

Прикладная биохимия и микробиология, 2020, T. 56, № 4, стр. 401-406
Разработка иммуноферментного анализа для определения соевого сырья в продуктах питания
Н. И. Смирнова 1, Е. А. Зверева 1, А. В. Жердев 1, Б. Б. Дзантиев 1, *
1 Институт биохимии им. А.Н. Баха, Федеральный исследовательский центр
“Фундаментальные основы биотехнологии” Российской академии наук
119071 Москва, Россия
* E-mail: dzantiev@inbi.ras.ru
Поступила в редакцию 26.12.2019
После доработки 04.02.2020
Принята к публикации 25.02.2020
Аннотация
Разработана методика иммуноферментного анализа для контроля присутствия и оценки содержания соевого сырья в мясных продуктах питания, основанная на связывании специфических антител с антигенными детерминантами соевого ингибитора трипсина. Из-за возможного влияния ферментативной и высокотемпературной обработок продуктов, которые могут нарушать нативную структуру ингибитора и приводить к его фрагментации, анализ основан на конкурентном взаимодействии с антителами антигена, содержащегося в пробе и иммобилизованного в лунках микропланшета. Установлены условия проведения анализа, обеспечивающие минимальный предел обнаружения и высокую точность измерений. Разработанный анализ имел продолжительность 60 мин, предел обнаружения 3.5 нг/мл и диапазон определяемых концентраций ингибитора 7.7–110 нг/мл. Показана возможность использования предложенной методики для тестирования мясных продуктов.
В настоящее время широко распространено добавление в мясные продукты растительных белков, что обусловлено технологическими и экономическими причинами. Самым популярным растительным компонентом является соя, благодаря высокому содержанию в ней белка (около 40%), способности к эмульгированию, впитыванию воды и жира, а также регулированию консистенции пищевых продуктов [1, 2]. Рецептуры различных мясных продуктов устанавливают предельное содержание в них немясных компонентов, превышение которых рассматривается как фальсификация. Кроме того, использование соевого сырья сопряжено с рисками аллергических реакций на его компоненты – прежде всего на глицинин, β-конглицинин, профилин, вакуолярный соевый белок и ингибитор трипсина типа Кунитца [1, 3]. В связи с этим для контроля соблюдения рецептуры пищевых продуктов крайне важно располагать достоверными производительными методами, выявляющими специфические компоненты соевого сырья и позволяющими оценивать его содержание в изготовленной продукции [4].
Для детектирования соевых добавок в продуктах питания предложены и применяются разнообразные методы [5, 6]. Однако большинство из них сопряжено с использованием специализированного оборудования и являются часто весьма трудоемкими и продолжительными. К таким методам относятся высокоэффективная жидкостная хроматография [7, 8], полимеразная цепная реакция [9–11], а также электрофоретический [12] и гистохимический [13] анализы.
Более перспективны для массового применения иммунохимические методы, характеризующиеся высокой производительностью, методической простотой и не требующие сложного приборного обеспечения [14, 15]. Описан ряд методов иммунодетекции соевых белков в продуктах питания [16], основанных, как правило, на принципах иммуноферментного [17–19] и иммунохроматографического [20] анализов. Однако выбор контролируемого биомаркера соевого сырья и формата иммуноанализа, обеспечивающих максимальную достоверность тестирования, остается дискуссионным. Так, в качестве кандидатных биомаркеров рассматривались β‑конглицинин [17, 20], глицинин [19], Gly m 4 [21], Gly m 8 [22] и ингибиторы трипсина Баумана-Бирка [23] и Кунитца [18]. При этом в описанных аналитических системах реализован сэндвич-формат иммуноанализа с образованием детектируемых тройных комплексов (иммобилизованное антитело – антиген, содержащийся в пробе, – меченое антитело). Такой выбор оправдан, если необходимо достижение максимальной чувствительности анализа [24] и аналит в пробе не подвергается деструкции. Однако изготовление пищевых продуктов сопряжено с ферментативной и тепловой обработкой, в результате чего в тестируемых пробах содержатся как нативные белковые молекулы, так и их фрагменты, препятствующие образованию детектируемых тройных комплексов.
