

Прикладная биохимия и микробиология, 2020, T. 56, № 6, стр. 595-601
Препаративная очистка бактериофагов Pseudomonas aeruginosa комбинацией гель-проникающей и анионообменной хроматографии
Н. Н. Ландышев 1, Я. Г. Воронько 1, Е. Е. Куликов 2, Н. Н. Сыкилинда 3, К. А. Мирошников 3, *
1 Российский университет дружбы народов, Медицинский институт
117198 Москва, Россия
2 Институт микробиологии им. Виноградского ФИЦ “Биотехнологии” РАН
117312 Москва, Россия
3 Институт биоорганической химии им. М.М. Шемякина и Ю.А. Овчинникова РАН
117997 Москва, Россия
* E-mail: kmi@ibch.ru
Поступила в редакцию 15.06.2020
После доработки 30.06.2020
Принята к публикации 02.07.2020
Аннотация
Разработана и описана схема очистки бактериофагов Pseudomonas aeruginosa, состоящая из последовательных этапов гель-фильтрационной и анионобменной хроматографии. Испытание схемы на пяти фагах различных размеров и морфологии показало одинаковые результаты, что позволяет рассматривать предложенный подход как универсальный и хорошо масштабируемый.
Бактериофаги (фаги), открытые в начале XX в., нашли свое применение во многих сферах. Они используются как противомикробные агенты, средства для фагового дисплея в изучении пептидов, инструменты эффективной вакцинации, доставки генов, а также специфического обнаружения и типирования бактерий. Чистота бактериофагов, необходимая для каждого применения, значительно различается. При этом фаговые препараты, предназначенные для медицинского и пищевого использования, должны содержать минимальное количество бактериальных токсинов [1–5]. Первые успешные попытки хроматографической очистки фагов были предприняты в 60–70 гг. прошлого столетия [6], однако впоследствии осаждение полиэтиленгликолем (ПЭГ) и ультрацентрифугирование в градиенте плотности CsCl стали общепринятыми методами на несколько десятилетий. Такие способы обеспечивают получение бактериофагов необходимой чистоты в лабораторном масштабе, однако они неприемлемы для промышленного применения из-за ограничений в масштабировании, невысокого выхода и необходимости последующего удаления CsCl [7].
В последние годы наблюдается возрождение интереса к применению хроматографических методов очистки фагов. Многочисленные исследования показали эффективность анионообменной хроматографии (АОХ) на метилметакрилатных монолитных смолах, однако высокая цена и сложность регенерации носителя остаются существенными препятствиями для массового внедрения метода [8–11]. Таким образом, потребность в доступных, масштабируемых и эффективных методах очистки бактериофагов остается открытой.
Цель работы – изучение возможности комбинированной хроматографической очистки (гель-фильтрация и анионобменная хроматография) для препаративной очистки бактериофагов.
МЕТОДИКА
Бактериальные штаммы и бактериофаги. В работе в качестве модельных использовали охарактеризованные бактериофаги Pseudomonas aeruginosa, относящиеся к различным группам порядка Caudovirales: phiKZ [12], SN [13], phiKMV [14], YuA [15], а также нитчатый (Inoviridae) фаг pFB, по размерам частицы и генома сходный с описанным фагом pF5 [16]. Морфология, размеры и другие параметры фагов приведены в табл. 1. Первые три бактериофага могут быть обнаружены в составе экспериментальных [17] и промышленных [18, 19] фаговых препаратов для лечения вторичных инфекций ран и ожогов, вызванных синегнойной палочкой. Для культивирования бактериофагов использовали лабораторный штамм P. aeruginosa PAO1 (ATCC 15692). Несмотря на отсутствие в этом штамме факторов вирулентности и пониженный уровень синтеза экзополисахаридов, лизаты PAO1 обладают высоким уровнем пирогенности [20]. Таким образом, выбранная модель позволяет адекватно верифицировать метод в контексте медицинского и ветеринарного применения бактериофагов.
Таблица 1.
