

Российский физиологический журнал им. И.М. Сеченова, 2019, T. 105, № 10, стр. 1203-1214
Глия в трехчастном нервно-мышечном синапсе
С. Е. Проскурина 1, *, К. А. Петров 2
1 Институт органической и физической химии им. А.Е. Арбузова ФИЦ КазНЦ РАН
Казань, Россия
2 Казанский институт биохимии и биофизики ФИЦ КазНЦ РАН
Казань, Россия
* E-mail: svetlana-proskurina@mail.ru
Поступила в редакцию 09.07.2019
После доработки 30.07.2019
Принята к публикации 03.08.2019
Аннотация
В данном обзоре рассматривается роль перисинаптических Шванновских клеток (ПШК) как активного компонента трехчастного синапса. Показана возможность модуляции синаптической передачи ПШК. Описываются механизмы, благодаря которым ПШК не только способна детектировать синаптическую активность, но и изменять ее. Кроме того, описываются изменения ПШК при патологиях периферической нервной системы и обсуждается возможность использования ПШК в качестве потенциальных мишеней для терапевтического воздействия.
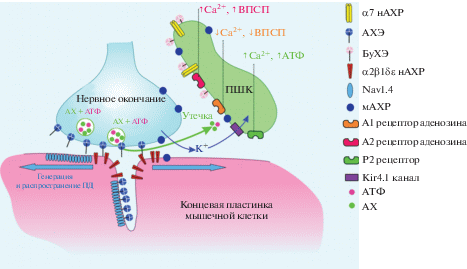
Нервно-мышечная синаптическая передача возбуждения является жизненно важным процессом, позволяющим трансформировать команды, поступающие из нервной системы, в мышечные сокращения. Нервно-мышечный синапс (НМС) представляет собой классический трехчастный синапс (“tripartite synapse”), т.е. синапс, в котором участие в передаче возбуждения принимают не только пресинаптическая и постсинаптическая клетки, но и перисинаптические глиальные клетки (рис. 1). В НМС в качестве пресинаптической клетки выступает окончание двигательного нерва, секретирующего в синаптическую щель нейротрансмиттер ацетилхолин (АХ). Постсинаптическая мембрана представляет собой участок мембраны мышечного волокна, специализированный на генерации возбуждающего постсинаптического потенциала (ВПСП). Глия в НМС представлена несколькими перисинаптическими Шванновскими клетками (ПШК), которые покрывают окончание двигательного нерва и в отличие от Шванновских клеток, покрывающих аксон, не образуют миелиновой оболочки [1, 2].
Рис. 1.
Трехчастный синапс: нервное окончание, мышечное волокно и перисинаптическая Шванновская клетка (ПШК). нАХР – никотиновый ацетилхолиновый рецептор; АХЭ – ацетилхолинэстераза; БуХЭ – бутирилхолинэстераза; Nav1.4 – потенциалуправляемый натриевый канал; мАХР – мускариновый ацетилхолиновый рецептор; А1, А2 рецепторы – аденозиновые рецепторы; Р2 рецептор – пуриновый рецептор; Kir4.1 – калиевый канал; АТФ – аденозинтрифосфат; АХ – ацетилхолин; ПД – потенциал действия; ВПСП – возбуждающий постсинаптический потенциал.
В настоящее время известно, что Шванновские клетки начинают формироваться на ранних стадиях эмбрионального развития из нервного гребня. На первом этапе возникают клетки-предшественники, которые мигрируют вместе с формирующимися аксонами, затем они преобразуются в незрелые Шванновские клетки, которые уже после рождения дифференцируются в зрелые миелинизирующие и немиелинизирующие клетки, к последним относятся ПШК [3, 4].
В процессе образования НМС ПШК формируются раньше, чем окончание двигательного нерва достигает постсинаптической мембраны [5–8]. В настоящее время показано, что ПШК играют важную роль в синаптогенезе, высвобождая множество синаптогенных факторов, либо факторов, опосредованно регулирующих конус роста мотонейрона, к ним относятся нейротрофины – NGF, нейрегулин-1 [9, 10]. Кроме того, ПШК являются источником фактора TGF-β [11], ретроградно активирующего рост пресинаптической терминали [12, 13]. Также было показано, что ПШК выделяют тромбоспондин 4 и α2δ-1, глиальный нейротрофический фактор GDNF, артемин, мозговой нейротрофический фактор BDNF, p75NTR и N-кадгерин – факторы, оказывающие влияние на формирование синапсов как в периферической нервной системе, так и в ЦНС [14, 15]. После завершения формирования НМС в процессе постнатального развития ПШК принимают участие в прекращении полинейрональной иннервации мышечных волокон, элиминируя лишние синаптические контакты [16–20].
Не являясь электровозбудимыми клетками, ПШК до недавнего времени изучались, главным образом, с точки зрения их участия в синаптогенезе, оставаясь за пределами интереса электрофизиологов, занимающихся исследованием синаптической передачи возбуждения с нерва на мышцу. Однако в последние десять лет были получены данные, позволяющие утверждать, что ПШК принимают непосредственное участие в регуляции процесса секреции АХ мотонейроном, что может оказывать значительное влияние, например, на эффективность терапии состояний патологической мышечной слабости.
В данном обзоре представлены современные представления о функциях ПШК в трехчастном НМС. Кроме того, проанализировано возможное участие ПШК в патогенезе заболеваний периферической нервной системы различной этиологии, что позволяет рассматривать ПШК в качестве перспективных мишеней при разработке потенциальных средств терапии.
РОЛЬ ПЕРИСИНАПТИЧЕСКИХ ШВАННОВСКИХ КЛЕТОК В СИНАПТИЧЕСКОЙ ПЕРЕДАЧЕ
В настоящее время НМС можно отнести к классическим объектам нейрофизиологии. Основные этапы процесса нервно-мышечной передачи возбуждения были описаны к середине XX века. Так, в классических работах показано, что электрический сигнал, распространяющийся по аксону мотонейрона, достигая нервного окончания, инициирует вход в него ионов Са2+ через потенциалзависимые кальциевые каналы и освобождает из нервного окончания АХ [21]. При этом АХ может выделяться как в процессе экзоцитоза везикул [22, 23], которые, сливаясь с мембраной клетки, выбрасывают свое содержимое в просвет синаптической щели [24, 25], так и невезикулярным образом [26, 27]. К сожалению, точный механизм и физиологическое значение невезикулярного способа секреции АХ на сегодняшний день не определены.
