировании “сигнала смерти”. Согласно одной из них, сополимеризация гемихромов с CDB3 и/или спектрином способствует агрегации Band3 и экспонированию антигена старения, с которыми связываются циркулирующие автоантитела (IgG), образуя сайты опсонизации [96–99]. Предполагается, что эритроциты распознаются макрофагами, как старые, в том случае, если размеры кластеров превышают некую пороговую величину. В норме примерно 0.1% эритроцитов содержат агрегированный Band3 [100]. Необходимым условием агрегации Band3 является сочетанное действие перекисного окисления мембранных липидов и повышенные уровни гемихромов [100].
Гемихромы связываются с цитоплазматической мембраной по двум сайтам: высокоаффинному (на CDB3) и низкоаффинному (на спектрине) [97]. Связывание с CDB3 обнаруживает следующую стехиометрию: на один димер белка полосы 3 приходится 2.5 тетрамерных гемихрома. С помощью гемихромов эритроциты могут “чувствовать” окислительный стресс (редокс-состояние) [37, 96].
Рассмотрим последовательность событий, приводящую к формированию сигнала о редокс-условиях внутри клетки. Начальным событием является обратимое окисление близкорасположенных остатков цистеина (Cys201 и Cys317) в Band3 с образованием межмолекулярной дисульфидной связи (ковалентно-сшитый димер Band3) [81]. Образование дисульфидной связи также происходит под действием гемихромов. Такая сшивка вызывает конформационные изменения в Band3, что делает возможным специфическое взаимодействие с Syk-киназой, фосфорилирующей остатки тирозина в цитоплазматическом домене CDB3 [37, 101]. Вызванное гемихромами гиперфосфорилирование Band3 снижает его сродство к анкирину и способствует диссоциации от спектрин-актинового цитоскелета [37, 101], что приводит к нарушению структурной целостности мембраны и, как следствие, к внутрисосудистому гемолизу. Напротив, обратимое фосфорилирование не вызывает лизис эритроцитов.
Гемихромы также индуцируют фосфорилирование остатков серина белков цитоскелета. По-видимому, это явление играет ключевую роль в высвобождении мембранных микрочастиц (экзовезикулообразование), нагруженных гемихромами [102]. В больших количествах такие частицы содержатся в крови больных талассемией [101]. В этом случае процесс образования везикул физиологически оправдан, поскольку направлен на удаление поврежденных мембранных фрагментов, тем самым сохраняя вполне жизнеспособные эритроциты [102].
До наступления физиологического старения эритроциты могут подвергаться повреждениям, которые нарушают их целостность и, таким образом, запускают программу их гибели – эриптоз. Эритроциты проявляют следующие признаки апоптоза: сморщивание клетки, везикулообразование, перераспределение липидов в мембране, которое приводит к экспонированию фосфатидилсерина на ее поверхности [103]. Фосфатидилсерин узнается макрофагами, утилизирующими такие эритроциты. Описано несколько механизмов эриптоза, которые могут запускаться одновременно. Один из таких механизмов может реализоваться при участии окисленных форм Hb – гемихромов, специфически связывающихся с Band3.
Усиленный эриптоз происходит при сахарном диабете, злокачественных новообразованиях, сердечной и почечной недостаточности, анемиях различного генеза, гемолитическом уремическом синдроме, сепсисе, микоплазменной инфекции [104]. Благодаря индукции эриптоза эритроциты, инфицированные малярийным плазмодием (Plasmodium falciparum), быстро удаляются из кровотока, тем самым защищая весь организм от распространения инфекции [56, 104].
Несмотря на то, что в целом эриптоз является полезным физиологическим механизмом, его чрезмерная активация может стать причиной обширного внутрисосудистого гемолиза. Стимуляция эриптоза при злокачественных опухолях дополнительно усугубляется цитостатическим лечением, что способствует возникновению анемии [105]. Для снижения анемии у онкологических больных целесообразно применение ингибиторов эриптоза, одним из которых является оксид азота [106]. Разработка различных NO-донорных соединений для коррекции нарушений в системе микроциркуляции является перспективным направлением в фармакологии. В частности, в качестве таких физиологических носителей NO-группы могут выступать динитрозильные комплексы железа (ДНКЖ, см. ниже).
Регуляция объема клетки. В нескольких работах было исследовано участие гемоглобина в регуляции объема клетки. Возможны два механизма такой регуляции. Согласно первому, Hb участвует в детекции объема посредством эффекта краудинга (неспецифического влияния скученности макромолекул) [107]. Концентрирование или разбавление внутриклеточного раствора Hb в результате флуктуации объема клетки может повлиять на активность протеинкиназ и фосфатаз, участвующих в регуляции объема. Альтернативный механизм состоит в том, что связывание Hb с мембраной активирует K+–Cl−-котранспортер, участвующий в регуляции клеточного объема [108]. В гипер- и гипотонической средах Hb снижает транспорт K+и Cl−. Оксигенация стимулирует перенос ионов (через активацию K+–Cl−-котранспортера), вызывая набухание, тогда как дезоксигенация вызывает сморщивание (через активацию каналов Na+–K+–2Cl− и Na+/H+) [109]. Набухание клеток, являясь аналогом напряжения сдвига, приводит к открытию механо-чувствительных катионных каналов, проницаемых для Ca2+, что запускает Ca2+-зависимые сигнальные каскады, регулирующие перестройку цитоскелета, белки которого имеют сайты связывания Ca2+ [110].
Регуляция заряда мембраны эритроцитов. Предложено несколько механизмов, объясняющих участие Hb в регуляции электрического поля мембраны эритроцитов [111]. Один из них состоит в адсорбции-десорбции Hb на мембране. Дезоксигемоглобин образует на мембране сплошной слой такой толщины, что он компенсирует примерно половину мембранных зарядов, тем самым вдвое ослабляя трансмембранную разность потенциалов. Присоединение кислорода к Hb переводит его в R-состояние, в котором он десорбируется с мембраны и электростатическое поле в ней возрастает. Второй механизм состоит в том, что протоны, высвободившиеся благодаря усилению кислотных свойств Hb при оксигенации, выходят из эритроцита, увеличивая отрицательный заряд цитоплазмы и, тем самым, увеличивая поле в мембране. Все это приводит к уменьшению проницаемости мембраны.
СТАБИЛИЗИРУЮЩЕЕ ДЕЙСТВИЕ ГЕМОГЛОБИНА НА ЭРИТРОЦИТАРНЫЕ МЕМБРАНЫ
Еще в 70 гг. прошлого столетия было показано стабилизирующее действие примембранного слоя гемоглобина на эритроциты, которое проявляется в снижении уровня гемолиза при механическом воздействии [112, 113]. Эксперименты с тенями эритроцитов показали, что разрушение липопротеиновой структуры эритроцитарных мембран с высоким содержанием Hb происходило в гораздо меньшей степени по сравнению с мембранами с малым содержанием Hb. Изучение молекулярных механизмов этого эффекта показало, что стабилизирующее действие на мембраны оказывают oxyHb и metHb, в то время как продукты его окисления и денатурации (обратимые, необратимые гемихромы и тельца Гейнца), накапливающиеся под действием окислительного стресса или в процессе старения эритроцита, нарушают взаимодействие мембраны с цитоскелетом [114]. Заметное стабилизирующее действие на примембранный цитоскелет связано с тем, что oxyHb и metHb способствуют самоорганизации спектринового димера в тетрамер [114, 115]. Константа связывания oxyHb со спектрином на два порядка превышает таковую для CDB3.