В настоящей работе рассматриваются возможности применения в качестве контролируемых компонентов молекул соевого ингибитора трипсина (СИТ) и их фрагментов. Данный белок является специфическим биомаркером для соевого сырья, который не имеет иммунохимического сходства с белками, содержащимися в мясе. Его содержание в сое высокое, что позволяет работать со значительными разбавлениями проб, исключая влияние компонентов матрикса на результаты анализа.
Установлено, что частичный гидролиз СИТ различными ферментами – алкалазой, трипсином, химотрипсином, бромелаином и папаином – не приводит к разрушению как минимум аллергенных детерминант, а в некоторых случаях даже усиливает связывание с ними [25]. Поэтому оправданным решением для контроля соевого сырья в мясопродуктах представляется реализация конкурентного иммуноанализа СИТ. Для повышения доступности антигенных детерминант были учтены рекомендации работы [26], предлагающие в качестве иммуногена подвергнутый структурной модификации СИТ. Такая модификация происходит, в частности, при конъюгировании СИТ с низкомолекулярными гаптенами. В связи с этим разработка анализа осуществлялась с использованием антисывороток против конъюгатов СИТ с гаптенами, отсутствующими в мясопродуктах.
Цель работы – разработка иммуноферментного анализа (ИФА) для определения соевого сырья в пищевых продуктах.
МЕТОДИКА
Реактивы. В работе использовали соевый ингибитор трипсина Кунитца (T9003), детергент тритон X-100 (“Sigma-Aldrich”, США), трис-основание (“Химмед”, Россия), однокомпонентный субстратный раствор тетраметилбензидина (ТМБ) (“НВО Иммунотех”, Россия), антивидовые антитела (коза против кролика), меченные пероксидазой (“Медгамал”, Россия). Кроличьи поликлональные антитела против СИТ (8 препаратов) были получены в рамках предыдущих исследований лаборатории иммунобиохимии ФИЦ биотехнологии РАН (Россия). Все вспомогательные реагенты (соли, кислоты, щелочи и органические растворители) были аналитической или химической чистоты. Буферные растворы готовили на воде, деионизированной с помощью установки Milli-Q (“Millipore”, США). ИФА проводили в 96-луночных прозрачных полистироловых микропланшетах Costar 9018 (“Corning Costar”, США).
Тестирование связывания антисывороток с СИТ. В лунках микропланшета сорбировали по 50 мкл раствора 0.1 мкг/мл СИТ в 50 мМ К-фосфатном буфере, рН 7.4, содержащем 0.1 М NaCl (ФБС), в течение ночи при 4°С. Затем четырехкратно отмывали микропланшет ФБС, содержащим 0.05% Тритона Х-100 (ФБСТ). Далее в лунки микропланшета вносили по 50 мкл антисывороток против СИТ (интервал разведений 1 : 200–1 : 107), инкубировали в течение 1 ч при 37°С, а затем четырехкратно отмывали ФБСТ и добавляли в лунки по 50 мкл антивидовых антител, меченных пероксидазой (разведение 1 : 3.000 в ФБСТ), и снова инкубировали 1 ч при 37°С. После отмывки определяли пероксидазную активность связавшейся с носителем ферментной метки. Для этого в лунки микропланшета вносили по 50 мкл раствора субстрата, инкубировали 15 мин при комнатной температуре и останавливали реакцию добавлением 25 мкл 1 М H2SO4. Оптическую плотность (D450) продукта реакции измеряли при 450 нм с использованием микропланшетного фотометра Zenyth 3100 (“Anthos Labtec Instruments”, Австрия).