Диффундируя через синаптическую щель, молекулы АХ достигают концевой пластинки – области мышечного волокна морфологически и функционально специализированной для генерации возбуждающего постсинаптического потенциала (ВПСП). В данной области мембрана мышечного волокна образует многочисленные инвагинации – синаптические складки. За счет подобного строения площадь поверхности многократно увеличивается, что позволяет разместить большое количество рецепторов и ионных каналов (рис. 1). На гребнях складок расположены никотиновые ацетилхолиновые рецепторы (нАХР) мышечного типа – (α1)2β1δε, активация которых приводит к открытию сопряженных ионных каналов. Ток катионов (преимущественно Nа+) через канал нАХР по электрохимическому градиенту приводит к кратковременному снижению мембранного потенциала в области постсинаптической мембраны – генерации ВПСП. В свою очередь ВПСП приводит к открытию расположенных на дне синаптических складок потенциалзависимых натриевых каналов, обеспечивающих генерацию потенциала действия (ПД) мышечного волокна. После генерации электрогенной мышечной мембраной ПД следует восстановление исходной проницаемости мембраны для ионов и ее потенциала покоя до исходного уровня (реполяризация). Скорость возвращения всей системы в исходное состояние определяет лабильность синапса, т.е. способность воспроизводить задаваемый ритм раздражения, и в значительной мере зависит от времени жизни АХ, которое контролируется ферментами ацетилхолинэстеразой (АХЭ) и бутирилхолинэстеразой (БуХЭ). Причем известно, что АХЭ расположена в синаптической щели и, таким образом, контролирует воздействия АХ на пост- и пресинаптическую мембраны. В то же время БуХЭ расположена за пределами синаптической щели, на поверхности ПШК и контролирует время жизни экстрасинаптического АХ [28]. Важно отметить, что помимо АХ в синаптических везикулах содержатся его котрансмиттеры – АТФ [29] и, по некоторым данным, глутамат [30–33].
Помимо нАХР, в НМС описано наличие мускариновых ацетилхолиновых рецепторов (мАХР) – метаботропных рецепторов, ассоциированных с G-белками. С помощью методов иммунногистохимии было показано, что в НМС позвоночных присутствуют мАХР по меньшей мере четырех из пяти известных на данный момент подтипов – М1, M2, M3 и М4 [34], однако на данный момент функциональная роль показана только для М1, M2 и М3 подтипов мАХР. Принято считать, что баланс между активацией М1/М2 подтипами помогает адаптировать уровень секреции АХ к изменению условий активности мотонейрона, предотвращая неоправданный расход данного нейротрансмиттера. Так, было показано, что активация М2 подтипа запускает каскад реакций в нервном окончании, снижающих уровень секреции АХ мотонейроном, а активация М1 подтипа, напротив, стимулирует секрецию АХ [35–38]. Для М3 мАХР, которые, предположительно, расположены на мышечных волокнах, было показано, что их стимуляция запускает секрецию эндоканнабиноида 2-арахидоноилглицерина, который посредством активации CB1-рецепторов запускает снижение секреции мотонейроном АХ [39].
В отличие от остальных двух компонентов трехчастного НМС (мотонейрона и мышечных волокон) ПШК не экспрессируют потенциалзависимых натриевых каналов, поэтому мембрана ПШК не способна генерировать ПД. Традиционно, основную роль ПШК в процессе синаптической передачи возбуждения связывают с контролем концентрации ионов К+. Хорошо известно, что нервные и мышечные клетки при генерации потенциала действия выделяют во внеклеточную среду ионы K+, накопление которых способно приводить к деполяризации мембраны электровозбудимой клетки и, как следствие, инактивации потенциалзависимых Nav1.4 натриевых каналов. Глиальные клетки экспрессируют Kir4.1 калиевые каналы, что позволяет ПШК выступать в качестве буфера ионов К+ в перисинаптическом пространстве [40]. Известно, что своевременное удаление ионов K+ предотвращает развитие мышечного утомления [41].
Помимо калиевых каналов ПШК экспрессирует целый ряд рецепторов нейротрансмиттеров, что позволяет ПШК детектировать синаптическую активность. Показано, что ПШК млекопитающих экспрессируют мАХР [42]. Функциональная значимость мАХР ПШК хорошо выявляется в условиях денервации, вызванной механическим повреждением аксона, либо патологическим процессом. При повреждении аксона активация мАХР на поверхности ПШК прекращается, так как нарушается синаптическая передача. Это запускает процесс де-дифференциации ПШК [43, 44], в ходе которого они теряют свою специфичность и увеличивают способность к миграции. В таком состоянии ПШК начинают синтезировать новые факторы, к примеру, фактор, присущий астроцитам – GFAP – глиальный фибриллярный кислый белок [45, 46], появление которого служит достоверным маркером повреждения аксонов и потери синаптических входов на уровне НМС [47–49]. После этого начинается инфильтрация культи аксона воспалительными клетками, которые представлены тучными клетками, макрофагами и T-лимфоцитами. Сами ПШК тоже начинают вести себя подобно макрофагам и захватывают фагоцитозом клеточные и аксональные остатки; этот процесс запускается митохондриальными факторами – аларминами [50], в частности, перекисью водорода, цитохромом С и митохондриальной ДНК, которые высвобождаются из разрушающегося аксона и участвуют в стимулировании и облегчении повторной иннервации. В ответ на наличие аларминов, ПШК формируют отростки от интактных синапсов к денервированным, которые служат мостиками для направленного прорастания аксона и приводят к спраутингу неповрежденных аксонов [51–53]. Таким образом, тоническая стимуляция мАХР ПШК в процессе синаптической активности играет важную роль в поддержании синаптического баланса.
Кроме мАХР, ПШК экспрессируют пуриновые рецепторы, которые могут быть активированы аденозинтрифосфатом (АТФ), выделяющимся совместно с секрецией АХ или же продуктом гидролиза АТФ – аденозином [42].