Деструктивное действие окисленных форм Hb обусловлено появлением гемина. Было показано, что гемин является мощным гемолитическим агентом, который неспецифически нарушает белок-белковые и белок-липидные взаимодействия [116, 117]. В эритроците гемин ослабляет межмолекулярные взаимодействия внутри комплекса спектрин-белок 4.1-актин [114]. Источником гемина являются гемихромы. По мере усиления окислительного стресса гемихромы образуют стабильные агрегаты − тельца Гейнца, которые прикрепляются к мембране эритроцитов и при прижизненном окрашивании метилвиолетом выявляются в виде фиолетовых включений. Этот процесс сопровождается повышением проницаемости для ионов калия, перекисным окислением липидов, сшиванием мембранных белков и снижением деформируемости клетки [114]. ПОЛ, индуцированное как самим Hb, так и продуктами его распада (железом и гемовой группой), также может вносить вклад в процесс разрушения эритроцитов [88, 91].
Таким образом, Hb оказывает разнонаправленное действие на устойчивость клетки: при незначительном и обратимом окислении – стабилизирующее, при глубоком окислении – дестабилизирующее (рис. 1). По мере окисления Hb, стабилизирующий эффект на цитоскелет постепенно исчезает, а негативный – возрастает. Представленная на рисунке кривая зависимости стабильности эритроцитов от уровня окислительного стресса является графическим выражением эффекта гормезиса [118]. Гормезис описывается U-образной или перевернутой U-образной кривой, показывающей изменение знака биологического эффекта при возрастании дозы химического вещества [118, 119]. Такая двухфазная кривая описывает компенсаторно-приспособительную реакцию живой системы.
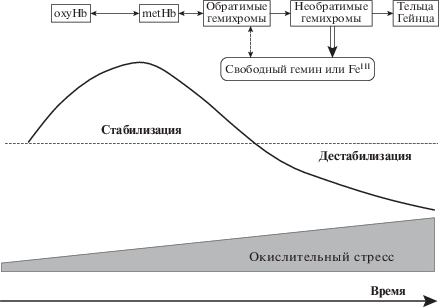
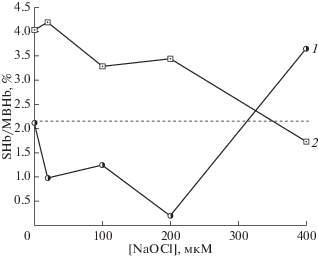
Фазное изменение уровня MBHb нами наблюдалось при обработке изолированных эритроцитов человека возрастающими дозами различных редокс-активных веществ (метилглиоксаль, гипохлорит, пероксид трет-бутил, нитрит натрия) [51, 120], причем повышенные уровни MBHb коррелировали с повышенной устойчивостью к окислительному гемолизу. На рис. 2 представлены две кривые, характеризующие изменение уровня MBHb и гемолитической устойчивости эритроцитов в зависимости от концентрации гипохлорита (NaOCl). Видно, что между двумя параметрами существует взаимосвязь. На графике можно выделить диапазон, которому соответствует повышение устойчивости клетки, который еще называют зоной гормезиса. Такие зависимости были получены и для других веществ.
Рис. 2.
Взаимосвязь между количеством Hb, вышедшего в раствор, ( 1) и уровнем MBHb ( 2) при действии возрастающих концентраций NaOCl на суспензию изолированных эритроцитов. Каждая точка — среднее из 3 опытов. SHb (Soluble Hb) – гемоглобин, вышедший в раствор в результате гемолиза, % от общего содержания Hb в эритроцитах.
Наблюдаемый в эксперименте стабилизирующий эффект низких доз окислителя может быть связан с образованием межбелковых сшивок, образованием примембранного белкового слоя, а также перестройкой цитоскелета. Так, например, сокращение цитоскелетного каркаса (“субмембранного ретикулума”) при истощении ATP обусловливает репарацию “дефектных зон”, или каналов утечки ионов, формирующихся в мембране под действием окислителей [121].
ЭНДОГЕННЫЕ ДОНОРЫ ОКСИДА АЗОТА КАК РЕГУЛЯТОРЫ УСТОЙЧИВОСТИ ЭРИТРОЦИТОВ
Одними из универсальных регуляторов различных биохимических и физиологических процессов являются метаболиты NO − нитрозотиолы и ДНКЖ, которые включают три компонента: NO-группу, двухвалентное железо и лиганды. В качестве лигандов ДНКЖ могут выступать различные эндогенные вещества: фосфаты, тиолы, имидазолят-анион, дипептид карнозин [122–125]. В эритроцитах наиболее вероятно образование низкомолекулярных ДНКЖ с глутатионовыми лигандами (ДНКЖ–GS).
В ряде работ было показано, что ДНКЖ–GS оказывают защитное действие на эритроциты в различных патологических ситуациях [106, 126, 127]. В основе цитопротекторного действия этих комплексов лежат следующие механизмы: 1) ингибирование реакций ПОЛ, 2) защита от окисления SH-групп белков эритроцитарных мембран, 3) адресная передача NO или [Fe–NO2]-фрагмента на белки плазматической мембраны и цитоскелета, 4) восстановление оксоферрильной формы Hb. Ранее нами было показано образование ДНКЖ, связанных с SH-группой остатка цистеина β-цепи гемоглобина [124]. В условиях индуцированного окислительного стресса эти комплексы защищали тиоловые группы Hb от окисления. Также Д-НКЖ–GS препятствовали образованию oxoferrylMb [128].
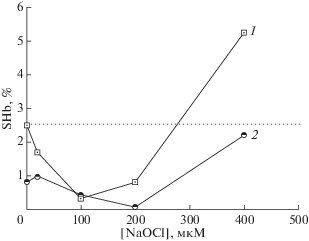
Было исследовано влияние низких физиологических доз ДНКЖ–GS на устойчивость суспензии изолированных эритроцитов к индуцированному NaOCl гемолизу [120]. Во всем диапазоне используемых концентраций окислителя в предобработанных динитрозильными комплексами эритроцитах уровень гемолиза был ниже, чем в контрольном образце (рис. 3). Как видно из рис. 3, ДНКЖ–GS снижали уровень автогемолиза, вызванного экспериментальными манипуляциями. Поскольку лизису подвергаются прежде всего старые и поврежденные эритроциты, то, скорее всего, ДНКЖ–GS оказывают стабилизирующее действие именно на эти клетки. Для сравнения было исследовано цитопротекторное действие веществ, образующихся при диссоциации ДНКЖ или их окислительном распаде: нитрозоглутатиона (GSNO), восстановленного глутатиона (GSH), двухвалентного железа и нитрит-ионов. Предварительная обработка суспензии эритроцитов перечисленными веществами, взятыми в концентрациях, эквивалентных их содержанию в ДНКЖ, приводила к ингибированию окислительного гемолиза эритроцитов: в случае GSH на 42, нитрита − 35 и GSNO – 20%. Двухвалентное железо практически не оказывало влияние на устойчивость клеток. Степень ингибирования ДНКЖ–GS в аналогичных условиях составляла 55%.