Конкурентный ИФА СИТ. В лунках микропланшета сорбировали по 50 мкл раствора 0.1 мкг/мл СИТ в ФБС в течение ночи при 4°С, после чего микропланшет четырехкратно отмывали ФБСТ. Далее в лунки вносили по 25 мкл раствора СИТ (интервал концентраций от 1 мкг/мл до 10 пг/мл) или экстрактов соевых продуктов (интервал разведений от 1 : 6 до 1 : 350.000) в ФБСТ и добавляли по 25 мкл антисывороток против СИТ в разведении 1 : 15.000 в ФБСТ. Микропланшет инкубировали 1 ч при 37°С, затем четырехкратно отмывали ФБСТ, добавляли в лунки по 50 мкл антивидовых антител, меченных пероксидазой (разведение 1 : 3.000 в ФБСТ), и снова инкубировали 1 ч при 37°С. После отмывки в лунки вносили по 50 мкл раствора субстрата, инкубировали 15 мин при комнатной температуре, останавливали реакцию добавлением 25 мкл 1 М H2SO4 и измеряли D450. Каждую пробу тестировали в трех повторностях.
Зависимости оптической плотности (y) от концентрации антигена в пробе (x) аппроксимировали 4-параметрической сигмоидной функцией y = (A − D)/(1 + (x/C)B) + D с помощью программного обеспечения Origin Pro 9.0 (“OriginLab”, США). Значение параметра С соответствовало концентрации антигена, ингибирующей связывание антител на 50% (IC50). Концентрацию антигена, вызывающую 10%-ное ингибирование (IC10), рассчитывали с использованием той же функции и рассматривали как предел обнаружения.
Подготовка экстрактов из соевых продуктов. Были протестированы препараты соевого белка и соевого изолята, любезно предоставленные Н.Л. Востриковой (Федеральный научный центр пищевых систем им. В.М. Горбатова РАН), а также приобретенные через розничную торговую сеть. К 15 мг сухих препаратов прибавляли 1.5 мл буферного раствора и тщательно перемешивали с помощью ротационного смесителя RM-1L (“Elmi”, Латвия) в течение 15–45 мин. После этого растворы центрифугировали при 12000 g 10 мин и отбирали для анализа надосадочную жидкость.
Подготовка экстрактов из мясных продуктов. Мясные продукты (колбаса, ветчина и сардельки) покупали в розничной торговой сети. Пробы (2 г) тщательно растирали в ступке с 4 мл буферного раствора, центрифугировали при 12000 g 10 мин и отбирали для анализа надосадочную жидкость.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Характеристика антисывороток. На первом этапе работы было изучено связывание препаратов кроличьих антисывороток с СИТ, иммобилизованным в лунках микропланшета. Из 8 охарактеризованных препаратов наиболее эффективно с антигеном взаимодействовали антисыворотки 3, 5, 6 и 8, кривые титрования которых представлены на рис. 1.
Рис. 1.
Регистрация методом ИФА взаимодействия антисывороток № 3 (1), 8 (2), 6 (3) и 5 (4) с иммобилизованным СИТ в 50 мМ К-фосфатном буфере, рН 7.4, содержащем 0.1 М NaCl и 0.05% Тритона Х-100.

Разработка методики ИФА для определения СИТ. Отобранные препараты были использованы для реализации конкурентного ИФА СИТ. Использовали разведения, найденные по кривым титрования, – 1 : 15 .000 (3), 1 : 5.000 (5), 1 : 20 .000 (6) и 1 : 15.000 (8).
Для этих разведений были получены концентрационные зависимости аналитического сигнала (D450) в диапазоне концентраций СИТ от 5 мкг/мл до 0.03 нг/мл. Пределы обнаружения СИТ варьировали от 0.24 (антисыворотка 8) до 22.9 нг/мл (антисыворотка 6). Следует отметить, что чувствительность ИФА не коррелировала с титром антисывороток, отражающим как аффинность специфических к СИТ антител, так и их содержание в антисыворотке. В дальнейших экспериментах использовали антисыворотку 8, обеспечивающую минимальный предел обнаружения СИТ в конкурентном ИФА.
