

Российский физиологический журнал им. И.М. Сеченова, 2019, T. 105, № 11, стр. 1349-1372
Новые подходы в исследовании молекулярного патогенеза спиноцеребеллярной атаксии 2-го типа
П. А. Егорова 1, *, И. Б. Безпрозванный 1
1 Санкт-Петербургский политехнический университет Петра Великого
Санкт-Петербург, Россия
* E-mail: bio_polya@mail.ru
Поступила в редакцию 08.08.2019
После доработки 15.08.2019
Принята к публикации 16.08.2019
Аннотация
Спиноцеребеллярная атаксия 2-го типа (СЦА2) представляет собой прогрессирующее наследственное заболевание, причиной которого на генетическом уровне является экспансия полиглутамина в белке атаксин-2. Эффективные способы терапевтического лечения, так же как и болезнь-модифицирующая терапия, все еще недоступны для пациентов с СЦА2. На настоящий момент пациентам предписывают только симптоматическое лечение вместе с методами паллиативной медицины. С целью поиска новых терапевтических мишеней для лечения заболевания СЦА2 многими научными группами были предприняты попытки исследовать физиологические, молекулярные и биохимические изменения нейронов мозжечка в случае пациентов, больных СЦА2, а также на различных модельных системах. Современные подходы в исследовании патогенеза СЦА2 позволили получить новые сведения о молекулярных механизмах заболевания и предложить возможные стратегии для потенциальной терапии данной болезни. В настоящей работе обобщены актуальные сведения о генетической основе СЦА2, описаны известные на настоящий момент свойства и функции белка атаксин-2, рассмотрены механизмы дегенерации клеток коры мозжечка, нарушения их физиологических функций, а также связанные с ними повреждения проводящих путей мозжечка, представлены сведения о современных модельных системах, позволяющих изучать основы СЦА2, а также приведена информация о новых подходах в исследовании молекулярных механизмов, лежащих в основе патологии СЦА2, таких как агрегация, окислительный стресс, нарушения клеточной сигнализации и кальциевого сигналинга, рассмотрена роль аутофагии и микроглии в молекулярном патогенезе СЦА2.
Спиноцеребеллярная атаксия 2-го типа (СЦА2) представляет собой генетическое заболевание с аутосомно-доминантным типом наследования, вызываемое экспансией триплета CAG в повсеместно экспрессируемом гене ATXN2, кодирующем белок атаксин-2 с удлиненным полиглутаминовым трактом [1–5]. Патофизиологические проявления СЦА2 включают в себя прогрессирующую атрофию мозжечка и последующую постепенно нарастающую атаксию [2]. На настоящий момент для СЦА2 не существует болезнь-модифицирующей терапии, для поддержки СЦА2 пациентов применяются только симптоматическое лечение и методы паллиативной медицины. Атаксин-2, белок, мутация в гене которого вызывает СЦА2, проявляет различные функции в клетке и вовлечен в метаболизм РНК [6]. С целью изучения основ патологии СЦА2 были разработаны различные модельные системы на клетках, дрожжах, червях, мухах, мышах, а также на нейронах, полученных из индуцированных плюрипотентных стволовых клеток (иПСК) СЦА2 пациентов. В случае СЦА2 мышей и пациентов в первую очередь наблюдается поражение клеток Пуркинье (КП) коры мозжечка и проводящих путей мозжечка. Исследования, проводимые на упомянутых модельных системах, выявили нарушения в морфологии, биохимии и нейрофизиологии КП и проводящих путей мозжечка [7, 8], что помогло сделать выводы о возможных молекулярных механизмах заболевания. Экспериментальные данные говорят о том, что в молекулярный патогенез СЦА2 вовлечены такие различные механизмы, как агрегация, окислительный стресс, нарушения клеточной сигнализации, дисрегуляция гомеостаза кальция, аномальная аутофагия и неправильный процессинг ДНК [9–16]. Современные подходы в исследовании патогенеза СЦА2 способствовали развитию нескольких перспективных терапевтических стратегий, что в будущем, вероятно, будет использовано для эффективного лечения СЦА2 пациентов. Уже сейчас многообещающие данные были получены в доклинических испытаниях антисенс-терапии [17, 18], а также в ранних клинических испытаниях иПСК [19] и рилузола [20]. Болезнь-модифицирующая терапия с использованием стабилизаторов кальция также показала многообещающие результаты в доклинических исследованиях на трансгенных СЦА2 мышах [21–23].