Показано, что в ответ на секрецию мотонейроном АХ и АТФ, в ПШК наблюдается выброс ионов Са2+ из внутриклеточных депо (т.н. кальциевые осцилляции). Данный процесс запускается активацией мАХР [42, 54], либо активацией P2-рецепторов ПШК [55, 56].
Показано, что ПШК не только пассивно детектируют нейротрансмиттеры, но и обладают способностью влиять на количество АХ, выделяющегося из двигательного нервного окончания [57]. Поскольку изменение количества АХ, секретируемого в синаптическую щель, оказывает влияние, в первую очередь, на амплитуду ВПСП, то ПШК способны изменять фактор надежности нервно-мышечной синаптической передачи. Так, в настоящее время известно, что увеличение уровня внутриклеточных ионов Са2+ в ПШК вызывает секрецию АТФ, который, деградируя до аденозина, активирует А1 и А2 подтипы аденозиновых рецепторов (рис. 1). Важно отметить, что параметры кальциевого ответа ПШК зависят от паттерна стимуляции двигательного нерва. Длительная непрерывная стимуляция (20 Гц, 1800 стимулов) вызывает относительно короткое по времени увеличение уровня ионов Са2+ в ПШК, что коррелирует с увеличением амплитуды ВПСП, данный эффект опосредуется активацией А2-подтипа аденозиновых рецепторов. Стимуляция двигательного нерва тремя сериями импульсов с частотой 20 Гц в течение 60 с, с двумя интервалами в 30 с между сериями, коррелирует с уменьшением амплитуды ВПСП, данный эффект опосредуется активацией А1-подтипа рецепторов к аденозину [58]. Поскольку как посттетаническая потенциация, так и посттетаническая депрессия не наблюдаются после инъекции в ПШК хелатора ионов Са2+, то можно говорить именно о ключевой роли ПШК в процессе модуляции нервно-мышечной синаптической передачи посредством баланса между активацией А1- и А2-подтипов аденозиновых рецепторов. Сходный механизм описан для новорожденных животных, у которых идет процесс перехода от полинейрональной иннервации мышечных волокон к условиям, характерным для взрослых животных, когда на каждом мышечном волокне имеется только один синаптический контакт. Важно отметить, что элиминации подвергаются менее активные синаптические контакты и ПШК принимают участие в процессе разделения конкурирующих синаптических контактов по уровню их активности. Так, ПШК, детектируя уровень синаптической активности посредством P2Y1-подтипа рецепторов к АТФ, реагируют увеличением уровня внутриклеточных ионов Са2+, секретируют дополнительный АТФ, который является источником аденозина. Активация пресинаптических А2а аденозиновых рецепторов приводит к синаптической потенциации, дополнительно выделяя активные синаптические контакты [58]. Совсем недавно было показано, что у новорожденных животных P2Y1-рецепторы и связанные с ними кальциевые осцилляции играют важную роль и в регуляции буферной функции ПШК в отношении ионов К+ и, как следствие, в развитии мышечного утомления [59].
Не только АХ и АТФ способны вызывать кальциевый ответ ПШК. Короткая экзогенная аппликация нейротрофина-3 или BDNF у новорожденных животных также приводит к увеличению уровня внутриклеточных ионов Са2+ в ПШК, что коррелирует с увеличением высвобождения медиатора путем активации пресинаптических рецепторов тропомиозин-рецепторной киназы (Trk) [60]. Уровень кальция в ПШК может изменяться и при прямом воздействии на G-белок-ассоциированные рецепторы: микроинъекция GTPγS (негидролизуемый аналог гуанозинтрифосфата) в ПШК снижает вызванное высвобождение нейромедиатора, в то время как микроинъекция GDPβS (негидролизуемый аналог гуанозиндифосфата) снижает синаптическую депрессию, вызываемую высокочастотной стимуляцией [61].
Также есть основания полагать, что увеличение уровня внутриклеточных ионов Са2+ в ПШК не является единственным механизмом секреции глиотрансмиттеров. Так, было показано, что ингибирование БуХЭ – фермента, который в НМС расположен исключительно на поверхности ПШК, приводит к снижению количества АХ, секретируемого в ответ на стимуляцию аксона двигательного нерва (рис. 1). Показано, что процесс снижения секреции АХ опосредуется активацией α7-подтипа нАХР [62]. Поскольку данный процесс ауторегуляции секреции АХ не сопровождается характерными для ПШК изменениями уровня внутриклеточных ионов Са2+, то можно говорить о существовании альтернативных изменению уровня внутриклеточных ионов Са2+ механизмов секреции глиотрансмиттеров ПШК.
Важно отметить, что данный процесс ауторегуляции секреции АХ, включающий α7-нАХР ПШК в качестве сенсора экстрасинаптического АХ, имеет функциональное значение при терапии состояний патологической мышечной слабости при помощи ингибиторов холинэстераз. Хорошо известно, что нарушения нервно-мышечной синаптической передачи, сопровождающие ряд заболеваний, приводят к развитию мышечной слабости, а в тех случаях, когда патология затрагивает дыхательную мускулатуру, то и к смерти. Причиной мышечной слабости чаще всего является уменьшение чувствительности постсинаптической мембраны к АХ из-за снижения плотности нАХР мышечного типа (миастения гравис, врожденные миастенические синдромы), либо снижение количества АХ, секретируемого мотонейроном (синдром Ламберта–Итона) [63]. Традиционные схемы терапии состояний патологической мышечной слабости, связанной со снижением плотности функциональных нАХР мышечного типа, включают частичное угнетение активности ферментов, гидролизующих АХ – АХЭ и БуХЭ, что увеличивает время жизни АХ. В свою очередь, увеличение времени жизни АХ в синаптической щели позволяет скомпенсировать снижение плотности нАХР и восстановить нормальную силу мышечных сокращений. Применяемые на данный момент для терапии миастений ингибиторы холинэстераз, равно эффективны в отношении как АХЭ, так и БуХЭ [64]. Последняя, как уже упоминалось, располагается за пределами синаптической щели, на поверхности ПШК и контролирует уровень АХ, активирующего α7-нАХР ПШК. Таким образом, только ингибирование АХЭ оказывает положительный эффект, позволяя скомпенсировать снижение плотности рецепторов в синаптической щели. Ингибирование БуХЭ увеличивает вклад активации α7-нАХР ПШК, что приводит к значимому снижению секреции АХ мотонейроном, снижая эффективность терапии состояний мышечной слабости [62].