Бактериофаги Pseudomonas aeruginosa, использованные в работе (таксономические данные представлены в соответствии с выпуском ICTV-2018b)
| Фаг | Семейство/подсемейство | Род | Размер (капсид/хвост) | Геном, п.н. | NCBI Genbank № | Ссылка |
|---|---|---|---|---|---|---|
| PhiKZ | Myoviridae | Phikzvirus | 145/200 | 280.334 | NC_004629 | [12] |
| SN | Myoviridae | Pbunavirus | 74/140 | 66.390 | NC_011756 | [13] |
| YuA | Siphoviridae | Yuavirus | 72 × 51/145 | 58.663 | AM_749441 | [15] |
| phiKMV | Podoviridae/Autographivirinae | Phikmvvirus | 60/20 | 42.519 | NC_005045 | [14] |
| pFB1* | Inoviridae | – | – | 12.409 | Родственнен pF5 | [16] |
Культивирование и первичное выделение бактериофагов. Фаголизаты были получены традиционным способом [21]. Клетки штамма PAO1 P. aeruginosa культивировали при 37°C и интенсивной аэрации в жидкой среде 2xTY (бакто-триптон – 16 г/л, дрожжевой экстракт–10 г/л, NaCl – 5 г/л ) до плотности 2 × 108 кл./мл, затем инфицировали суспензией фага и инкубировали до полного лизиса клеток. Дополнительно клетки лизировали обработкой 0.5%-ным (об./об.) раствором хлороформа в течение 2 ч при 4°C. Затем фаголизат обрабатывали ДНКазой I (“Sigma”,США, 20 г/мл) в течение 1 ч. Фрагменты клеток удаляли центрифугированием при 4000 g (Megafuge 2.0 R, “Heraeus”, Германия) в течение 20 мин. Титр фагов в фаголизате составил 1010–1011 БОЕ/мл
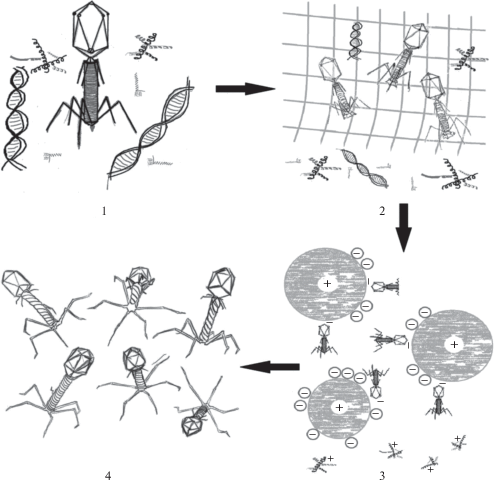
Жидкостная хроматография. Хроматографию проводили на полуавтоматической хроматографической системе Pharmacia FPLC (“GE Healthcare”, США) с проточной детекцией продуктов по поглощению УФ-излучения с длиной волны 280 нм (А280). Для оценки воспроизводимости и точности метода, а также емкости колонки, все эксперименты проводили для каждого фага трижды. Схематическое представление эксперимента показано на рис. 1.
Рис. 1.
Схематическое изображение эксперимента: 1 этап – лизат с бактериофагами и примесями клеточных метаболитов, белков, ДНК; 2 этап – гель-фильтрационная хроматография (задержка бактериофага и крупных белков/ДНК в матрице стекла с контролируемым размером пор); 3 этап – анионобменная хроматография (показано взаимодействие отрицательно заряженных бактериофагов с положительно заряженной смолой); 4 этап – очищенные бактериофаги.
Гель-проникающая хроматография. Микропористое стекло с контролируемым размером пор 500 Å, mesh 120/200 (“Serva”, Германия) ресуспендировали в воде и заполняли колонку 1.5 × 75 см, снабженную верхним адаптером (“Bio-Rad”, США). Сорбент уравновешивали буферным раствором А (20 мM трис–HCl, pH 8.0) и промывали 3%-ным (вес./об.) полиэтиленгликолем (ПЭГ) 20.000 (“Fluka Analytics”, США) в буферном растворе А до стабилизации базовой линии. Бактериофаги осаждали центрифугированием при 24 000 g (Sorvall RS 5, ротор SA600, “Thermo Fisher Scientific”, США) в течение 1 ч при 4°C. Осадок ресуспендировали в 2 мл среды 2хTY и наносили на колонку. Изократическую элюцию буферным раствором А проводили при скорости 4 мл/мин. После каждой хроматограммы колонку стерилизовали 2 об. 30%-ного этанола в буферном растворе А.