МУТАЦИЯ В ГЕНЕ ATXN2
Генетически СЦА2 вызывается экспансией CAG триплета в кодирующей области гена ATXN2, локализованного на хромосоме 12q24.1 [24]. Патология СЦА2 проявляется в случае пациентов с количеством повторов CAG ≥ 33 [25]. В соответствии с доминантным типом наследования экспансия CAG повторов действует как мутация приобретения функции [26]. В целом, неустойчивость мутации СЦА2 в локусе ATXN2 между поколениями зависит от длины экспансии CAG повтора, пола и возраста зачатия родителем-носителем мутации [27, 28].
Атаксин-2 представляет собой цитоплазматический белок массой 140 кДа, продукт гена ATXN2. Данный белок локализуется на гранулярном эндоплазматическом ретикулуме (ЭПР) [29], а также – на других компонентах клетки, включая аппарат Гольджи, стрессовые гранулы, тельца включения и собственно цитоплазму. Недостаток атаксина-2 приводит к ожирению, нарушениям липидного обмена, инсулинорезистентности [30, 31], а также – к нарушениям циркадной системы у мышей [32] и плодовых мушек [33, 34]. Исследования атаксина-2 с удлиненным полиглутаминовым трактом с помощью гранулярного цитоплазматического окрашивания и анализа частоты нейрональных включений в области ствола мозга СЦА2 пациентов обнаружило агрегацию мутантного атаксина-2 в тканях этих больных [35]. Белковая структура атаксина-2 содержит РАМ2 мотив, который принимает участие в ассоциации с поли(А)-связывающим белком (PABP) [36], что, вероятно, приводит к ассоциации атаксина-2 с полирибосомами [6]. Атаксин-2 также содержит два глобулярных Lsm домена и один Lsm-ассоциированный домен (Lsm-AD), которые, как было показано, напрямую взаимодействуют с РНК [6], таким образом, атаксин-2 скорее всего вовлечен в РНК метаболизм. В самом деле, in vitro исследования на нейронах и на фибробластах мышиных эмбрионов показали, что в ответ на недостаток питательных веществ наблюдается повышение экспрессии атаксина-2 в зависимой от сигналинга мишени рапамицина у млекопитающих (mTOR) манере. При этом отсутствие атаксина-2 приводило к адаптивным изменениям фосфоинозитид-3-киназы (PI3K)/mTOR-опосредованного фосфорилирования рибосомального белка S6 и белка 1, связывающего эукариотический фактор инициации трансляции 4Е (4E-BP1) в качестве регуляторных компонентов 48S комплекса инициации трансляции мРНК [37].
Другое исследование показало, что внутриклеточный уровень атаксина-2 влияет на образование стрессовых гранул (СГ) и Р-телец, таким образом происходит регуляция метаболизма мРНК включая трансляцию, стабильность и деградацию [38]. Было показано, что Lsm/Lsm-AD домен атаксина-2 взаимодействует с DEAD/H-Box РНК хеликазой DDX6, которая вовлечена в формирование СГ и Р-телец. Уменьшение уровня атаксина-2 приводило к аномальной сборке СГ, а также к повышению уровня РАВР, являющимся еще одним компонентом СГ [38]. Недавно было показано, что неупорядоченные домены атаксина-2 приводят к формированию гранул, содержащих мРНК и РНК-связывающие белки, а также способствуют нарушениям долговременной синаптической пластичности и проявляют цитотоксичность [39].