Представленный на рис. 2 эксперимент был проведен при иммобилизации в лунках микропланшета 0.1 мкг/мл СИТ. Снижение концентрации иммобилизуемого в лунках СИТ, потенциально способствующее дополнительному увеличению чувствительности анализа, существенно снижало амплитуду изменений D450 и, соответственно, ухудшало точность определения его содержания в пробах (рис. 3).
Рис. 2.
Градуировочные кривые ИФА СИТ, полученные с использованием антисывороток № 3 (1), 8 (2), 6 (3) и 5 (4).

Рис. 3.
Градуировочные кривые ИФА СИТ, полученные для 0.5 (1), 0.2 (2), 0.1 (3) и 0.05 (4) мкг/мл СИТ, иммобилизованных в лунках микропланшета.

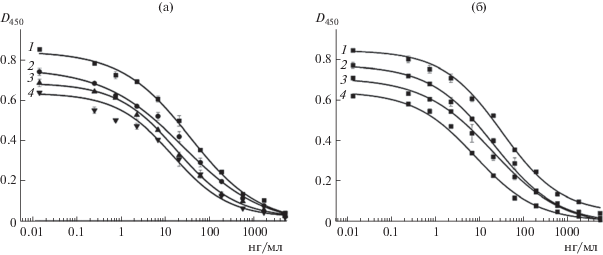
Показано, что сокращение продолжительности конкурентной стадии (рис. 4а) и стадии взаимодействия с антивидовыми антителами (рис. 4б), меченными пероксидазой, с 1 ч до 15 мин не приводило к потере чувствительности ИФА и амплитуды изменений D450, что позволяло сократить продолжительность анализа с 2 ч 30 мин до 1 ч.
Рис. 4.
Градуировочные кривые ИФА СИТ, полученные при изменении продолжительности конкурентной стадии от 60 (1), 45 (2), 30 (3) до 15 (4) мин (а) и стадии взаимодействия с мечеными антивидовыми антителами (б).
На рис. 5 представлен полученный в оптимизированных условиях градуировочный график зависимости оптической плотности при 450 нм от логарифма концентрации СИТ. Предел обнаружения СИТ составил 3.5 нг/мл, а диапазон определяемых концентраций – 7.7–110 нг/мл. Относительное отклонение сигнала в рабочем диапазоне анализа изменялось в интервале от 0.1 до 2.9%.

Пробоподготовка. Переход к анализу реальных проб сопряжен с необходимостью выбора режима пробоподготовки, прежде всего, состава экстрагирующего раствора, позволяющего, с одной стороны, максимально извлечь СИТ из препарата, а с другой – минимизировать влияние других компонентов матрикса на результаты ИФА. Вопросы пробоподготовки для иммуноанализа препаратов сои неоднократно рассматривались в предшествующих работах и суммированы в сравнительных исследованиях [27, 28]. Однако вопросы о рН экстрагирующей среды и содержании в ней детергента оставались открытыми и потребовали дополнительного изучения.
Для экстракции СИТ из соевых препаратов, используемых при производстве продуктов питания (соевый изолят и соевый белок), были протестированы растворы с рН 5.0 (50 мМ ацетатный буфер), 7.4 (50 мМ ФБС) и 9.0 (50 мМ Трис-HCl).
Показано, что ФБС и Трис-HCl обладали одинаковой экстрагирующей способностью, тогда как с помощью ацетатного буфера за то же время экстрагировалось в 7–8 раз меньше иммуноактивных компонентов. Для дальнейших экспериментов был выбран ФБС, оптимальный для проведения ИФА.
Увеличение концентрации детергента Тритон Х-100, добавляемого к экстрагирующему буферному раствору, в диапазоне от 0.05 до 1% не приводило к увеличению количества экстрагируемых из соевых препаратов иммуноактивных компонентов. Поэтому для дальнейшей работы использовали ФБС, содержащий 0.05% детергента.