Рис. 3.
Индуцированный гипохлоритом гемолиз эритроцитов. 1 – контрольные образцы, 2 – предобработанные ДНКЖ–GS. Каждая точка − среднее из 3 опытов. Горизонтальная пунктирная прямая − уровень автогемолиза в контроле. SHb (Soluble Hb) – гемоглобин, вышедший в раствор в результате гемолиза, % от общего содержания Hb в эритроцитах.
Отчасти стабилизирующее действие ДНКЖ–GS можно объяснить передачей [Fe–NO2]-фрагмента на SH-группы мембранных рецепторов, что обратимо блокирует их активность и, таким образом, понижает чувствительность клетки к внешним воздействиям. Необходимо учитывать, что модификация белков может вызывать и структурные перестройки в мембране, увеличивающие ее жесткость.
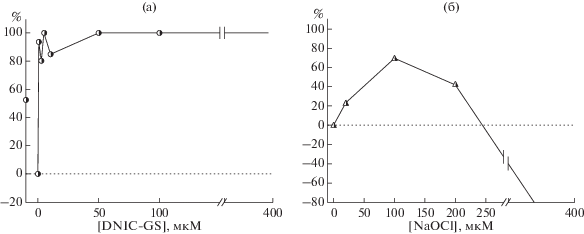
Особенность ДНКЖ как биорегуляторов состоит в том, что у этих соединений разница между биологически активной дозой и повреждающей намного больше, чем у нефизиологических раздражителей. В наших экспериментах [129] ДНКЖ–GS в концентрации 400 мкМ все еще оказывали стабилизирующее действие на клетки, в то время как гипохлорит в такой же концентрации вызывал гемолиз (рис. 4).
Рис. 4.
Степень ингибирования автогемолиза эритроцитов (%) возрастающими концентрациями ДНКЖ–GS (а) и HOCl (б). За ноль принята степень ингибирование гемолиза в контроле. Каждая точка — среднее из 3 опытов.
Полученные результаты согласуются с ранее установленными фактами положительного влияния ДНКЖ на метаболизм эритроцитов [127].
ГИПОТЕЗА: ГЕМОГЛОБИН КАК КОМПОНЕНТ ПРОГРАММИРУЕМОГО ГЕМОЛИЗА
Как было отмечено выше, эритроциты проявляют индуцированные гипоксией реакции, направленные на регуляцию кровотока. В последнее время набирает популярность точка зрения о важности внутрисосудистого гемолиза единичных эритроцитов как источника ATФ для местной пуринергической регуляции кровотока [70–72, 84]. Согласно этой концепции, внутрисосудистый гемолиз стареющих клеток может быть физиологически оправдан в условиях гипоксии и при длительной физической нагрузке. Расчеты показали, что даже малочисленная фракция стареющих эритроцитов содержит количество ATФ, достаточное для оказания регуляторного действия на кровоток [72].
Была обнаружена положительная корреляция между степенью гемолиза эритроцитов (при напряжении сдвига, гипоксии и гипотоническом шоке) и содержанием ATФ в плазме [70, 71]. Возникает вопрос, существуют ли молекулярные механизмы, дестабилизирующие мембрану в перечисленных ситуациях. Многие факты указывают, что такой внутрисосудистый гемолиз – процесс не случайный [72] и существует специальная программа ослабления мембраны и образования в ней литических микропор. В качестве возможных пусковых механизмов гемолиза обсуждается снижение уровня ATФ, поддерживающего структуру цитоскелета, и/или увеличение внутриклеточной концентрации Ca2+ [72]. Возможным фактором нарушения стабильности мембраны также может быть ослабление мембранно-цитоскелетных взаимодействий при длительной дезоксигенации, что увеличивает предрасположенность мембраны к разрыву [72]. Следует подчеркнуть, что гемолизу подвержены преимущественно стареющие эритроциты со множеством микродефектов в мембране [130].
Поскольку речь идет о гемолизе, индуцируемом прежде всего гипоксией, то вполне закономерно предположить участие в этом процессе Hb, который, как известно, является не только датчиком pO2, но и источником АФК. На рис. 5 схематично изображена последовательность событий, приводящая к гемолизу в условиях длительной гипоксии. Самые первые этапы обусловлены автоокислением гемоглобина, в результате чего образуются АФК и окисленные формы Hb [9]. Как было отмечено выше, избыточное накопление этих форм дестабилизирует мембрану (рис. 1). Мы полагаем вполне вероятным наличие специальной программы гемолиза, запускаемой гемоглобином в результате изменения его окислительно-восстановительного состояния вследствие изменения pO2 или редокс-условий. Это тем более возможно, что между уровнем гемолиза и содержанием МBHb обнаружена положительная корреляция [71].
Рис. 5.
Схема участия Hb в программируемом гемолизе. HO-1 – гемоксигеназа, GSH – восстановленный глутатион. ДНКЖ с глутатионовыми лигандами, включает NO, образованный в результате стимуляции пуринэргических рецепторов эндотелия и/или активации макрофагов, например, при воспалении. Стрелками обозначено усиление процесса (активация), тупичками – ингибирование.
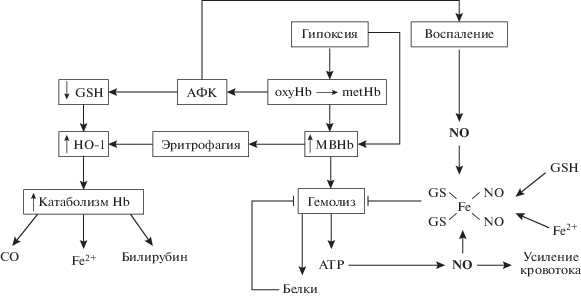
Как было описано в предыдущем разделе, ДНКЖ–GS оказывают стабилизирующее действие на популяцию эритроцитов, предрасположенных к лизизу. Поэтому не исключено, что эти комплексы являются частью механизма, регулирующего распад эритроцитов в кровеносном русле (рис. 5). На возможность ДНКЖ участвовать в регуляции программируемого гемолиза указывает и тот факт, что уровень этих метаболитов NO в кровотоке является интегральным показателем кислородных и редокс-условий. Молекулярный механизм регуляторного действия ДНКЖ–GS в настоящее время изучается в нашей лаборатории.
Все вышесказанное позволило сделать предположение, что незначительный внутрисосудистый гемолиз можно рассматривать как физиологически детерминированный адаптивный ответ организма на умеренный окислительный стресс и гипоксию. О пользе гемолиза свидетельствуют и полезные физиологические эффекты продуктов распада Hb (гемина, CO, Fe2+ и биливердина) [131–133]. Подчеркнем, что эти продукты могут быть полезны только в низких концентрациях.