Недавние исследования показали, что белок Pbp1, дрожжевой ортолог атаксина-2 человека, регулирует ретротранспозонную активность, поскольку изменения его экспрессии подавляли ретротранспозоны TY1 с помощью определенных механизмов [40]. Анализ общего транскриптома в крови СЦА2 пациентов показал, что атаксин-2 является фактором процессинга цитозольной РНК и выявил его роль в регуляции транскрипции при формировании ответа на митохондриальный стресс, включая транскрипцию фактора контроля качества митохондрий PINK1 [41]. Упомянутые физиологические функции атаксина-2, вероятно, нарушаются при патологии СЦА2, в таком случае увеличение концентрации атаксина-2 с нормальным количеством CAG повторов могло бы иметь потенциальный терапевтический эффект в случае СЦА2.
Исследования на тканях мозга СЦА2 пациентов postmortem, на СЦА2 фибробластах, СЦА2 и ПСК, а также на мышиных моделях СЦА2 показали, что локус ATXN2 транскрибируется двунаправленно, при этом антисмысловой транскрипт ATXN2-AS с экспансией CUG повторов проявляет нейротоксичную активность в случае клеточной модели СЦА2, а также формирует РНК агрегаты в СЦА2 КП [42]. Таким образом, антисмысловой транскрипт ATXN2-AS может быть вовлечен в добавочный механизм патогенеза СЦА2 и может рассматриваться в качестве новой терапевтической мишени при СЦА2.
Вопрос о том, является ли мутация атаксина-2 при СЦА2 мутацией приобретения функции или мутацией потери функции является противоречивым. Многие исследования показали, что потеря атаксина-2 приводит к нарушениям мРНК метаболизма, включая трансляцию, стабильность и деградацию мРНК [38], опосредованно провоцирует повреждения путей биогенеза рибосом, начала трансляции, секреции ЭПР, липидного обмена [43], патологически изменяет процессы усвоения питательных веществ и метаболизма [44], а также негативно воздействует на экспрессию генов регуляции баланса кальция [45]. Таким образом, физиологическое уменьшение уровня атаксина-2, обуславливаемое мутацией в гене ATXN2 и последующей наработкой мутантного белка с нарушенными функциями, в самом деле может быть рассмотрено в качестве мутации потери функции. Однако следует отметить, что проявление функций мутантного атаксина-2 в качестве мутации приобретения функции также является значимым. Выявление агрегатов мутантного атаксина-2 в мозжечковой ткани пациентов СЦА2 [35] вместе с результатами экспериментов на планарных липидных бислоях, показавших, что атаксин-2 с увеличенным количеством CAG повторов связывается с рецептором инозитол-1,4,5-трисфосфата (IP3R) и значительно увеличивает его активацию молекулами IP3 [21], свидетельствуют о том, что мутация, вызывающая СЦА2, приводит к приобретению новой патологической функции. Можно только предположить, что правда о типе мутации находится где-то посередине: в то время, как при СЦА2 наблюдается потеря нормальной физиологической функции атаксина-2, мутантный атаксин-2 с увеличенным количеством CAG повторов проявляет свойства цитотоксичности.
МОЛЕКУЛЯРНЫЕ ИЗМЕНЕНИЯ, ВЕДУЩИЕ К ПОТЕРЕ КП
В случае СЦА2 пациентов на клеточном уровне в первую очередь наблюдается поражение КП. Исследования postmortem на СЦА2 пациентах обнаружили гетеротопные КП, чьи клеточные тела были смещены в сторону молекулярного слоя мозжечка [46].