ПШК ПРИ ПАТОЛОГИЧЕСКИХ СОСТОЯНИЯХ
На момент написания данного обзора наибольшее количество информации о возможном вкладе ПШК в патогенез заболеваний периферической нервной системы накоплено в отношении бокового амиотрофического склероза, однако имеются отдельные данные и в отношении других патологических состояний.
Боковой амиотрофический склероз (БАС) – тяжелое нейродегенеративное заболевание, характеризующееся потерей функциональности периферических мотонейронов. На ранних стадиях больные БАС страдают от мышечной слабости, по мере прогрессирования заболевания наступает потеря моторики и инвалидизация. Течение болезни неблагоприятное и в большинстве случаев приводит к смерти. В 90% случаях БАС не наблюдается генетической предрасположенности к заболеванию (спорадический патогенез) и лишь у 10% заболевших выявляется генетически наследуемая форма БАС (генетический патогенез). Известно, что мутация по гену Cu/Zn-супероксид дисмутазы 1 (mSOD-1) является доказанной причиной развития генетически-наследуемой формы БАС [65, 66].
Было показано, что если ПШК не экспрессирует mSOD-1, то процесс нейродегенерации замедляется [67]. Однако необходимо отметить, что не во всех исследованиях удаление мутантного гена mSOD-1 из глиальных клеток приводило к положительному влиянию на течение БАС. Так, на мышах SOD1G73R было показано, что удаление мутантного SOD1G37R из Шванновских клеток только ускорило развитие заболевания [68], в то время как нокдаун по гену SOD-1 в Шванновских клетках трансгенных мышей SOD1G85R задерживал начало заболевания и увеличивал их выживаемость, что указывает на то, что экспрессия SOD1G85R является нейротоксичной [69]. Данные противоречивые результаты свидетельствуют о том, что различные мутации по гену SOD-1 в глиальных клетках вероятно дают разные варианты цитотоксичности.
Разрушение НМС – один из основных показателей прогрессирования заболевания, наблюдается даже в том случае, если сам мотонейрон удается спасти от действия апоптотических факторов, основными из которых являются активные формы кислорода [70–75]. Разрушению НМС предшествует изменение морфологии ПШК. При исследовании ультраструктурными методами образцов, полученных от пациентов с БАС, было обнаружено, что во многих синапсах ПШК образовывали отростки, аномально проникающие в синаптическую щель [76].
Интересно отметить, что у мышей с мутацией mSOD-1 в возрасте 120 дней, когда функциональных нарушений в работе НМС еще не отмечается, ПШК имеют повышенную способность увеличивать уровень Са2+ в ответ на активацию мАХР ПШК эндогенным АХ [75]. Таким образом, постоянная активация мАХР ПШК может быть одной из причин дефектов в морфологии и функции ПШК при прогрессировании БАС. Кроме того, учитывая количество и разнообразие внутриклеточных путей, которые регулируются изменением цитоплазматической концентрации Са2+, можно предположить, что их избыточная активация в ПШК, может привести к перегрузке митохондрий, последующему усилению окислительного стресса и продукции свободных радикалов [77].
В условиях модели БАС на мышах было показано, что еще до начала процесса денервации, у мутантных мышей SOD-1 наблюдается активация апоптоза в ПШК и, как следствие, уменьшение количества этих клеток [73]. Уменьшение общего количества ПШК может привести к изменению сигнальных путей, которые важны для восстановления НМС и его правильного функционирования, например, таких, как путь нейрегулин-ErbB. Показано, что нейрегулин и его рецепторы (ErbB) специфически локализованы на плазматической мембране ПШК. Данные нескольких исследований показали, что целостность сигнального пути нейрегулина в ПШК критична для поддержания стабильности НМС, в то время как его чрезмерная активация, так и его подавление при БАС приводили к потере синапсов [77]. Показано, что экспрессия нейрегулина (NRG) снижается как у пациентов с БАС, так и в условиях модели БАС на животных, а генная терапия вирусом, увеличивающим уровень экспрессии нейрегулина, способна существенно продлить жизнь мышей с мутацией гена SOD-1 [78].
Важно отметить, что сходные с БАС изменения ПШК описаны и при других патологиях. Показано, что изменения морфологии НМС, связанные с ПШК, сопровождают не только БАС, но и некоторые врожденные миастенические синдромы. Так, при миастениях, вызванных мутациями гена Lamb2 или агрина, наблюдаются инвазии ПШК в синаптическую щель [79, 80].
Также известно, что потеря ПШК сопровождает не только БАС, но и такие заболевания как спинальные мышечные атрофии (СМА), а также характерна для синдрома Миллера Фишера.
СМА – группа генетически-детерминированных заболеваний, характеризующихся прогрессирующей атрофией мышц тела и дыхательной мускулатуры из-за постепенной гибели нейронов в спинном мозге и стволе головного мозга [81]. Генетический дефект локализуется на хромосоме 5q с вовлечением гена Smn1 (survival motor neuron gene 1). На трансгенных мышах с моделью СМА была показана прогрессирующая во времени потеря ПШК [82], что аналогично БАС; это поднимает вопрос об их возможной роли в развитии СМА.
Синдром Миллера Фишера – редкий вариант острой воспалительной демиелинизирующей полинейропатии, характеризующийся триадой симптомов, включающих офтальмоплегию, атаксию и отсутствие сухожильных рефлексов. Это аутоиммунное заболевание, вызываемое выработкой антител к ганглиозидам (анти-GQ1b). На модели заболевания у мышей было показано, что данные антитела связываются с ганглиозидом GD3, что вызывает исключительно гибель ПШК, но не влияет на другие компоненты НМС [83–85]. Данные эксперименты поднимают вопрос о вкладе специфической абляции ПШК в патогенез синдрома Миллера Фишера.