Известно, что гипоксия является универсальным неспецифическим синдромом при многих патологических состояниях. В частности, она может быть связана с развитием анемии, возникающей в результате как нарушения гомеостаза железа, так и развития хронических воспалительных состояний. При железодефицитной анемии повышенный уровень MBHb можно объяснить необходимостью усиленной отдачи гемоглобином кислорода (за счет высвобождения связанного с мембраной 2.3-БФГ) и необходимостью увеличения кровоснабжения капилляров (за счет действия внеклеточных NO и ATФ). В этом случае связывание Hb носит обратимый характер. При анемиях, вызванных хроническими заболеваниями и воспалениями, повышенные уровни MBHb обусловлены образованием окисленных и денатурированных форм Hb, которые необратимо связываются с мембраной и вызывают образование везикул, трансформацию и разрушение эритроцитов. Таким образом, контроль кислородсвязывающих свойств Hb, реологических характеристик эритроцитов и продолжительности его жизни является адаптивным регуляторным механизмом.
ИСПОЛЬЗОВАНИЕ МЕБРАНОСВЯЗАННОГО ГЕМОГЛОБИНА В КЛИНИКО-БИОХИМИЧЕСКОЙ ДИАГНОСТИКЕ
Эритроциты являются высокочувствительными датчиками метаболического состояния системы крови, способными воспринимать и аккумулировать повреждения, а реактивность эритроцитов можно использовать в качестве критерия оценки тяжести состояния организма и эффективности фармакотерапии. Она изменяется под влиянием физической нагрузки, гипоксии, эндотоксинов, фармакологических препаратов. Существуют различные способы оценки реактивности эритроцитов: по устойчивости их суспензии к окислительному или осмотическому гемолизу, по сорбционной способности и по электрофоретической подвижности эритроцитов [134], по реактивности плазматических мембран и их липидному составу, по способности связывать краситель альциановый синий компонентами гликокаликса [111]. В работе [135] сообщалось о существовании нескольких адаптивных вариантов эритроцитов с различной реактивностью клеточных мембран.
Наличие прямой корреляции между уровнем окислительного стресса и количеством MBHb позволило использовать этот показатель для оценки окислительного повреждения эритроцитов [23, 136, 137]. Увеличение MBHb наблюдалось при метаболическом синдроме [138, 139], коронарном атеросклерозе [49], инфаркте миокарда [140], ишемии [49, 139–142], гипертонии [139], псориазе [143], хронической почечной недостаточности [144], хронической обструктивной болезни легких [50], атипичном/тяжелом алкогольном делирии [145], сахарном диабете [146]. Повышенное содержание MBHb также было зафиксировано в эритроцитах больных талассемией и различными гемоглобинопатиями [147–149]. Повышенные уровни MBHb отмечены не только при патологических, но и при некоторых физиологических процессах, среди которых можно отметить беременность [150], тяжелые мышечные нагрузки [41, 151] и естественное старение эритроцитов [97]. В наибольшей степени связывание Hb с мембранами происходило в стареющих клетках с низким уровнем антиоксидантных ферментов. Во всех ситуациях наблюдается прямая корреляция между тяжестью патологии и уровнем MBHb.
Уровень MBHb может быть еще одним достаточно простым критерием реактивности эритроцитов. Приведем несколько примеров, иллюстрирующих потенциальную возможность использования MBHb в клинической диагностике. В работах Пивоварова с соавт. [47, 139] было предложено использовать данные о содержании MBHb для расчета коэффициента устойчивости эритроцитов к функциональной нагрузке − тканевой гипоксии. Прямая корреляция наблюдалась между уровнем MBHb и тяжестью заболевания ишемической болезнью сердца (ИБС) [49, 139]. Также существенные различия были отмечены в изменении содержания MBHb после функциональной нагрузки: у здоровых доноров происходило снижение MBHb (с 3.6 до 0.9%), а у больных, наоборот, повышение (с 8.2 до 10.9%) [47]. Таким образом, уровень MBHb является дополнительной характеристикой тяжести ИБС, позволяющей оценить степень напряжения адаптивных систем организма к ишемии у больного со стенокардией. Было предложено учитывать данные об уровне MBHb и для расчета интегрального индекса структурно-функционального состояние мембран эритроцитов у больных хронической обструктивной болезнью легких [50].
Отмечена положительная корреляция между уровнем MBHb и интенсивностью окислительного стресса в эритроцитах. В работе [143] предложено рассматривать уровень MBHb в качестве одного из интегральных маркеров окислительного и протеолитического стресса при псориазе. У больных повышенный уровень MBHb положительно коррелировал с увеличением лейкоцитов, перекисным окислением липидов и осмотической хрупкостью эритроцитов. У больных хронической почечной недостаточностью увеличение MBHb коррелировало с увеличением C-реактивного белка [144].
С помощью простого метод определения MBHb в пробах крови [51] установлен диапазон нормальных значений для МBHb: 3.3%–4.9% [152], измерен уровень MBHb у пациентов Национального медицинского исследовательского центра онкологии им. Н.Н. Блохина. Выборка пациентов включала группу больных со злокачественными опухолями различной локализации (n = 125). Среди обследованных онкологических больных несоответствие норме уровня МBHb наблюдалось у 61%, в то время как в контрольной группе (n = 26) эта величина составляла 38%. Различия носили достоверный характер (t = 1.7; p < 0.05). Среди пациентов с диагностированной анемией (анемия хронических заболеваний, железодефицитная анемия, микроангиопатическая гемолитическая анемия) (n = 23) несоответствие норме было у 74%, в контрольной группе (n = 23) − у 35% (t = 2.05; p < 0.05). Данные о содержании MBHb в группе онкологических больных были включены в разработанную пилотную версию компьютерной экспертной системы для диагностики анемий при онкологических заболеваниях [153]. Такая система, наряду с использованием искусственных нейросетей [154], признана одним из наиболее перспективных направлений гематологической диагностики [155].
Несмотря на многочисленные исследования мембраносвязанного Hb, этот показатель еще не стал общеупотребительным в диагностической практике. Основные причины этого кроются в отсутствии стандартизированной методики определения MBHb и утвержденного представления о границах нормы. Необходимо также исследовать динамику изменения MBHb как в условиях нормы, так и при патологии. Наиболее эффективно использование определения MBHb для оценки функционального состояния спортсменов [41, 111] и для совершенствования диагностики и лечения ИБС [47, 49, 55, 139, 142].
* * *
Несмотря на длительную историю изучения Hb, его сигнальная функция долгое время не была очевидной, хотя в научной литературе газосенсорные свойства гемоглобинподобных белков обсуждаются давно. Если для эритроцитарного Hb сигнальная функция является дополнительной, то для некоторых представителей гемоглобинового суперсемейства участие в редокс-сигнальных путях – главное предназначение [156], причем свойства этих Hb модулируются модификацией (как правило, окислением) поверхностных цистеинов.
Проблема кислородзависимой регуляции метаболизма эритроцитов обсуждается уже на протяжении двух десятилетий. За это время был накоплен огромный массив экспериментальных данных, подтверждающих ключевую роль Hb в этом процессе. В недавней работе [157] в экспериментах на мышах были получены прямые доказательства того, что комплекс deoxyHb–Band3 является триггером различных внутриклеточных процессов, адаптирующих свойства эритроцитов к кислородным условиям внутри организма.