Сравнение результатов ИФА при разной продолжительности экстракции показало, что основная часть (85–90%) белка экстрагировалась из соевых препаратов в первые 15 мин. Дальнейшее увеличение времени экстракции не приводило к существенному увеличению содержания экстрагируемого белка.
Для оценки эффективности выбранного таким образом протокола пробоподготовки были охарактеризованы результаты, получаемые при нескольких последовательных циклах экстракции. После проведения первого цикла к полученному осадку добавлялась новая порция экстрагирующего буфера, смесь перемешивали, центрифугировали и отбирали супернатант, а полученный осадок подвергали очередной экстракции. При тестировании полученного ряда супернатантов методом ИФА показано, что 89.3% СИТ экстрагируется уже после первого цикла (табл. 1).
Таблица 1.
Определение СИТ с помощью разработанной методики ИФА в пробах, полученных после нескольких последовательных экстракций из соевого изолята
| Цикл экстракции | СИТ | |
|---|---|---|
| мкг/мл | % | |
| 1 | 29 | 89.3 |
| 2 | 2.7 | 8.3 |
| 3 | 0.4 | 1.2 |
| 4 | 0.3 | 0.9 |
| 5 | 0.1 | 0.3 |
Использование разработанной методики ИФА для анализа пищевых продуктов. Разработанная методика была применена для анализа 6 препаратов сои. Для двух соевых изолятов содержание СИТ составило 309 и 137 мкг на мл экстракта, для четырех препаратов соевого протеина – 269, 154, 4.0 и 1.2 мкг на мл экстракта. Все величины, полученные методом ИФА, соответствовали данным изготовителей о номинальном содержании соевого сырья.
Разработанная методика была также применена для анализа 6 мясных продуктов. Содержание СИТ составило: для сарделек – 1.0%, для колбасы ливерной – 0.05%, для ветчины из бедра индейки – 0.15%. В двух препаратах колбасы и препарате буженины СИТ не был выявлен. Как следует из полученных данных, несмотря на температурную и ферментативную обработки, сохранялись антигенные компоненты СИТ, которые позволили провести количественную оценку содержания соевого сырья. В отличие от соевых препаратов в данном случае достоверные сведения о составе продуктов были получены лишь для препарата колбасы, изготовленной в ФНЦ пищевых систем им. В.М. Горбатова РАН (Москва, Россия). Тем не менее полученные результаты позволили сделать вывод как о пригодности разработанного аналитического метода, так и соответствии содержания соевого сырья в протестированных продуктах его допустимому уровню для соответствующей продукции.
Принципиальными преимуществами ИФА как средства характеристики состава пищевых продуктов являлись его малая продолжительность, производительность благодаря возможности одновременной характеристики нескольких десятков проб, доступность недорогого оборудования для проведения тестирования и регистрации результатов, автоматизация основных манипуляций и возможность количественной характеристики содержания контролируемых компонентов. Важно, что, несмотря на особенности технологии получения мясопродуктов, вызывающие частичную деструкцию СИТ, сохраняющиеся антигенные детерминанты позволили осуществить достоверную иммунодетекцию.
Работа выполнена при финансовой поддержке Российского научного фонда (проект № 19-16-00108).
Список литературы
L’Hocine L., Boye J.I. // Crit. Rev. Food Sci. Nutr. 2007. V. 47. № 2. P. 127–143.
Choi M.S., Rhee K.C. // Med. Food. 2006. V. 9. № 1. P. 1–10.
He L., Han M., Qiao S., He P., Li D., Li N., Ma X. // Curr. Protein Pept. Sci. 2015. V. 16. № 7. P. 613–621.
Piccolo F., Vollano L., Base G., Girasole M., Smaldone G., Cortesi M.L. // Ital. J. Food Safety. 2016. V. 5. № 3(5780). P. 161–164.
Jiang T.L., Cai Q.F., Shen J.D., Huang M.J., Zhang L.J., Liu G.M., Cao M.J. // LWT – Food Sci. Technol. 2015. V. 64. № 1. P. 344–349.