Анионоообменная хроматография. Для хроматографии использовали картриджи на 5 мл EconoPac High Q (“Bio Rad”, США). Фракции, полученные после гель-проникающей хроматографии, соответствующие частично очищенному фагу, наносили на колонку, уравновешенную буферным раствором А. Сорбированный материал элюировали линейным градиентом (0–1.0 М) NaCl в буферном растворе А. Фракции, содержащие инфекционные фаговые частицы, диализовали против фагового буферного раствора (10 мM Трис-HCl pH 7.5; 200 мM NaCl, 2 мM MgSO4) и, при необходимости, концентрировали с помощью центрифужного ультраконцентратора Centricon YM 100 (“Millipore”, США).
Активность бактериофагов. Инфекционную активность в лизатах и фракциях при хроматографии определяли традиционным титрованием с использованием двуслойного агара на культуре штамма PAO1 P. aeruginosa [21].
Электрофорез в полиакриламидном геле. Чистоту и целостность частиц бактериофагов оценивали посредством разделения структурных белков электрофорезом в 12%-ном полиакриламидном геле в денатурирующих условиях с последующей окраской Coomassie Blue [22] и визуальным сравнением профилей белковых полос с образцовыми структурными электрофореграммами препаратов бактериофагов, очищенных ультрацентрифугированием в градиенте CsCl.
Электронная микроскопия. Препараты бактериофагов наносили на покрытую коллодийной пленкой и напыленную аморфным углеродом сетку и контрастировали 1%-ным раствором уранил-ацетата в метаноле [23]. Изображения бактериофагов были получены методом просвечивающей электронной микроскопии с использованием микроскопа Jeol-JEM CX100 (“Jeol”, Япония) с ускоряющим напряжением 100 кВ.
Контроль концентрации эндотоксина. Концентрацию эндотоксина в образцах бактериофагов оценивали в абсолютных значениях, выраженных в единицах эндотоксина/мл (ЕЭ/мл), с помощью фармакопейного полуколичественного LAL-теста согласно инструкции производителя (“Merck”, Германия).
Статистический анализ проводили в системе Microsoft Excel 2016.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Очистка бактериофагов с использованием комбинированного подхода. В настоящем исследовании была использована комбинация хроматографических методов (гель-фильтрации и анионобменной хроматографии) для очистки бактериофагов. Применение такой системы позволило достичь снижения концентрации эндотоксинов в препарате до <150 ЕЭ/мл, вне зависимости от размера и морфологии вируса. Чистоту полученных препаратов подтверждали результатами электронной микроскопии, убедительно показавшими просветление фона и уменьшение количества загрязняющих везикулярных частиц (рис. 2). В то же время инфекционность фаговых частиц оставалась на высоком уровне 1–510–11 БОЕ/мл, выход метода составил >59% в зависимости от вида очищаемого вируса. Параметры чистоты и выхода конкретных бактериофагов представлены в табл. 2. Несмотря на использование в эксперименте бактериофагов с различной морфологией и размером капсида, выход метода показал высокую сходимость (коэффициент вариации 8.3%), что позволило утвержать, что такой подход может быть универсальным.
Рис. 2.
Электронная микрофотография фага SN с негативным контрастированием: а– после гель-фильтрации, б – очищенный фаг(шкала – 100 нм).

Таблица 2.
Результаты гель-фильтрационной и анионобменной хроматографии
| Фаг | PhiКZ | SN | YuA | phiKMV | pFB1 |
|---|---|---|---|---|---|
| Титр в лизате БОЕ*1011/мл | 1.7 | 7 | 3 | 4 | 1 |
| Титр после гель-фильтрации БОЕ*1011/мл | 1.3 | 5.8 | 3 | 3 | 0.8 |
| Выход после гель-фильтрации, % | 76 | 84 | 100 | 75 | 80 |
| Титр после анионобменной хроматографии БОЕ *1011/мл | 1 | 5 | 2.1 | 2.5 | 0.7 |
| Выход после анионобменной хроматографии, в % | 77 | 86 | 70 | 83 | 88 |
| Выход после комбинированной очистки,% | 59 | 71 | 70 | 62 | 70 |
| Концентрация эндотоксина | <150 ЕЭ/мл | ||||
Применение хроматографии представляет собой перспективную альтернативу существующим методам в контексте крупномасштабного промышленного получения медицинских препаратов. Однако на сегодняшний день не существует метода, который удовлетворял бы всем требованиям “золотого стандарта”, дающего возможность получения препаратов высокой степени очистки, имеющих невысокую стоимость, универсально подходящих для очистки фагов разной морфологии и размера, экологичных и масштабируемых. Предложенная нами комбинация хроматографических подходов направлена на достижение оптимального баланса между указанными требованиями.