В заключение хочется отметить, что данные, приведенные в этом обзоре, свидетельствуют о том, что ПШК подвержены значительным изменениям при заболеваниях абсолютно различной этиологии. Примеры нарушения структуры и функции ПШК при различных патологиях суммированы в табл. 1. Таким образом, существует вероятность, что ПШК вносят свой вклад в патогенез большого числа заболеваний, так или иначе затрагивающих НМС. Поэтому, лучшее понимание особенностей и закономерностей работы ПШК с высокой долей вероятности сможет способствовать как лучшему пониманию патогенеза большого количества заболеваний, так и открытию новых возможностей по созданию методов их терапии.
Таблица 1.
Примеры нарушения структуры и функции ПШК при патологических состояниях
| Аномалии ПШК | Патология | Источник информации |
|---|---|---|
| Нарушение способности ПШК детектировать активность синапса, усиление способности генерировать кальциевые осцилляции | Модель БАС на животных | [71] |
| Спраутинг ПШК, инвазии ПШК в синаптическую щель | БАС (биопсия пациентов) | [72] |
| Активация апоптоза в ПШК | Модель БАС на животных | [70] |
| Потеря иммуноокрашивания на белок S100 | Модель СМА на животных | [86] |
| Прогрессирующая гибель ПШК | Модель СМА на животных | [87] |
| Гибель ПШК | Модель синдрома Миллера Фишера на животных |
[88] |
| Потеря способности ПШК к образованию мостиков для направленного прорастания аксона | Мышечная дистрофия | [89, 90] |
| Инвазия ПШК в синаптическую щель | Модель врожденного миастеничного синдрома (мутация ColQ) на животных | [91] |
| Инвазия ПШК в синаптическую щель, гибель ПШК | Врожденные миастеничные синдромы (биопсия пациентов) | [92, 93] |
Список литературы
Sanes J.R., Lichtman J.W. Induction, assembly, maturation and maintenance of a postsynaptic apparatus. Nat. Rev. Neurosci. 2(11): 791–805. 2001.
Wu H., Xiong W.C., Mei L. To build a synapse: Signaling pathways in neuromuscular junction assembly. Development. 137(7): 1017–33. 2010.
Le Douarin N. M. Cell line segregation during peripheral nervous system ontogeny. Science. 231(4745). 1515–1522. 1986.
Jessen K.R., Brennan A., Morgan L., Mirsky R., Kent A.,Hashimoto Y. The Schwann cell precursor and itsfate: A study of cell death and differentiation during gliogenesis in rat embryonic nerves. Neuron. 12(3). 509–527. 1994.
Barik A., Li L., Sathyamurthy A., Xiong W.C., Mei L. Schwann Cells in Neuromuscular Junction Formation and Maintenance. J. Neurosci. 36(38): 9770–81. 2016.
Bastiani M.J., Goodman C.S. Guidance of neuronal growth cones in the grasshopper embryo. III. Recognition of specific glial pathways. J. Neurosci. 6(12): 3542–51. 1986.
Sepp K.J., Schulte J., Auld V.J. Peripheral glia direct axon guidance across the CNS/PNS transition zone. Dev. Biol. 238(1): 47–63. 2001.
Aigouy B., Van de Bor V., Boeglin M., Giangrande A. Time-lapse and cell ablation reveal the role of cell interactions in fly glia migration and proliferation. Development. 131(20): 5127–38. 2004.
Bandtlow C.E., Heumann R., Schwab M.E., Thoenen H. Cellular localization of nerve growth factor synthesis by in situ hybridization. EMBO J. 6(4): 891–899. 1987.
Carroll S.L., Miller M.L., Frohnert P.W., Kim S.S., Corbett J.A. Expression of neuregulins and their putative receptors, ErbB2 and ErbB3, is induced during Wallerian degeneration. J. Neurosci. 17(5): 1642–1659. 1997.
Feng Z., Ko C.P. Schwann cells promote synaptogenesis at the neuromuscular junction via transforming growth factor-beta1. J. Neurosci. 28(39): 9599–9609. 2008.
Fuentes-Medel Y., Ashley J., Barria R., Maloney R., Freeman M., Budnik V. Integration of a retrograde signal during synapse formation by glia-secreted TGF-β ligand. Curr. Biol. 22(19): 1831–1838. 2012
Kerr K.S., Fuentes-Medel Y., Brewer C., Barria R., Ashley J., Abruzzi K.C., Sheehan A., Tasdemir-Yilmaz O.E., Freeman M.R., Budnik V. Glial wingless/Wnt regulates glutamate receptor clustering and synaptic physiology at the Drosophila neuromuscular junction. J. Neurosci. 34(8): 2910–2920. 2014.
Arber S., Caroni P. J. Thrombospondin-4, an extracellular matrix protein expressed in the developing and adult nervous system promotes neurite outgrowth. Cell Biol. 131(4): 1083–1094. 1995.
Arikkath J., Campbell K.P. Auxiliary subunits: Essential components of the voltage-gated calcium channel complex. Curr. Opin. Neurobiol. 13(3): 298–307. 2003.
Bishop D.L., Misgeld T., Walsh M.K., Gan W.B., Lichtman J.W. Axon branch removal at developing synapses by axosome shedding. Neuron. 44(4): 651–661. 2004.
Tapia J.C., Wylie J.D., Kasthuri N., Hayworth K.J., Schalek R., Berger D.R., Guatimosim C., Seung H.S., Lichtman J.W. Pervasive synaptic branch removal in the mammalian neuromuscular system at birth. Neuron. 74(5): 816–829. 2012.
Darabid H., Arbour D., Robitaille R., Glial cells decipher synaptic competition at the mammalian neuromuscular junction. J. Neurosci. 33(4): 1297–1313. 2013.
Fuentes-Medel Y., Logan M.A., Ashley J., Ataman B., Budnik V., Freeman M.R. Glia and muscle sculpt neuromuscular arbors by engulfing destabilized synaptic boutons and shed presynaptic debris. PLoS Biol. 7.8: e1000184. 2009.
Smith I.W., Mikesh M., Lee Y. and Thompson W.J. Terminal Schwann Cells Participate in the Competition Underlying Neuromuscular Synapse Elimination. J. Neurosci. 33(45) 17724–17736. 2013.
Katz B., Miledi R. The release of acetylcholine from nerve endings by graded electric pulses. Proc. R. Soc. B. 167: 23–38. 1967.