Подводя итоги, можно с уверенностью констатировать, что гемоглобин – это настоящий газо- и редокс-сенсорный белок, который воспринимает изменение внешней и внутренней среды, формирует информационные сигналы, передает их метаболическим структурам и, тем самым, осуществляет саморегуляцию внутриклеточных метаболических процессов. С его помощью осуществляется взаимодействие между такими параметрами, как связывание кислорода, синтез ATФ, регуляция рН, деформируемость и редокс-состояние клетки. Сигнальная функция Hb реализуется за счет его обратимого связывания с мембраной. Благодаря малому различию в свободных энергиях растворимого и связанного Hb, между этими двумя формами возможны взаимные равновесные переходы. Этот древний механизм саморегуляции живых систем в эритроцитах является основным, поскольку в них отсутствует ядерный аппарат и изменение метаболизма через синтез новых белков невозможно. С помощью белок-мембранных взаимодействий регулируются даже такие процессы, как программируемая гибель клетки (в случае эритроцита – эриптоз), клеточные часы, задающие продолжительность жизни клетки [96], и ее циркадные ритмы [158]. Изучение этих процессов подтверждает участие цитоплазмы в процессах регуляции метаболизма.
Эритроцит является примером хорошо развитого механизма формирования адаптивного ответа с помощью белков и мембранных структур. Здесь возникает вопрос – где заканчивается адаптивный ответ и начинается патология? Обратимое встраивание Hb в мембрану направлено на оптимизацию доставки кислорода к тканям, необратимое – с ремонтом клетки и элиминацией старых и поврежденных клеток. Примембранный белковый слой, образованный продуктами окисления Hb, может быть фактором, поддерживающим целостность нативной мембраны. Возможно, такие эритроциты, жертвуя функциональностью, приобретают большую стабильность. С другой стороны, высокий уровень MBHb приводит к нарушению ряда характеристик эритроцитов: зарядового баланса, барьерных свойств мембран, реологических свойств. Но даже дестабилизация мембраны и последующий лизис единичных клеток могут быть физиологически оправданы, например, в условиях гипоксии. Поэтому необходимо учитывать, что наблюдаемые при различных болезненных состояниях сдвиги в уровне MBHb не всегда имеют патологическую направленность и могут отражать компенсаторно-приспособительные процессы, реализующиеся с целью защитить организм и/или клетку в изменившихся условиях. Знание диапазона изменения MBHb, соответствующего зонам адаптации, может повысить информационную значимость этого показателя для диагностики.
Работа выполнена при частичной финансовой поддержке Российского фонда фундаментальных исследований (грант № 18-34-00561_мол_а).
Список литературы
Kurganov B.I. // Organized Multienzyme Systems: Catalytic Properties. N.Y.: Acad. Press, 1985. P. 263–267.
Курганов Б.И., Любарев А.Е. // Биохимия. 1991. Т. 56. № 1. С. 19–32.
Капрельянц А.С. // Биол. науки. 1988. Т. 6. С. 5–13.
Oparin A.I., Manskaja S.M., Magaram M. // Biochem. Z. 1933. B. 265. S. 21–28.
Chung H.S., Wang S.B., Venkatraman V., Murray C.I., Van Eyk J.E. // Circulation Research. 2013. V. 112. P. 382–392.
Космачевская О.В., Шумаев К.Б., Топунов А.Ф. // Успехи биологической химии. 2019. Т. 58. С. 63–91.
Yang J., Carroll K.S., Liebler D.C. // Mol. Cell. Proteomics. 2016. V. 15. Article ID 1. doi 10.1074/mcp.O115.056051
Benesch R.E., Benesch R. // Biochemistry. 1962. V. 1. P. 735–738.
Kannan R., Labotka R., Low P.S. // J. Biol. Chem. 1988. V. 263. P. 13766–13773.
Зайцева О.И., Терещенко В.П., Манчук В.Т., Прахин Е.И., Эверт Л.С., Нягашкина Е.И. // Фундаментальные исследования. Биологические науки. 2004. № 6. С. 8–21.
Petibois C., Déléris G. // Arch. Med. Res. 2005. V. 36. P. 524–531.
Martusevich A.A., Deryugina A.V., Martusevich A.K. // J. Stress Physiol. Biochem. 2016. V. 12. P. 5–11.
Hoffman J.F. // J. Gen. Physiol. 1958. V. 42. C. 9–28.
Anderson H.M., Turner J.C. // Nature. 1959. V. 183. P. 112–113.
Anderson H.M., Turner J.C. // J. Clin. Invest. 1960. V. 39. P. 1–7.
Fung L.W. // Biochemistry. 1981. V. 20. P. 7162–7166.
Eisinger J., Flores J., Salhany J.M. // Proc. Natl. Acad. Sci. USA. 1982. V. 79. P. 408–412.
Walder J.A., Chatterjee R., Steck T.L., Low P.S., Musso G.F., Kaiser E.T., Rogers P.H., Arnone A. // J. Biol. Chem. 1984. V. 259. P. 10238–10246.
Chu H., Breite A., Ciraolo P., Franco R.S., Low P.S. // Blood. 2008. V. 111. P. 932–938.
Cao Z., Bell J.B., Mohanty J.G., Nagababu E., Rifkind J.M. // Am. J. Physiol. Heart. Circ. Physiol. 2009. V. 297. P. 1494–1503.
Sega M.F., Chu H., Christian J.A., Low P.S. // Blood Cells Mol. Dis. 2015. V. 55. P. 266–271.
Chan E., Desforges J.F. // Blood. 1974. V. 44. P. 926.
Sharma R., Premachandra B.R. // Biochem. Med. Metab. Biol. 1991. V. 46. P. 33–44.
Chaimanee P., Yuthavong Y. // FEBS Lett. 1977. V. 78. P. 119–123.
Mishra K., Chakrabarti A., Das P.K. // J. Phys. Chem. B. 2017. V. 121. P. 7797–7802.
Shaklai N., Yguerabide J., Ranney H.M. // Biochemistry. 1977. V. 16. P. 5593–5597.
Rauenbuehler P.B., Cordes K.A., Salhany J.M. // Biochim. Biophys. Acta – Biomembranes. 1982. V. 692. P. 361–370.
Datta P., Chakrabarty S., Chakrabarty A., Chakrabarty A. // Biochim Biophys Acta. 2008. V. 1778. P. 1–9.
Lebbar I., Stetzkowski-Marden F., Mauffret O., Cassoly R. // Eur. J. Biochem. 1987. V. 170. P. 273–277.
Tuvia S., Levin S., Korenstein R. // Biophys. J. 1992. V. 63. P. 599–602.
Reithmeier R.A., Casey J.R., Kalli A.C., Sansom M.S.P., Alguel Y., Iwata S. // Biochim. Biophys. Acta. 2016. V. 1858. P. 1507–1532.
Spring F.A., Bruce L.J., Anstee D.J., Tanner M.J.A. // Biochem. J. 1992. V. 288. P. 713–716.
Ferru E., Giger K., Pantaleo A., Campanella E., Grey J., Ritchie K., Vono R., Turrini F., Low P.S. // Blood. 2011. V. 117. P. 5998–6006.
Pantaleo A., Ferru E., Pau M.C., Khadjavi A., Mandili G., Mattè A., Spano A., De Franceschi L., Pippia P., Turrini F. // Oxid. Med. Cell. Longev. 2016. Article ID 6051093. doi 10.1155/2016/6051093
Walder J.A., Chatterjee R., Steck T.L., Low P.S., Musso G.F., Kaiser E.T., Rogers P.H., Arnone A. // J. Biol. Chem. 1984. V. 259. P. 10238–10246.