Недавний анализ, проведенный на 20-ти семьях с СЦА с помощью комплексного подхода, включающего в себя методы секвенирования всего экзома, направленного ресеквенирования, а также анализ генетической сети, выявил пять новых генов, ассоциированных с СЦА, вовлеченных в такие процессы как развитие центральной нервной системы (ЦНС), аксональное наведение, регуляция транскрипции, контроль функции митохондрий, аутофагия и синаптическая передача [47]. Нарушения в этих важных физиологических процессах, возможно, объясняют дегенерацию КП коры мозжечка в патологии СЦА2 и будут рассмотрены нами далее в разделе, посвященном молекулярным механизмам СЦА2.
Литературный анализ данных говорит о том, что во время своего роста и развития мозжечок проявляет повышенную чувствительность к разрушающим эффектам активных форм кислорода. При этом, при индуцированном окислительном стрессе наблюдаются структурные изменения КП, их дендритов, гранулярных клеток, астроцитов, микроглии, аксонов и олигодендроглии мозжечка [48]. Типичные для СЦА2 пациентов изменения мозжечка, обусловленные окислительным стрессом, могут быть вызваны нарушением гомеостаза различных элементов в крови и сыворотке пациентов [49, 50]. Опубликованные данные говорят о том, что в крови СЦА2 пациентов наблюдаются изменения уровня меди, марганца, цинка и ванадия [49]. В случае СЦА2 пациентов, не проявлявших значительного клинического фенотипа, было обнаружено значительное уменьшение ферментной активности супероксиддисмутазы (SOD3) [51]. Недавние исследования выявили связь полиморфизма омега 2 глутатион-S-трансферазы и СЦА2 [52]. Было показано, что другой полиморфизм, A10398G в митохондриальной ДНК, вовлечен в ухудшение когнитивных функций на начальных этапах проявления СЦА2 [53]. В образцах крови СЦА2 пациентов также наблюдалось значительное увеличение уровня общего 5-гидроксиметилцитозина, обнаруживая таким образом общее метилирование ДНК в качестве биомаркера данного заболевания [54]. Также сообщалось, что тонкий нейрофиламент сыворотки является еще одним многообещающим периферическим биомаркером СЦА [55].
МОДЕЛИРУЮЩИЕ СЦА2 СИСТЕМЫ
Для изучения основ СЦА2 были разработаны различные модельные системы. С целью исследования молекулярных механизмов и тестирования потенциальных терапевтических стратегий были созданы различные модели заболевания на дрожжах, червях, мушках и мышах. Вначале были разработаны простые модели СЦА2, использующие для модуляции заболевания организмы дрожжей, червей C. elegans, и плодовых мушек D. melanogaster. Данные модели позволили определить основные функции атаксина-2, однако последующее создание мышиных моделей СЦА2 предоставило исследователям возможность тестировать терапевтические стратегии, которые впоследствии могут быть перенесены в клиническую практику [7].
Среди мышиных моделей СЦА2 первой была создана трансгенная линия SCA2-58Q. В данных мышах была достигнута КП-специфичная экспрессия полноразмерного человеческого гена Atxn2 с 58 CAG повторами под контролем промотора гена белка КП 2 (Pcp2) [56]. Для данных мышей характерно прогрессирующее ухудшение координации движений, проявляющееся начиная с возраста 32 недели согласно результатам тестов на моторную активность. Методы оценки моторной активности включали в себя метод “прогулка по перекладине” и ротарод тест и показали увеличение времени пробега по перекладине, а также увеличение количества соскальзывания лап в случае СЦА2 мышей по сравнению с диким типом того же возраста [21]. В возрасте 24-х недель наблюдалось значительное уменьшение количества КП коры мозжечка вместе с прогрессирующей потерей кальбиндина-28К, представляющего собой белковый маркер для оценки нейрональной дисфункции [21]. Для данных мышей также характерно нарушение электрофизиологических функций, выявленное в исследованиях на срезах мозжечка [22, 57], а также в исследованиях in vivo [58, 59].