De Robertis E.D., Bennett H.S. Some features of the submicroscopic morphology of synapses in frog and earthworm. J. Biophys. Biochem. Cytol. 1(1): 47–58. 1955.
Robertson J.D. The ultrastructure of a reptilian myoneural junction. J. Biophys. Biochem. Cytol. 2(4): 381–94. 1956.
Fatt P., Katz B. The effect of sodium ions on neuromuscular transmission. J. Physiol. 118(1): 73–87. 1952.
Del Castillo J., Katz B. Localization of active spots within the neuromuscular junction of the frog. J. Physiol. 132(3): 630–49. 1956.
Katz B, Miledi R. Transmitter leakage from motor nerve endings. Proc. R. Soc. Lond. B Biol. Sci. 196 (1122): 59–72. 1977.
Vyskocil F, Illés P. Non-quantal release of transmitter at mouse neuromuscular junction and its dependence on the activity of Na+-K+ ATP-ase. Pflugers Arch. 370 (3): 295–7. 1977.
Davis R., Koelle G. Electron microscopic localization of acetylcholinesterase and nonspecific cholinesterase at the neuromuscular junction by the gold-thiocholine and gold-thiolacetic acid methods. J. Cell Biol. 34(1): 157–171. 1967.
Silinsky E., Redman R. ATP released together with acetylcholine as the mediator of neuromuscular depression at frog motor nerve endings. J. Physiol. 477: 117–127. 1994.
Boulland J.L., Qureshi T., Seal R.P., Rafiki A., Gundersen V., Bergersen L.H., Fremeau R.T.Jr., Edwards R.H., Storm-Mathisen J., Chaudhry F.A. Expression of the vesicular glutamate transporters during development indicates the widespread corelease of multiple neurotransmitters. J. Comp. Neurol. 480: 264–280. 2004.
Malomouzh A.I., Mukhtarov M.R., Nikolsky E.E., Vyskocil F., Lieberman E.M., Urazaev A.K. Glutamate regulation of non-quantal release of acetylcholine in the rat neuromuscular junction. J. Neurochem. 85: 206–213. 2003.
Nishimaru H., Restrepo C.E., Ryge J., Yanagawa Y., Kiehn O. Mammalian motor neurons corelease glutamate and acetylcholine at central synapses. Proc. Natl. Acad. Sci. U. S. A. 102(14): 5245–5249. 2005.
Petrov K., Malomouzh A., Kovyazina I., Krejci E., Nikitashina A., Proskurina S. Regulation of acetylcholinesterase activity by nitric oxide in rat neuromuscular junction via N-methyl-D-aspartate receptor activation. Eur. J. Neurosci. 37: 181–189. 2013.
Garcia N., Santafé M., Salon I., Lanuza M., Tomàs J. Expression of muscarinic acetylcholine receptors (M1-, M2-, M3- and M4-type) in the neuromuscular junction of the newborn and adult rat. Histol. Histopathol. 20(3): 733–743. 2005.
Slutsky I., Silman I., Parnas I., Parnas H. Presynaptic M(2) muscarinic receptors are involved in controlling the kinetics of ACh release at the frog neuromuscular junction. J. Physiol. 536 (Pt 3): 717–25. 2001.
Oliveira L., Timóteo M., Correia-de-Sá P. Modulation by adenosine of both muscarinic M1-facilitation and M2-inhibition of [3H]-acetylcholine release from the rat motor nerve terminals. Eur. J. Neurosci. 15(11): 1728–1736. 2002.
Oliveira L., Timóteo M., Correia-de-Sá P. Negative crosstalk between M1 and M2 muscarinic autoreceptors involves endogenous adenosine activating A1 receptors at the rat motor endplate. Neurosci. Lett. 459(3): 127–131. 2009.
Dudel J. The time course of transmitter release in mouse motor nerve terminals is differentially affected by activation of muscarinic M1 or M2 receptors. Eur. J. Neurosci. 26(8): 2160–2168. 2007
Newman Z., Malik P., Wu T.-Y., Ochoa C., Watsa N., Lindgren C. Endocannabinoids mediate muscarine-induced synaptic depression at the vertebrate neuromuscular junction. Eur. J. Neurosci. 25(6): 1619–1630. 2007.
Newman E.A., Frambach D.A., Odette L.L. et al. Control of extracellular potassium levels by retinal glial cell K+ siphoning. Science. 225(4667): 1174–1175. 1984.
Heredia D.J., Feng C.Y., Hennig G.W., Renden R.B., Gould T.W. Activity-induced Ca2+ signaling in perisynaptic Schwann cells of the early postnatal mouse is mediated by P2Y1 receptors and regulates muscle fatigue. eLife. 7: e30839. 2018.
Rochon D., Rousse I., Robitaille R. Synapse-glia interactions at the mammalian neuromuscular junction. J. Neurosci. 21(11): 3819–3829. 2001.
Chen Z-L., Yu W-M., Strickland S. Peripheral Regeneration. Annu. Rev. Neurosci. 30: 209–233. 2007.
Jessen K.R., Mirsky R. Negative regulation of myelination: Relevance for development, injury, and demyelinating disease. Glia. 56(14): 1552–1565. 2008.
Hall E.D., Oostveen J.A., Gurney M.E. Relationship of microglial and astrocytic activation to disease onset and progression in a transgenic model of familial ALS. Glia 23: 249–256. 1998.
McGeer P.L., McGeer E.G., Kawamata T., Yamada T., Akiyama H. Reactions of the immune system in chronic degenerative neurological diseases. Can. J. Neurol. Sci. 18 (3 Suppl.): 376–379. 1991.
Cheng C., Zochodne D.W. In vivo proliferation, migration and phenotypic changes of Schwann cells in the presence of myelinated fibers. Neuroscience.115 (1): 321–329. 2002.
Georgiou J., Robitaille R., Trimble W.S., Charlton M.P. Synaptic regulation of glial protein expression in vivo. Neuron. 12 (2): 443–455. 1994.
Triolo D., Dina G., Lorenzetti I., Malaguti M., Morana P., Del Carro U., Comi G., Messing A., Quattrini A., Previtali S.C. Loss of glial fibrillary acidic protein (GFAP) impairs Schwann cell proliferation and delays nerve regeneration after damage. J. Cell. Sci. 119 (Pt 19): 3981–3993. 2006.