Гель-проникающая хроматография на стекле с контролируемыми порами. Выбор такого метода хроматографии в качестве первого этапа очистки был обусловлен необходимостью максимального удаления ионогенных биомолекул ДНК и РНК перед анионобменной хроматографией. Согласно полученным результатам, выход фага показал высокую сходимость (коэффициент вариации 12%), хроматографические профили также были сопоставимы (рис. 3), что позволило утверждать, что гель-проникающая хроматография может быть использована для первичной очистки бактериофагов разной морфологии и размера. Корреляционный анализ показал, что выход метода слабо зависит от размера капсида (r = –0.34) – слабая отрицательная связь согласно шкале Чеддока и очень слабо зависит от размера хвоста и общей длины вируса (r = 0.25 и 0.05 соответственно) – очень слабая положительная связь согласно шкале Чеддока.
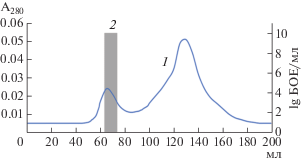
Рис. 3.
Хроматограмма, полученная при гель-фильтрации фаголизата на стекле с контролируемым размером пор. 1 – A280; 2 – инфекционность фага в собранных фракциях, lg БОЕ/мл.
Каркасы из пористого стекла с контролируемым размером пор (controlled-pore glass, GPG) являются распространенными матрицами для синтеза хроматографических сорбентов. Такие матрицы могут быть использованы для хроматографии и без модификации. Разделение на стеклянных носителях основано на сочетании гель-фильтрации и обратимой сорбции компонентов на поверхности. Термодинамические основы сорбции вирусов описаны в работе [24]. Адсорбирующая поверхность пор может быть уменьшена за счет обработки ПЭГ [25]. На этапе планирования эксперимента предполагали, что неспецифическое связывание фагов частицами смолы значительно снизит выход метода, однако в ходе эксперимента гипотеза о необходимости добавления ПЭГ была опровергнута. Вероятно, низкий уровень необратимого связывания фагов объясняется более высоким сродством низкомолекулярных компонентов лизата к частицам сорбента, что препятствовало адсорбции фага.
Успех хроматографической очистки бактериофагов зависит от понимания того, как эндотоксины бактерий ведут себя на различных сорбентах. Благодаря амфифильной природе молекулы липополисахарида, а также его ассоциации с мембранными липидами в водном растворе в присутствии двухвалентных катионов линейные эндотоксины формируют мицеллы и везикулы с молекулярной массой более 1000 кДа [26]. Получившиеся структуры отличаются по своему размеру от фаговой частицы меньше, чем мономеры эндотоксинов, что усложняет выбор среды для эффективной очистки от липополисахаридов методом гель-фильтрации [27]. В настоящем исследовании показано, что размер пор 500 Å оптимален для такого применения.
Большинство исследований, посвященных применению хроматографии со стеклом с контролируемым размером пор, были проведены в 70 гг прошлого века. Последующее развитие синтетической химии привело к появлению сорбентов для гель-фильтрационной хроматографии, имеющих высокую стабильность и выраженную зависимость объема элюции от объема и массы элюата. Таким образом, хроматография с использованием CPG была признана устаревшей для использовании в биологии. Однако такой вид разделения имеет множество преимуществ: низкую себестоимость, высокие механическую и химическую стабильность по сравнению с агарозой, целлюлозой, декстраном и поливиниловыми смолами и, кроме того, позволяет легко масштабировать процесс и применять высокую скорость элюции без заметного воздействия на разрешение. Отдельным преимуществом является возможность использования доступных и экологичных реагентов для очистки и стерилизации колонки.