Tsuneshige A., Imai K., Tyuma I. // J. Biochem. 1987. V. 101. P. 695–704.
Demehin A.A., Abugo O.O., Jayakumar J.R., Rifkind J.M. // Biochemistry. 2002. V. 41. P. 8630–8637.
Shaklai N., Ranney H.R. // Isr. J. Med. Sci. 1978. V. 14. P. 1152–1156.
Kumar S., Bandyopadhyay U. // Toxicol. Lett. 2005. V. 157. P. 175–188.
Kriebardis A.G., Antonelou M.H., Stamoulis K.E., Economou-Petersen E., Margaritis L.H., Papassideri I.S. // J. Cell. Mol. Med. 2007. V. 11. P. 148–155.
Welbourn E.M., Wilson M.T., Yusof A., Metodiev M.V., Cooper C.E. // Free Radic. Biol. Med. 2017. V. 103. P. 95–106.
Giardina B., Scatena R., Clementi M.E., Ramacci M.T., Maccari F., Cerroni L., Condò S.G. // Adv. Exp. Med. Biol. 1991. V. 307. P. 75–84.
Топунов А.Ф., Голубева Л.И. // Успехи биологической химии. 1989. Т. 30. С. 239–252.
Nikolić M., Stanić D., Antonijević N., Niketić V. // Clin. Biochem. 2004. V. 37. P. 22–26.
Drabkin D.R. // Am. J. Med. Sci. 1945. V. 209. P. 268–270.
Токтамысова З.С., Биржанова Н.Х. // Биофизика. 1990. Т. 35. № 5. С. 1019–1020.
Пивоваров Ю.И., Кузнецова Э.Э., Горохова В.Г., Корякина Л.Б. Патент РФ. 2008. № 2008108370/15.
Сергеева А.С., Пивоваров Ю.И., Курильская Т.Е., Кузнецова Э.Э. // Российский кардиологический журн. 2014. Т. 115. № 11. С. 13–18.
Чуйко Е.С., Орлова Г.М., Кузнецова Э.Э., Горохова В.Г., Корякина Л.Б. // Сибирский медицинский журн. (Иркутск). 2015. № 7. С. 101–104.
Давыдкин И.Л., Селезнев А.В., Ромашева Е.П., Мишина Н.А. // Основы клинической гемостазиологии. Самара: Офорт, 2009. С. 77–96.
Насыбуллина Э.И., Космачевская О.В., Топунов А.Ф. // Труды Карельского научного центра РАН. 2018. № 4. С. 93–104.
Созарукова М.М., Владимиров Г.К., Измайлов Д.Ю. // Science and Practice: New Discoveries. Чехия, Карловы Вары – Россия, Москва, Proceedings of materials the international scientific conference / Ed. Shvec I.M., Ismagilova L.A., Gur’eva V.A., Telegina E.A., Sedenko V.I. Киров: Международный центр научно-исследовательских проектов, 2015. С. 771–781.
Sears D.A., Lewis P.C. // J. Lab. Clin. Med. 1980. V. 96. P. 318–327.
Громов П.С., Захаров С.Ф., Шишкин С.С., Ильинский Р.В. // Биохимия. 1988. Т. 53. № 8. С. 1316–1326.
Brazhe N.A., Abdali S., Brazhe A.R., Luneva O.G., Bryzgalova N.Y., Parshina E.Y., Sosnovtseva O.V., Maksimov G.V. // Biophys. J. 2009. V. 97. P. 3206–3214
Giardina B., Messana I., Scatena R., Castagnola M. // Crit. Rev. Biochem. Mol. Biol. 1995. V. 30. P. 165–196.
De Rosa M.C., Carelli Alinovi C., Galtieri A., Scatena R., Giardina B. // Gene. 2007. V. 398. P. 162–171.
Huang Z., Louderback J.G., Goyal M., Azizi F., King S.B., Kim-Shapiro D.B. // Biochim. Biophys. Acta. 2001. V. 1568. P. 252–560.
Vaughn M.W., Huang K.-T., Kuo L., Liao J.C. // J. Biol. Chem. 2000. V. 275. P. 2342–2234.
Han T.H., Hyduke D.R., Vaughn M.W., Fukuto J.M., Liao J.C. // Proc. Natl. Acad. Sci. U.S.A. 2002. V. 99. P. 7763–7768.
Campanella M.E., Chu H., Low P.S. // Proc. Natl. Acad. Sci. U.S.A. 2005. V. 102. P. 2402–2407.
Курганов Б.И., Любарев А.Е. // Молекулярная биология. 1988. Т. 22. № 6. С. 1605–1613.
Puchulu-Campanella E., Chu H., Anstee D.J., Galan J.A., Tao W.A., Low P.S. // J. Biol. Chem. 2013. V. 288. P. 848–858.
Low P.S., Rathinavelu P., Harrison M.L. // J. Biol. Chem. 1993. V. 68. P. 14627–14631.
Chu H., Low P.S. // Biochem. J. 2006. V. 400. P. 143–151.
Messana I., Orlando M., Cassiano L., Pennacchietti L., Zuppi C., Castagnola M., Giardina B. // FEBS Lett. 1996. V. 390. P. 25–28.
Weber R.E., Voelter W., Fago A., Echner H., Campanella E., Low P.S. // Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004. V. 287. P. 454–464.
Иржак Л.И. Гемоглобины и их свойства. M.: Наука, 1975. 240 с.
Конев С.В. Структурная лабильность биологических мембран и регуляторные процессы. Минск: Наука и техника, 1987. 238 с.
Sikora J., Orlov S.N., Furuya K., Grygorczyk R. // Blood. 2014. V. 124. P. 2150–2157.
Luneva O.G., Sidorenko S.V., Ponomarchuk O.O., Tverskoy A.M.,Cherkashin A.A., Rodnenkov O.V., Alekseeva N.V., Deev L.I., Maksimov G.V., Grygorczyk R., Orlov S.N. // Cell Physiol. Biochem. 2016. V. 39. P. 81–88.
Grygorczyk R., Orlov S.N. // Front. Physiol. 2017. V. 8. Article ID 1110. doi 10.3389/fphys.2017.01110
Jia L., Bonaventura C., Bonaventura J., Stamler J.S. // Nature. 1996. V. 380. P. 221–226.
Stamler J.S., Singel D.J., Piantadosi C.A. // Nature Medicine. 2008. V. 14. № 10. P. 1008–1009.
Huang K.T., Keszler A., Patel N., Patel R.P., Gladwin M.T., Kim-Shapiro D.B., Hogg N. // J. Biol. Chem. 2005. V. 280. P. 31126–31131.
Gladwin M.T., Raat N.J., Shiva S., Dezfulian C., Hogg N., Kim-Shapiro D.B., Patel R.P. // Am. J. Physiol. Heart Circ. Physiol. 2006. V. 291. P. 2026–2035.
Shiva S. // Redox Biology. 2013. V. 1. P. 40–44.
Bergfeld G.R., Forrester T. // Cardiovasc. Res. 1992. V. 26. P. 40–47.