Duregotti E., Negro S., Scorzeto M., Zornetta I., Dickinson B.C., Chang C.J., Montecucco C., Rigoni M., Mitochondrial alarmins released by degenerating motor axon terminals activate perisynaptic Schwann cells. Proc. Natl. Acad. Sci. U. S. A. 112(5): E497–505. 2015.
Dave V., Gordon, C. W., and McCarthy, K. D. Cerebral type 2 astroglia are heterogeneous with respect to their ability to respond to neuroligands linked to calcium mobilization. Glia. 4: 440–447. 1991.
Pearce B., Cambray-Deakin M., Morrow C., Grimble J., Murphy S. Activation of muscarinic and of a-adrenergic receptors on astrocytes results in the accumulation of inositol phosphates. J. Neurochem. 45: 1534–1540. 1985.
Todd K., Darabid H., Robitaille R. Perisynaptic glia discriminate patterns of motor nerve activity and influence plasticity at the neuromuscular junction. J. Neurosci. 30(35): 11870–11882. 2010.
Ko C.P., Robitaille R. Perisynaptic Schwann Cells at the Neuromuscular Synapse: Adaptable, Multitasking Glial Cells. Cold Spring Harb. Perspect. Biol. 7(10): a020503. 2015.
Kang H., Tian L., Thompson W. Terminal Schwann cells guide the reinnervation of muscle after nerve injury. J. Neurocytol. 32(5–8): 975–985. 2003.
Madison R.D., Sofroniew M.V., Robinson G.A. Schwann cell influence on motor neuron regeneration accuracy. Neuroscience. 163(1): 213–221. 2009.
Magill C.K., Tong A., Kawamura D., Hayashi A., Hunter D.A., Parsadanian A., Mackinnon S.E., Myckatyn T.M. Reinnervation of the tibialis anterior following sciatic nerve crush injury: A confocal microscopic study in transgenic mice. Exp. Neurol. 207(1): 64–74. 2007.
Darabid H., St-Pierre-See A., Robitaille R. Purinergic-Dependent Glial Regulation of Synaptic Plasticity of Competing Terminals and Synapse Elimination at the Neuromuscular Junction. Cell Rep. 25(8): 2070–2082. 2018.
Heredia D.J., Feng C.Y., Hennig G.W., Renden R.B., Gould T.W. Activity-induced Ca2+ signaling in perisynaptic Schwann cells of the early postnatal mouse is mediated by P2Y1 receptors and regulates muscle fatigue. eLife. 7: e30839. 2018.
Lohof A.M., Ip N.Y., Poo M.M. Potentiation of developing neuromuscular synapses by the neurotrophins NT-3 and BDNF. Nature. 363(6427): 350–353. 1993.
Robitaille R. Modulation of synaptic efficacy and synaptic depression by glial cells at the frog neuromuscular junction. Neuron. 4: 847–855. 1998.
Petrov K.A., Girard E., Nikitashina A.D., Colasante C., Bernard V., Nurullin L., Leroy J., Samigullin D., Colak O., Nikolsky E., Plaud B., Krejci E. Schwann cells sense and control acetylcholine spillover at the neuromuscular junction by α7 nicotinic receptors and butyrylcholinesterase. J. Neurosci. 34(36): 11870–11883. 2014.
Ruff R.L., Endplate contributions to the safety factor for neuromuscular transmission. Muscle Nerve. 44(6): 854–861. 2011.
Leader H., Wolfe A.D., Chiang P.K. Gordon R.K. Pyridophens: binary pyridostigmine−aprophen prodrugs with differential inhibition of acetylcholinesterase, butyrylcholinesterase, and muscarinic receptors. J. Med. Chem. 45: 902–910. 2002.
Rosen D.R., Siddique T., Patterson D., Figlewicz D.A., Sapp P., Hentati A., Donaldson D., Goto J., O’Regan J.P., Deng H.X. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 362(6415): 59–62. 1993.
Gurney M.E., Pu H., Chiu A.Y., Dal Canto M.C., Polchow C.Y., Alexander D.D., Caliendo J., Hentati A., Kwon Y.W., Deng H.X. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 264(5166): 1772–1775. 1994.
Clement A.M., Nguyen M.D., Roberts E.A., Garcia M.L., Boillée S., Rule M., McMahon A.P., Doucette W., Siwek D., Ferrante R.J., Brown R.H. Jr, Julien J.P., Goldstein L.S., Cleveland D.W. Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science. 302(5642): 113–117.
Lobsiger C.S., Boillee S., McAlonis-Downes M., Khan A.M., Feltri M.L., Yamanaka K., Cleveland D.W. Schwann cells expressing dismutase active mutant SOD1 unexpectedly slow disease progression in ALS mice. Proc. Natl. Acad. Sci. U. S. A. 106(11): 4465–4470. 2009.
Wang L., Pytel P., Feltri M.L., Wrabetz L., Roos R.P. Selective knockdown of mutant SOD1 in Schwann cells ameliorates disease in G85R mutant SOD1 transgenic mice. Neurobiol. Dis. 48(1): 52–57. 2012.
De Winter F., Vo T., Stam F.J., Wisman L.A., Bär P.R., Niclou S.P., van Muiswinkel F.L., Verhaagen J. The expression of the chemorepellent Semaphorin 3A is selectively induced in terminal Schwann cells of a subset of neuromuscular synapses that display limited anatomical plasticity and enhanced vulnerability in motor neuron disease. Mol. Cell Neurosci. 32(1–2): 102–117. 2006.
Keller A.F., Gravel M., Kriz J. Live imaging of amyotrophic lateral sclerosis pathogenesis: disease onset is characterized by marked induction of GFAP in Schwann cells. Glia. 57(10): 1130–1142. 2009.
Carrasco D.I., Bichler E.K., Seburn K.L., Pinter M.J. Nerve terminal degeneration is independent of muscle fiber genotype in SOD1 mice. PLoS One. 5(3): e9802. 2010.
Carrasco D.I., Seburn K.L., Pinter M.J. Altered terminal Schwann cell morphology precedes denervation in SOD1 mice. Exp. Neurol. 275: 172–181. 2016.