Другая мышиная СЦА2 модель трансгенной линии SCA2-75Q экспрессирует полноразмерный человеческий ген Atxn2 под контролем эндогенного СЦА2 промотора. Несмотря на то, что экспрессия трансгена наблюдалась во всех органах и тканях мышиного организма, нейропатологический анализ показал, что только КП коры мозжечка подверглись дегенерации. Методами оценки моторной активности, а именно – с помощью теста ротарод, было продемонстрировано, что значительное ухудшение моторных функций гетерозиготных мышей линии SCA2-75Q происходит в возрасте 12-ти недель, а гомозиготных – в возрасте 6-ти недель по сравнению с мышами дикого типа того же возраста [60].
На настоящий момент были опубликованы данные только об одной нок-ин (KI) мышиной модели СЦА2 [61]. У мышей данной линии SCA2-42KI экспансия 42-х CAG повторов была вставлена в мышиный ген Atxn2. Для фенотипа SCA2-42KI мышей было характерно уменьшение массы и незначительное ухудшение моторных функций. Статистически значимые различия в координации движений при прохождении теста ротарод определялись для гомозиготных мышей в возрасте 72-х недель, при этом гетерозиготные мыши не проявили различий во всех исследуемых возрастах по сравнению с мышами дикого типа того же возраста. Нейропатологический анализ также выявил незначительные и поздно проявляющиеся изменения, соответствующие результатам оценки моторной активности [61].
Другие мыши-модели СЦА2 трансгенной линии SCA2-127Q имеют 127 CAG повторов под контролем Pcp2 промотора [62]. Данные мыши проявляют гораздо более тяжелые симптомы патологии из-за более длинной экспансии полиглутамина. Так, присутствие агрегатов атаксина-2 в клеточных телах КП наблюдалось уже в возрасте 4-х недель, далее статистически значимые различия в электрофизиологических свойствах КП проявлялись в возрасте 6-ти недель, и, наконец, прогрессирующие нарушения моторных функций при прохождении теста ротарод становились значимыми в возрасте 8-ми недель. Нарушения биохимических, электрофизиологических и моторных функций постепенно ухудшались в период между 8-ью и 40-ка неделями, однако потеря КП наблюдалась в случае данной модели только, начиная с возраста 40 недель [62]. Анализ взвешенных сетей коэкспресии генов выявил ранние и прогрессирующие нарушения в клеточной экспрессии генов, в первую очередь, в генах, ассоциированных с сигналингом ГТФазы, кальциевой сигнализацией и клеточной смертью, при этом временные изменения в экспрессии мРНК, начиная с первого дня существования, показали, что апрегуляция генов была связана с ацетилированием гистонов и ремоделированием хроматина, в то время как даунрегуляция генов была связана с клеточной адгезией и компонентами внеклеточного матрикса [63].
Наиболее недавняя трансгенная мышиная модель СЦА2, линия BAC-72Q, была создана с помощью искусственной бактериальной хромосомы (ВАС), включающей в себя ген Atxn2 целиком, в котором экзон 1 содержит вставку в 72 CAG повтора под контролем эндогенного человеческого промотора [64]. Для мышей линии BAC-SCA2-72Q характерны потеря массы, прогрессирующее ухудшение моторной координации согласно тесту ротарод, наблюдаемое начиная с возраста 16 недель, а также – утончение дендритного дерева КП и уменьшение уровней экспрессии белков кальбиндина и Рср2 в мозжечке [64].
Говоря об исследованиях, проводимых на мышиных моделях СЦА2, следует упомянуть мышей, нокаутных (KO) по гену атаксина-2. Первые мутантные мыши, нокаутные по атаксину-2, были получены методом электропорации целевого конструкта в эмбриональные стволовые клетки [30]. Морфологический анализ данных КО мышей не выявил никаких значимых ухудшений, между тем, у взрослых особей наблюдалось ожирение. Авторы данной работы сделали заключение о том, что белок атаксин-2 не является необходимым для развития и выживания мышей [30]. Однако, несмотря на то, что морфология и общее благополучие КО мышей не были значительно изменены, у них наблюдались нарушения биохимической и клеточной машинерии. Так, у атаксин-2 КО мышей наблюдалось повышенное фосфорилирование рибосомального белка S6, а также патологические изменения в синтезе белка [43]. Недавние исследования также выявили, что для КО мышей характерны нарушения метаболических путей жирных кислот, аминокислот лейцина, валина, изолейцина [44], а также гомеостаза кальция [45].