Ramdani G., Langsley G. // Biomed. J. 2014. V. 37. P. 284–292.
Pawloski J.R., Hess D.T., Stamler J.S. // Proc. Natl. Acad. Sci. USA. 2005. V. 102. P. 2531–2536.
Thevenin B.J.-M., Willardson B.M., Low P.S. // J. Biol. Chem. 1989. V. 264. P. 15886–15892.
Salhany J.M. // Biochemistry. 2008. V. 47. P. 6059–6072.
Sridharan M., Adderley S.P., Bowles E.A., Egan T.M., Stephenson A.H., Ellsworth M.L., Sprague R.S. // Am. J. Physiol. Heart Circ. Physiol. 2010. V. 299. P. 1146–1152.
Лунева О.Г., Сидоренко С.В., Максимов Г.В., Григорчик Р., Орлов С.Н. // Биологические мембраны. 2015. Т. 32. № 4. С. 223–234.
Wei H.S., Kang H., Rasheed I.Y., Zhou S., Lou N., Gershteyn A., McConnell E.D., Wang Y., Richardson K.E., Palmer A.F., Xu C., Wan J., Nedergaard M. // Neuron. 2016. V. 91. P. 851–862.
Stefanovic M., Puchulu-Campanella E., Kodippili G., Low P.S. // Biochem. J. 2013. V. 449. P. 143–150.
Ito H., Murakami R., Sakuma S., Tsai C.D., Gutsmann T., Brandenburg K., Pöschl J.M., Arai F., Kaneko M., Tanaka M. // Sci Rep. 2017. V. 7. Article ID 43134. doi 10.1038/srep43134
Nagababu E., Mohanty J.G., Bhamidipaty S., Ostera G.R., Rifkind J.M. // Life Sci. 2010. V. 86. P. 133–138.
Miyazawa T., Suzuki T., Fujimoto K., Kinoshita M. // Mech. Ageing Dev. 1996. V. 86. P. 145–150.
Лунева О.Г., Браже Н.А., Фадюкова О.Е., Алахая М.Я., Байжуманов А.А., Паршина Е.Ю., Демидова А.Е., Кошелев В.Б., Максимов Г.В. // Доклады Академии наук. 2005. Т. 405. № 6. С. 834–836.
Rifkind J.M., Nagababu E. // Antioxid. Redox Signal. 2013. V. 18. P. 2274–2283.
Dutra F.F., Bozza M.T. // Front. Pharmacol. 2014. V. 5. Article ID 115. doi 10.3389/fphar.2014.00115
Шперлинг И.А., Рязанцева Н.В., Новицкий В.В., Михаленко А.Н., Шевцова Н.М., Наследникова И.О., Миллер А.А., Филиппова О.Н. // Вестник Российской Военно-медицинской академии. 2011. Т. 3. № 35. С. 156–162.
Kiefmann R., Rifkind J.M., Nagababu E., Bhattacharya J. // Blood. 2008. V. 111. P. 5205–5214.
Klei T.R., Meinderts S.M., van den Berg T.K., van Bruggen R. // Front. Immunol. 2017. V. 8. Article ID 73. doi 10.3389/fimmu.2017.00073
Badior K.E., Casey J.R. // IUBMB Life. 2018. V. 70. P. 32–40.
Waugh S.M., Low P.S. // Biochemistry. 1985. V. 24. P. 34–39.
McPherson R.A., Sawyer W.H., Tilley L. // Biochemistry. 1992. V. 31. P. 512–518.
Bosman G.J., Lasonder E., Groenen-Döpp Y.A., Willekens F.L., Werre J.M., Novotný V.M. // J. Proteomics. 2010. V. 73. V. 396–402.
Arashiki N., Kimata N., Manno S., Mohandas N.,Takakuwa Y. // Biochemistry. 2013. V. 52. P. 5760–5769.
Ferru E., Pantaleo A., Carta F., Mannu F., Khadjavi A., Gallo V., Ronzoni L., Graziadei G., Cappellini M.D., Turrini F. // Haematologica. 2014. V. 99. P. 570–578.
Willekens F.L., Werre J.M., Groenen-Döpp Y.A., Roerdinkholder-Stoelwinder B., de Pauw B., Bosman G.J. // Br. J. Haematol. 2008. V. 141. P. 549–556.
Белевич Е.И., Костин Д.Г., Слобожанина Е.И. // Успехи современной биологии. 2014. Т. 134. № 2. С. 149–157.
Briglia M., Rossi M.A., Faggio C. // Current Medicinal Chemistry. 2017. V. 24. P. 937–942.
Lang E., Bissinger R., Qadri S.M., Lang F. // Int. J. Cancer. 2017. V. 141. P. 1522–1528.
Nicolay J.P., Liebig G., Niemoeller O.M., Koka S., Ghashghaeinia M., Wieder T., Haendeler J., Busse R., Lang F. // Pflugers Arch. 2008. V. 456. P. 293–305.
Colclasure G.C., Parker J.C. // J. Gen. Physiol. 1992. V. 100. P. 1–10.
Vitoux D., Beuzard Y., Brugnara C. // J. Membr. Biol. 1999. V. 167. P. 233–240.
Gibson J.S., Cossins A.R., Ellory J.C. // J. Exp. Biol. 2000. V. 203. P. 1395–1407.
Barvitenko N.N., Adragna N.C., Weber R.E. 2005. // Cell Physiol. Biochem. 2005. V. 15. P. 1–18.
Боровская М.К., Кузнецова Э.Э., Горохова В.Г., Корякина Л.Б., Курильская Т.Е., Пивоваров Ю.И. // Бюллетень ВСНЦ СО РАМН. 2010. Т. 73. С. 334–354.
Knutton S., Finean J.B., Coleman R., Limbrick A.R. // J. Cell Sci. 1970. V. 7. P. 357–371.
Комиссарчик Я.Ю., Левин С.В., Свиридов Б.Е., Сабаляускас И.Ю., Айдитите Г.С. // Общие механизмы клеточных реакций на повреждающие воздействия. Л.: Институт цитологии, 1977. С. 29–31.
Jarolim P., Lahav M., Liu S.C., Palek J. // Blood. 1990. V. 76. P. 2125–2131.
Liu S.C., Palek J. // J. Biol. Chem. 1984. V. 259. P. 11556–11562.
Kirschner-Zilber I., Rabizadeh E., Shaklai N. // Biochim. Biophys. Acta. 1982. V. 690. P. 20–30.
Dadosh N., Shaklai N. // J. Muscle Res. Cell. Motil. 1987. V. 9. P. 86–92.
Calabrese E.J. // Microbial Cell. 2014. V. 1. P. 145–149.
Насонов Д.Н. Местная реакция протоплазмы и распространяющееся возбуждение. М.-Л.: АкадемИздат, 1962. 426 с.
Космачевская О.В., Насыбуллина Э.И., Блиндарь В.Н., Топунов А.Ф. // Актуальные вопросы экспериментальной биологии и медицины. Сухум: Дом печати, 2017. С. 421–429.
Белоус А.М., Бондаренко В.А., Бондаренко Т.П., Бабийчук Л.А. // Криобиология и криомедицина. 1983. № 12. С. 13–24.