В литературе приведено множество примеров успешного применения CPG хроматографии для очистки крупных мицелл [28], токсинов [29], а также вирусов растений [30, 31], животных и бактерий [32]. Применение данного метода для вирусов позволяет добиться чистоты, сопоставимой с таковой при ультрацентрифугировании в градиенте сахарозы, что подтверждается параметрами оптической плотности, тестами на инфекционность и серологическими методами.
Анионобменная хроматография (АОХ). Выбор АОХ в качестве второго этапа очистки бактериофагов обусловлен большим опытом применения данного метода для очистки вирусов, что было подробно рассмотрено в обзоре [33]. Согласно полученным данным, выход фага после этого метода составил более 70% (табл. 2). Коэффициент вариации выхода, равный 9%, и сходимость полученных хроматограмм (рис. 4) позволили утверждать, что АОХ применима для очистки фагов разной морфологии и размеров.
Рис. 4.
Анионобменная хроматография образца частично очищенных фракций после гель-фильтрации. 1 – A280, 2 ‒ градиент NaCl от 0 до 1.0 М, 3 – инфекционность фага в собранных фракциях, lg БОЕ/мл.

Несмотря на ряд преимуществ, у данного метода есть и некоторые ограничения: распределение частиц сорбента по размерам не оптимально для прохождения между ними больших частиц, клеточные лизаты содержат большое количество заряженных примесей (особенно нуклеиновых кислот), которые конкурируют с бактериофагами за участки связывания. Первый недостаток может быть преодолен использованием расширенной упаковки колонок [34] или монолитных сорбентов с большим размером каналов, которые улучшают прохождение вирусов через колонку и повышают степень иммобилизации [11]. Использование метилметакрилатных монолитов, однако, не удовлетворяло условиям идеальной колонки из-за высокой цены и, как следствие, трудностей масштабирования. Второе ограничение может быть преодолено предварительной очисткой препарата от нуклеиновых кислот. В данной работе большая часть бактериальной ДНК и мРНК была удалена во время первого этапа хроматографии, что позволило достичь удовлетворительной емкости колонки (~1010 фаговых частиц/мл смолы). Сравнительные эксперименты с сорбентами, модифицированными четвертичными аминогруппами, от различных производителей: Q-Sepharose Fast Flow (“GE Healthcare”, США), Toyopearl S-uperQ 650M (“Tosoh biosciences”, Япония), MacroPrep HighQ (“Bio Rad”, США), не показали значимых преимуществ какого-то продукта по емкости или разрешению.
Также при сравнении хроматограмм была отмечена интересная особенность: во всех экспериментах концентрации NaCl, при которых проходила элюция фага, были близки вне зависимости от размера или морфологии фаговых частиц (рис. 3), что открыло возможности для изолирования сообществ нитчатых и хвостатых фагов с последующей одновременной элюцией с колонки.
Контроль содержания бактериальных эндотоксинов. Клеточные лизаты содержат большое количество компонентов среды, фрагментов клеточных стенок бактерий, эндо- и экзотоксинов, пептидогликанов, органелл и других потенциально иммуногенных факторов. Потенциальная опасность конкретных загрязнителей зависит от вида и штамма бактерий. В то время как компоненты лизата клеток Escherichia coli не вызывают, как правило, серьезных проблем и даже повышают противомикробный ответ, лизаты клеток Salmonella enteridis pv typhimurium крайне токсичны из-за наличия в них фрагментов полисахаридов клеточных стенок. В настоящей работе не была исследована концентрация эндотоксинов в процессе хроматографической очистки, однако экспериментально определенное конечное содержание эндотоксинов в очищенном препарате составило <150 ЕЭ/мл раствора бактериофага с концентрацией ~1011 БОЕ/мл. Растворы с таким значением могут применяться как местно, так и внутрь без дополнительной очистки [35, 36].