Мыши-модели СЦА2 различных трансгенных линий являются удобным инструментом для проведения фундаментальных исследований биохимических, электрофизиологических, и особенно моторных изменений, наблюдаемых при патологии СЦА2, однако даже несмотря на то, что большинство мышиных линий содержат ген человеческого атаксина-2, организм мышей значительно отличается от организма человека, а терапевтические подходы, показавшие многообещающие результаты в опытах на мышах, часто проваливаются в ходе клинических испытаний на людях. Недавний прогресс в области технологии плюрипотентного репрограммирования клеток позволил исследователям получить болезнь-специфические иПСК из СЦА2 пациентов. Данные клетки могут быть дифференцированы в нейроны, таким образом предоставляя возможность исследования механизмов заболевания СЦА2 и патологического развития болезни in vitro на нейронах, полученных из пациентов [7, 65]. Клеточные линии иПСК были успешно получены из человеческих фибробластов кожи СЦА2 пациентов [66], из периферических мононуклеарных клеток крови [67] или из обоих этих источников [65]. СЦА2-иПСК-полученные нейроны проявляли признаки СЦА2-ассоциированного патологического фенотипа in vitro, такие, как агрегация белков с увеличенным полиглутаминовым трактом, а также нарушенные микроструктуры митохондрий [65].
МОЛЕКУЛЯРНЫЙ МЕХАНИЗМ СЦА2
Молекулярный механизм СЦА2 обсуждается годами, многие исследовательские группы сделали значимый вклад в понимание патологии СЦА2, много различных подходов было использовано в попытке построить ясную картину молекулярного патогенеза СЦА2. Только четкое понимание молекулярных основ заболевания может дать нам шанс модифицировать патологические пути и, наконец, найти способ лечения данной болезни.
Агрегация
Для мутантного атаксина-2 с увеличенным полиглутаминовым трактом характерны конформационные изменения в сторону структуры, обогащенной β-листами, что способствует формированию нерастворимых агрегатов с амилоидными фибриллярными структурами, богатыми β-листами, и приводит к накоплению агрегатов в виде телец включения в нейронах [9]. Роль данных агрегатов и включений в дегенеративных процессах в мозжечке с патологией СЦА2 до сих пор остается неясной [68]. Изучение патологической агрегации в стволе мозга СЦА2 пациентов показало значимую положительную корреляцию между наличием гранулярного цитоплазматического окрашивания и более тяжелыми патологическими процессами, в то время как ядерные включения в нейронах играли защитную роль [35]. При помощи метода 1С2-иммунореактивного типирования в исследованиях на СЦА2 пациентах было показано, что гранулярный цитоплазматический паттерн наблюдался на ранних этапах заболевания, цитоплазматический и ядерный паттерн – в период активной стадии, и ядерный паттерн с включениями – во время конечной стадии [69]. Исследования поведения олигомеров с увеличенным полиглутаминовым трактом в мозге мышей обнаружили, что присутствие данных олигомеров приводило к разрыву мембраны ЭПР посредством вставки белка регуляции апоптоза Bax в мембрану ЭПР и последующей активации каспазы-7 [70]. Предотвращение формирования агрегатов и аккумуляции неправильно свернутых белков с увеличенным полиглутаминовым трактом может быть рассмотрено в качестве многообещающего терапевтического подхода для разработки болезнь-модифицирующей терапии заболеваний полиглутаминового тракта [9].