Ванин А.Ф. 2015. Динитрозильные комплексы железа с тиолсодержащими лигандами: физикохимия, биология, медицина. М.: Ин-т комп. исследований, 2015. 219 с.
Shumaev K.B., Gubkin A.A., Serezhenkov V.A., Lobysheva I.I., Kosmachevskaya O.V., Ruuge E.K., Lankin V.Z., Topunov A.F., Vanin A.F. // Nitric Oxide. 2008. V. 18. P. 37–46.
Shumaev K.B., Kosmachevskaya O.V., Timoshin A.A., Vanin A.F., Topunov A.F. // Methods in Enzymology. 2008. V. 436. P. 445–461.
Shumaev K.B., Kosmachevskaya O.V., Nasybullina E.I., Gromov S.V., Novikov A.A., Topunov A.F. // J. Biol. I-norg. Chem. 2017. V. 22. P. 153–160.
Мартусевич А.К., Соловьева А.Г., Перетягин С.П., Давыдюк А.В. // Биофизика. 2014. Т. 59. № 6. С. 1173–1179.
Шамова Е.В., Бичан О.Д., Дрозд Е.С., Горудко И.В., Чижик С.А., Шумаев К.Б., Черенкевич С.Н., Ванин А.Ф. // Биофизика. 2011. Т. 56. № 2. С. 265–271.
Shumaev K.B., Petrova N.E., Zabbarova I.V., Vanin A.F., Topunov A.F., Lankin V.Z., Ruuge E.K. // Biochemistry (Moscow). 2004. V. 69. № 5. P. 569–574.
Шумаев К.Б., Горудко И.В., Космачевская О.В., Панасенко О.М., Пугаченко И.С., Топунов А.Ф., Рууге Э.К. // Актуальные вопросы экспериментальной биологии и медицины. Сухум: Дом печати, 2017. С. 445–452.
Orbach A., Zelig O., Yedgar S., Barshtein G. // Transfus. Med. Hemother. 2017. V. 44. P. 183–187.
Stec D.E., Drummond H.A., Vera T. // Hypertension. 2008. V. 51. P. 597–604.
Otterbein L.E., Soares M.P., Yamashita K., Bach F.H. // Trends Immunol. 2003. V. 24. P. 449–455.
Konrad F.M., Zwergel C., Ngamsri K.C., Reutershan J. // Front. Immunol. 2017. V. 8. Article ID 1874. doi 10.3389/fimmu.2017.01874
Шурхина Е.С., Нестеренко В.М., Цветаева Н.В., Никулина О.Ф. // Клиническая лабораторная диагностика. 2014. № 7. С. 41–46.
Зайцева О.И., Терещенко В.П., Манчук В.Т., Прахин Е.И., Эверт Л.С., Нягашкина Е.И. // Фундаментальные исследования. Биологические науки. 2004. № 6. С. 18–21.
Sullivan S.G., Stern A. // Biochim. Biophys. Acta. 1984. V. 774. P. 215–220.
Rocha S., Costa E., Coimbra S., Nascimento H., Catarino C., Rocha-Pereira P., Quintanilha A., Belo L., Santos-Silva A. // Blood Cells, Molecules and Diseases. 2009. V. 43. P. 68–73.
Сергеева А.С., Пивоваров Ю.И., Бабушкина И.В. // Acta Biomedica Scientifica. 2015. № 4. С. 12–17.
Пивоваров Ю.И., Кузнецова Э.Э., Горохова В.Г., Сергеева А.С., Бабушкина И.В., Корякина Л.Б., Андреева Е.О. // Бюллетень ВСНЦ СО РАМН. 2016. Т. 1. № 4. С. 61–67.
Santos-Silva A., Castro E.M.B., Teixeira N.A., Guerra F.C., Quintanilha A. // Atherosclerosis. 1995. V. 116. P. 199–209.
Santos-Silva A., Rebelo I., Castro E., Belo L., Catarino C., Monteiro I., Almeida M.D., Quintanilha A. // Clin. Chim. Acta. 2002. V. 320. P. 29–35.
Пивоваров Ю.И., Кузнецова Э.Э., Корякина Л.Б., Горохова В.Г., Курильская Т.Е. // Тромбоз, гемостаз и реология. 2013. № 2. С. 39–45.
Rocha-Pereira P., Santos-Silva A., Rebelo I., Figneiredo A., Quintanilha A., Teixeira F. // Br. J. Dermatol. 2004. V. 150. P. 232–244.
Costa E., Rocha S., Rocha-Pereira P., Castro E., Miranda V., do Sameiro Faria M., Loureiro A., Quintanilha A., Belo L., Santos-Silva A. // Open Clin. Chem. J. 2008. V. 1. P. 57–63.
Виноградов Д.Б. Паначев И.В., Изаровский Б.В., Козочкин Д.А., Цейликман О.Б. // Вопросы наркологии. 2010. № 5. С. 44–50.
Bryszewska M. // J. Clin. Chem. Clin. Biochem. 1988. V. 26. P. 809–813.
Sears D.A., Luthra M.G. // J. Lab. Clin. Med. 1983. V. 102. P. 694–698.
Scott M.D., van den Berg J.J., Repka T., Rouyer-Fessard P.,Hebbel R.P., Beuzard Y., Lubin B.H. // J. Clin. Invest. 1993. V. 91. P. 1706–1712.
Hebbel R.P. // Blood. 1991. V. 77. P. 214–237.
Catarino C., Rebelo I., Belo L., Rocha-Pereira P., Rocha S., Bayer Castro E., Patrício B., Quintanilha A., Santos-Silva A. // J. Perinat. Med. 2009. V. 37. P. 19–27.
Santos-Silva A., Rebelo I., Castro E.M.B., Belo L., Guerra A., Rego C., Quintanilha A. // Clin. Chim. A-cta. 2001. V. 306. P. 119–126.
Космачевская О.В., Насыбуллина Э.И., Блиндарь В.Н., Шумаев К.Б., Топунов А.Ф. // Новые информационные технологии в медицине, биологии, фармакологии и экологии. М.: Ин-т новых информационных технологий, 2016. С. 172–180.
Nasybullina E.I., Nikitaev V.G., Pronichev A.N., Blindar V.N., Kosmachevskaya O.V., Topunov A.F. // Bull. Lebedev Phys. Inst. 2015. V. 42. № 7. P. 206–208.
Amendolia S.R., Brunetti A., Carta P., Cossu G., Ganadu M.L., Golosio B., Mura G.M., Pirastru M.G. // Med. Decis. Making. 2002. V. 22. P. 18–26.
Lippi G., Plebani M. // J. Lab. Precis. Med. 2018. V. 3. Article ID 68. doi 10.21037/jlpm.2018.07.09
Reeder B.J. // Antioxid. Redox. Signal. 2017. V. 26. P. 763–776.
Chu H., McKenna M.M., Krump N.A., Zheng S., Mendelsohn L., Thein S.L., Garrett L.J., Bodine D.M., Low P.S. // Blood. 2016. V. 128. P. 2708–2716.
Bayer S.B., Low F.M., Hampton M.B., Winterbourn C.C. // Free Radic. Res. 2016. V. 50. P. 1329–1339.
Дополнительные материалы отсутствуют.
Инструменты
Прикладная биохимия и микробиология