Carrasco D.I., Bahr B.A., Seburn K.L., Pinter M.J. Abnormal response of distal Schwann cells to denervation in a mouse model of motor neuron disease. Exp. Neurol. 278: 116–126. 2016.
Arbour D., Tremblay E., Martineau É., Julien J.P., Robitaille R. Early and persistent abnormal decoding by glial cells at the neuromuscular junction in an ALS model. J. Neurosci. 35(2): 688–706. 2015.
Bruneteau G., Bauché S., Gonzalez de Aguilar J.L., Brochier G., Mandjee N., Tanguy M.L., Hussain G., Behin A., Khiami F., Sariali E., Hell-Remy C., Salachas F., Pradat P.F., Lacomblez L., Nicole S., Fontaine B., Fardeau M., Loeffler J.P., Meininger V., Fournier E., Koenig J., Hantaï D. Endplate denervation correlates with Nogo-A muscle expression in amyotrophic lateral sclerosis patients. Ann. Clin. Transl. Neurol. 2(4): 362–372. 2015.
Arbour D., Vande Velde C., Robitaille R. New perspectives on amyotrophic lateral sclerosis: the role of glial cells at the neuromuscular junction. J. Physiol. 595(3): 647–661. 2017.
Lasiene J., Komine O., Fujimori-Tonou N., Powers B., Endo F., Watanabe S., Shijie J., Ravits J., Horner P., Misawa H., Yamanaka K. Neuregulin 1 confers neuroprotection in SOD1-linked amyotrophic lateral sclerosis mice via restoration of C-boutons of spinal motor neurons. Acta Neuropathol. Commun. 4: 15. 2016
Maselli A., Ng J., Anderson A., Cagney O., Arredondo J., Williams C., Wessel B., Abdel-Hamid H., Wollmann L. Mutations in LAMB2 causing a severe form of synaptic congenital myasthenic syndrome. J. Med. Genetics. 46(3): 203–208. 2009.
Nicole S., Chaouch A., Torbergsen T., Bauche S., de Bruyckere E., Fontenille J., Horn A., van Ghelue M., Loseth S., Issop Y., Cox D., Muller S., Evangelista T., Stalberg E., Ioos C., Barois A., Brochier G., Sternberg D., Fournier E., Hantai D., Abicht A., Dusl M., Laval H., Griffin H., Eymard B., Lochmuller H. Agrin mutations lead to a congenital myasthenic syndrome with distal muscle weakness and atrophy. Brain. 137: 2429–2443. 2014.
D’Amico A., Mercuri E., Tiziano D., Bertini E. Spinal muscular atrophy. Orphanet J. Rare Dis. 6(1): 71. 2011.
Murray M., Beauvais A., Bhanot K., Kothary R. Defects in neuromuscular junction remodelling in the Smn(2B/−) mouse model of spinal muscular atrophy. Neurobiol. Dis. 49: 57–67. 2013.
Halstead K., O’Hanlon M., Humphreys D., Morrison B., Morgan P., Todd J., Plomp J., Willison J. Anti-disialoside antibodies kill perisynaptic Schwann cells and damage motor nerve terminals via membrane attack complex in a murine model of neuropathy. Brain. 127(Pt 9): 2109–2123. 2004.
O’Hanlon M., Plomp J., Chakrabarti M., Morrison I., Wagner R., Goodyear S., Yin X., Trapp D., Conner J., Molenaar C., Stewart S., Rowan G., Willison J. Anti-GQ1b ganglioside antibodies mediate complement-dependent destruction of the motor nerve terminal. Brain. 124 (Pt 5): 893–906. 2001.
Wirguin I., Ifergane G., Almog Y., Lieberman D., Bersudsky M., Herishanu O. Presynaptic neuromuscular transmission block in Guillain-Barre syndrome associated with anti-GQ1b antibodies. Neuromuscul. Disord. 12(3): 292–293. 2002.
Neve A., Trüb J., Saxena S., Schümperli D. Central and peripheral defects in motor units of the diaphragm of spinal muscular atrophy mice. Mol. Cell Neurosci. 70: 30–41. 2016.
Murray L.M., Beauvais A., Bhanot K., Kothary R. Defects in neuromuscular junction remodelling in the Smn(2B/–) mouse model of spinal muscular atrophy. Neurobiol. Dis. 49: 57–67. 2013.
Halstead S.K., Humphreys P.D., Goodfellow J.A., Wagner E.R., Smith R.A., Willison H.J. Complement inhibition abrogates nerve terminal injury in Miller Fisher syndrome. Ann. Neurol. 58(2): 203–210. 2005.
Personius K.E., Sawyer R.P. Terminal Schwann cell structure is altered in diaphragm of mdx mice. Muscle Nerve. 32(5): 656–663. 2005.
Marques M.J., Pereira E.C., Minatel E., Neto H.S. Nerve-terminal and Schwann-cell response after nerve injury in the absence of nitric oxide. Muscle Nerve. 34(2): 225–231. 2006.
Feng G., Krejci E., Molgo J., Cunningham J.M., Massoulié J., Sanes J.R. Genetic analysis of collagen Q: Roles in acetylcholinesterase and butyrylcholinesterase assembly and in synaptic structure and function. J. Cell Biol. 144(6): 1349–1360. 1999.
Maselli R.A., Ng J.J., Anderson J.A., Cagney O., Arredondo J., Williams C., Wessel H.B., Abdel-Hamid H., Wollmann R.L. Mutations in LAMB2 causing a severe form of synaptic congenital myasthenic syndrome. J. Med. Genet. 46(3): 203–208. 2009.
Nicole S., Chaouch A., Torbergsen T., Bauché S., de Bruyckere E., Fontenille M.J., Horn M.A., van Ghelue M., Løseth S., Issop Y., Cox D., Müller J.S., Evangelista T., Stålberg E., Ioos C., Barois A., Brochier G., Sternberg D., Fournier E., Hantaï D., Abicht A., Dusl M., Laval S.H., Griffin H., Eymard B., Lochmüller H. Agrin mutations lead to a congenital myasthenic syndrome with distal muscle weakness and atrophy. Brain. 137(Pt 9): 2429–2443. 2014.
Дополнительные материалы отсутствуют.
Инструменты
Российский физиологический журнал им. И.М. Сеченова