Spychaj A., Pospiech E., Iwańska E., Montowska M. // J. Sci. Food Agric. 2018. V. 98. № 13. P. 4807–4815.
Hoffmann B., Münch S., Schwägele F., Neusüß C., Jira W. // Food Control. 2017. V. 71. P. 200–209.
Rosso M.L., Shang C., Edgar Correa E., Zhang B. // Crop Sci. 2018. V. 58. № 4. P. 1616–1623.
Espiñeira M., Santaclara F.J. // Methods Mol. Biol. 2017. V. 1620. P. 173–181.
Soares S., Amaral J.S., Oliveira M.B., Mafra I. // Meat Sci. 2014. V. 98. № 1. P. 41–46.
Costa J., Amaral J.S., Grazina L., Oliveira M.B.P.P., Mafra I. // Food Chem. 2017. V. 221. P. 1843–1850.
Woychik J.H., Happich M.C., Trinh H., Seilers R. // J. Food Sci. 1987. V. 52. № 6. P. 1532–1534.
Randulová Z., Tremlová B., Řezáčová–Lukášková Z., Pospiech M., Straka I. // Czech J. Food Sci. 2011. V. 29. № 4. P. 318–321.
Dzantiev B.B., Byzova N.A., Urusov A.E., Zherdev A.V. // TrAC Trends Anal. Chem. V. 55. P. 81–93.
Alves R.C., Barroso M.F., González-García M.B., Oliveira M.B., Delerue-Matos C. // Crit. Rev. Food Sci. Nutr. 2016. V. 56. № 14. P. 2304–2319.
Schubert-Ullrich P., Rudolf J., Ansari P., Galler B., Führer M., Molinelli A., Baumgartner S. // Anal. Bioanal. Chem. 2009. V. 395. № 1. P. 69–81.
You J., Li D., Qiao S., Wang Z., He P., Ou D., Dong B. // Food Chem. 2008. V. 106. P. 352–360.
Xi J., Shi Q. // Food Anal. Methods. 2016. V. 9. № 11. P. 2998–3005.
Ma X., Sun P., He P., Han P., Wang J., Qiao S., Li D. // Food Chem. 2010. V. 121. P. 546–551.
Wang Y., Li Z., Pei Y., Li Q., Sun Y., Yang J., Yang Y., Zhi Y., Deng R., Hou Y., Hu X. // Food Anal. Methods. 2017. V. 10. № 7. P. 2429–2435.
Geng T., Liu K., Frazier R., Shi L., Bell E., Glenn K., Ward J.M. // J. Agric. Food. Chem. 2015. V. 63. № 20. P. 4947–4953.
Ueberham E., Spiegel H., Havenith H., Rautenberger P., Lidzba N., Schillberg S., Lehmann J. // J. Agric. Food Chem. 2019. V. 67. № 31. P. 8660–8667.
Wang Y., Li Y., Wu J., Pei Y., Chen X., Sun Y. // Food Agric. Immunol. 2019. V. 30. № 1. P. 1202–1211.
Wild D. The Immunoassay Handbook. 4th Ed. Amsterdam: Elsevier Science, 2013. 1036 p.
Panda R., Tetteh A.O., Pramod S.N., Goodman R.E. // J. Agric. Food Chem. 2015. V. 63. № 43. V. 9629–9639.
Cucu T., Devreese B., Kerkaert B., Rogge M., Vercruysse L., De Meulenaer B. // Food Anal. Methods. 2012. V. 5. № 5. P. 1121–1130.
Luthria D.L., John K.M.M., Marupaka R., Natarajan S. // J. Sci. Food Agric. 2018. V. 98. № 15. P. 5572–5580.
Amponsah A., Nayak B. // J. Sci. Food Agric. 2018. V. 98. № 6. P. 2378–2384.
Дополнительные материалы отсутствуют.
Инструменты
Прикладная биохимия и микробиология