В настоящем исследовании продемонстрирована оптимальная комбинация хроматографических методов для препаративной очистки бактериофагов из клеточных лизатов. Оценку метода проводили на 5 бактериофагах P. aeruginosa, отличающихся по морфологии и размерам. Во всех случаях двухстадийная очистка позволяла избавиться от нежелательных примесей без значительных потерь титра и инфекционной способности фаговых частиц. Выход метода превышал 60%, а чистота полученных фаговых препаратов была равна или превосходила чистоту образцов, полученных методом градиентного ультрацентрифугирования В процессе очистки использовались недорогие хроматографические сорбенты, масштабируемость которых была продемонстрирована зарубежными исследователями [37], использовалось простое аппаратурное оформление (перистальтический насос и УФ-монитор). Например, фракции, полученные при хроматографии на стекле с контролируемым размером пор, могли быть сразу же нанесены на колонку для анионообменной хроматографии. Отдельно стоит отметить, что значения pH буферов, используемых при очистке, близки к физиологическим, что позволило сохранить инфекционную способность бактериофагов. Полученные нами результаты были подтверждены и другими авторами [11, 38].
Необходимо отметить, что разработанный метод имеет и недостатки. Например, нам не удалось избежать этапа центрифугирования из-за необходимости концентрирования лизата перед нанесением на колонку CPG. Использование буферных растворов с высокой ионной силой (600–700 мМ NaCl) не было оптимальным для хранения и применения бактериофагов, так как требовало последующего диализа. Однако разработанный протокол требует меньше времени и более универсален, по сравнению с широко используемым методом преципитации ПЭГ и ультрацентрифугирования в градиенте CsCl, что позволяет применять его для очистки фагов различной морфологии.
Авторы благодарят Е.Д. Шибанову (ИБХ РАН) за проведение LAL-теста.
Список литературы
Abedon S.T., Kuhl S.J., Blasdel B.G., Kutter E.M. // Bacteriophage. 2011. V. 1. № 2. P. 66–85.
Górski A., Miȩdzybrodzki R., Borysowski J., Weber-Dąbrowska B., Łobocka M., Fortuna W., Letkiewicz S., Zimecki M., Filby G. //Curr. Opin. Investig. Drugs. 2009. V. 10. № 8. P. 766–774.
Skurnik M., Pajunen M., Kiljunen S. //Biotechnol. Lett. 2007. V. 29. № 7. P. 995–1003.
Cooper C.J., Denyer S.P., Maillard J.Y. //Lett. Appl. Microbiol. 2014. V. 58. № 2. P. 118–122.
Gill J.J., Hyman P. //Curr. Pharm. Biotechnol. 2010. V. 11. № 1. P. 2–14.
Bachrach U., Friedmann A. //Appl. Microbiol. 1971. V. 22. № 4. P. 706–715.
Mutti M., Corsini L. //Front. Microbiol. 2019. V. 10. P. 2289
Adriaenssens E.M., Lehman S.M., Vandersteegen K., Vandenheuvel D., Philippe D.L., Cornelissen A., Clokie M.R.J., García A.J., De Proft M., Maes M., Lavigne R. // Virology. 2012. V. 434. № 2. P. 265–270.
Vandenheuvel D., Rombouts S., Adriaenssens E.M. Purification of Bacteriophages Using Anion-Exchange Chromatography. In: Methods in Molecular Biology. N.Y.: Humana Press, 2018. P. 59–69.
Oksanen H.M., Domanska A., Bamford D.H. // Virology. 2012. V. 434. № 2. P. 271–277.
Smrekar F., Ciringer M., Štrancar A., Podgornik A. // J. Chromatogr. A. 2011. V. 1218. № 17. P. 2438–2444.
Mesyanzhinov V.V, Robben J., Grymonprez B., Kostyuchenko V.A., Bourkaltseva M.V, Sykilinda N.N., Krylov V.N., Volckaert G. // J. Mol. Biol. 2002. V. 317. № 1. P. 1–19.
Ceyssens P.J., Miroshnikov K., Mattheus W., Krylov V., Robben J., Noben J. P., Vanderschraeghe S., Sykilinda N., Kropinski A.M., Volckaert G., Mesyanzhinov V., Lavigne R. // Environ. Microbiol. 2009. V. 11. № 11. P. 2874–2883.
Lavigne R., Burkal’tseva M.V, Robben J., Sykilinda N.N., Kurochkina L.P., Grymonprez B., Jonckx B., Krylov V.N., Mesyanzhinov V.V, Volckaert G. // Virology. 2003. V. 312. № 1. P. 49–59.
Ceyssens P.J., Mesyanzhinov V., Sykilinda N., Briers Y., Roucourt B., Lavigne R., Robben J., Domashin A., Miroshnikov K., Volckaert G., Hertveldt K. // J. Bacteriol. 2008. V. 190. № 4. P. 1429–1435.