Окислительный стресс
Исследование митохондриального окислительного стресса на фибробластах, полученных из СЦА2 пациентов, обнаружили повышенную экспрессию SOD, а также уменьшенную экспрессию каталазы как на уровне транскрипта, так и на белковом уровне [71]. Повышенный уровень внутриклеточной перекиси водорода, производимой увеличенной SOD из супероксида, не может быть утилизирован из-за пониженного уровня каталазы, приводя таким образом к увеличению окислительного стресса, нарушениям в антиоксидантной системе, изменениям в системе окислительного фосфорилирования, а также к повреждениям митохондриальной активности, которые были обнаружены на фибробластах, полученных из СЦА2 пациентов. При добавлении к СЦА2 фибробластам антиоксиданта кофермента Q10 наблюдалась нормализация повышенного окислительного стресса [71]. У СЦА пациентов длительный прием кофермента Q10 приводил к улучшениям, подтвержденным шкалой оценки и ранжирования атаксии (SARA) и унифицированной шкалой оценки болезни Хантингтона во время двухлетнего перекрестного исследования у СЦА1 и СЦА3 пациентов, при этом улучшения не были статистически значимыми у СЦА2 пациентов, что скорее всего объясняется малым объемом экспериментальной группы [72]. Таким образом, клеточная система антиоксидантов может быть вовлечена в патогенез СЦА2, а ее регуляция может быть потенциальной терапевтической мишенью.
Нарушенная клеточная сигнализация
Процессы передачи информации между клетками являются исключительно важными для нормального развития, нейрогенеза, репарации, гомеостаза, иммунного ответа и других значимых клеточных процессов. Нарушения в клеточной сигнализации могут вести к различным патологическим процессам и нейродегенерации. Представители Src-семейства нерецепторных тирозиновых киназ (SFK) являются необходимыми для поддержания адекватного функционирования нервной системы и могут быть вовлечены в процессы нейродегенерации. Недавние исследования показали, что белок MTSS1, являющийся ингибитором SFK киназ, вовлечен в патологию СЦА [73]. На мышах-химерах было показано, что недостаток белка MTSS1 приводит к увеличению ферментной активности SFK киназ, которое сопровождается нарушениями электрофизиологической активности КП и, в конечном итоге, ведет к клеточной смерти. Лечение мутантных MTSS1 мышей известным противоопухолевым препаратом дазатиниб, являющемся ингибитором SFK киназ, предотвращало SFK-зависимое нарушение электрофизиологических функций КП и замедляло прогрессию симптомов атаксии у этих мышей. Интересно также заметить, что у СЦА2 мышей трансгенной линии SCA2-127Q наблюдается пониженный уровень белка MTSS1 и повышенная активность SFK киназ [73].
Другая киназа, протеинкиназа С (PKC), крайне важна для адекватного функционирования КП коры мозжечка, поскольку мутация в гене, кодирующем данную киназу, приводит к аномальному развитию дендритов КП и мозжечковой атаксии, а именно – СЦА14 [74]. На мышиной модели СЦА14 было показано, что в условиях патологии СЦА14 наблюдается апрегуляция генов, кодирующих карбоангидраза-относящийся-белок 8 (CAR8) и IP3R1 [75]. В результате недавних исследований на СЦА2 мышах трансгенной линии SCA2-127Q было показано, что повышенное фосфорилирование субстратов РКС играет роль защитного модификатора при наблюдаемой дегенерации КП, поскольку нормализация активности РКС в данных мышах приводила к усилению дегенеративных процессов [76]. Активация РКС в КП является необходимой для долговременной депрессии (LTD) синаптической передачи в синапсе параллельное волокно (ПВ)-КП, данные исследования также продемонстрировали, что повышенная активность РКС может опосредованно приводить к защитному эффекту в случае дегенерации КП посредством ограничения гипервозбудимости мембраны КП [76].