Mooij M.J., Drenkard E., Llamas M.A., Vandenbroucke-Grauls C.M.J.E., Savelkoul P.H.M., Ausubel F.M., Bit-ter W. // Microbiology. 2007. V. 153. № 6. P. 1790–1798.
Merabishvili M., Pirnay J.P., Verbeken G., Chanishvili N., Tediashvili M., Lashkhi N., Glonti T., Krylov V., Mast J., Van Parys L., Lavigne R., Volckaert G., Mattheus W., Verween G., De Corte P., Rose T., Jennes S., Zizi M., De Vos D., Vaneechoutte M. // PLoS One. 2009. V. 4. № 3. P. e4944.
Мирошников К.А., Куликов Е.Е., Дарбеева О.С., Лысько К.А., Игнатьев Г.М. // Прикл. биохимия и микробиология. 2014. Т. 50. № 3. С. 338–344.
McCallin S., Sarker S.A., Sultana S., Oechslin F., Brüssow H. // Environ. Microbiol. 2018. V. 20. № 9. P. 3278–3293.
Devleeschouwer M.J., Cornil M.F., Dony J. // Appl. Environ. Microbiol. 1985. V. 50. № 6. P. 1509–1511.
Sambrook J., Russell D.W. Molecular Cloning-Sambrook & Russel. Cold Spring Harbor, N.Y.: Cold Springs Harb. Lab. Press, 2001. P. 200–214.
Brunelle J.L., Green R. //Methods Enzymol. 2014. V. 541. P. 151–159.
Clokie M.R.J., Kropinski A.M. Bacteriophages: Methods and Protocols. 2009. V. XXII. P. 113–127.
Loveland J.P., Ryan J.N., Amy G.L., Harvey R.W. // Colloids Surfaces A Physicochem. Eng. Asp. 1996. V. 107. P. 205–221.
Hawk G.L., Cameron J.A., Dufault L.B. // Prep. Biochem. 1972. V. 2. № 2. P. 193–203.
Ongkudon C.M., Chew J.H., Liu B., Danquah M.K. // ISRN Chromatogr. 2012. V. 2012. P. 1–9.
Lee S.H., Kim J.S., Kim C.W. // Process Biochem. 2003. V. 38. № 7. P. 1091–1098.
Mcneili G.P., Donnelly W.J. // J. Dairy Res. 1987. V. 54. № 1. P. 19–28.
Cassidy P., Harshman S. // Infect. Immun. 1976. V. 13. № 3. P. 982–986.
Marcinka K. //Acta Virol. 1972. V. 16. № 1. P. 53–62.
Krasilnikov I.V., Elbert L.B., Borisova V.N., Nakhapetyan L.A. // Acta Virol. 1985. V. 29. № 4. P. 273–278.
Gschwender H.H., Haller W., Hofschneider P.H. // BBA Sect. Nucleic Acids Protein Synth. 1969. V. 190. № 2. P. 460–469.
Trilisky E.I., Lenhoff A.M. // J. Chromatogr. A. 2007. V. 1142. № 1 SPEC. ISS. P. 2–12.
Anspach F.B., Curbelo D., Hartmann R., Garke G., Deckwer W.D. // J. Chromatogr. A. 1999. V. 865. № 1–2. P. 129–144.
Бактериальные эндотоксины. ОФС.1.2.4.0006.15 Государственная фармакопея Российский Федерации IV издания. Т. 1. М.: Министерство Здравоохранения Российской Федерации, 2018. С. 1233–1250.
Bacterial endotoxins test In: European Pharmacopoeia (Ph. Eur.) 2019. P. 161–168. https://gmpua.com/Validation/Method/LAL/EUPHARMACOPOEIA.pdf
Langer P., Schnabel R. Separation with Porous Glass Membranes and Controlled Pore Glass (CPG) Chromatography In: Separations for Biotechnology 2. Dordrecht, Netherlands: Springer, 1990. P. 371–380.
Smrekar F., Ciringer M., Peterka M., Podgornik A., Štrancar A. // J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2008. V. 861. № 2. P. 177–180.
Дополнительные материалы отсутствуют.
Инструменты
Прикладная биохимия и микробиология