Киназы не являются единственными участниками, вовлеченными в нейрональный клеточный сигналинг и отвечающими за правильное выполнение функций клеточного аппарата, различные транскрипционные факторы также играют важную роль в регуляции клеточных ответов. Ядерный фактор транскрипции NF-kB принимает участие в контроле транскрипции ДНК, выработки цитокинов, клеточного выживания, а также в процессах синаптической пластичности и памяти [77]. Недавно было показано, что NF-kB сигналинг необходим для увеличения количества микроглии, а также для выработки фактора некроза опухоли TNF-α в мозжечке СЦА1 мышей, вдобавок он вовлечен в процессинг синапсов во время развития мозжечка [78]. На мышиной СЦА3 модели было выявлено, что NF-kB является энхансером дегенерации, опосредуя активацию астроцитов, что приводило к нейродегенерации, в то время как специфическое ингибирование NF-kB в астроцитах способствовало улучшению показателей выживаемости и увеличению продолжительности жизни [79]. Несмотря на то, что на настоящий момент не было опубликовано данных о вовлечении NF-kB в патогенез СЦА2, анализ взвешенных сетей коэкспресии генов, выполненный на мышах-моделях СЦА2 трансгенной линии SCA2-127Q, выявил даун регуляцию пути NF-kB в этих мышах [63].
Кальциевая дисрегуляция
Ионы кальция представляют собой необходимую часть клеточной сигнализации и благодаря своему аллостерическому эффекту способны контролировать активность многих ферментов и регуляторных белков [80]. Кальций способен активировать кальций-зависимые калиевые каналы и влиять на форму и частоту потенциала действия (ПД), изменения в концентрации кальция могут контролировать такие важные физиологические процессы как обучение, память и поведение [80]. Высокий уровень содержания кальция в цитоплазме вызывает апоптоз [11, 14, 81]. Нарушенный нейрональный кальциевый сигналинг может приводить к изменениям внутриклеточного баланса, потере синапсов и нарушению их функций и, в конечном итоге, к клеточной смерти. Кальциевая дисрегуляция наблюдается на моделях болезни Хантингтона (БХ), в случае различных видов СЦА, а также при болезни Альцгеймера [11–14].
Кальциевая сигнализация играет особую роль в функционировании КП коры мозжечка, поскольку КП экспрессируют огромное количество различных кальций-зависимых белков и ферментов для поддержания внутриклеточного баланса кальция. Например, КП коры мозжечка имеют высокие концентрации кальбиндина D-28k (CB) и парвальбумина (PV) в своих аксонах, соме , дендритах и шипиках. Данные белки принадлежат к объемному семейству кальций-связывающих белков (CaBPs) EF-руки [82], а их потеря приводит к изменениям функции ионных каналов Cav2.1 (P/Q-тип потенциал-зависимых кальциевых каналов (VDCC)), кодируемых геном CACNA1A [83].
Регуляция входа кальция в КП через VDCC-каналы крайне важна для правильного формирования синапса лазящее волокно (ЛВ)-КП во время послеродового развития [84]. Нарушения в синапсе ЛВ-КП вносят свой вклад в нейрональную патологию при СЦА [85]. КП также экспрессируют в большом количестве кальмодулин-связывающий активатор транскрипции 1 (CAMTA1), при этом делеция гена CAMTA1 в мышах приводит к тяжелой атаксии, сопровождающейся дегенерацией КП и атрофией мозжечка [86]. Главной основой моторного обучения считается LTD ПВ на КП. КП экспрессируют кальций/кальмодулин-зависимую протеинкиназу II (CaMKII), чья активация приводит к продолжительному увеличению уровня циклического гуанозинмонофосфата, таким образом, индукция сигнального механизма LTD опосредованно осуществляется молекулами CaMKII [87].
Внутри клетки кальций хранится в ЭПР и митохондриях. Было показано, что деление митохондрий и митохондриальный транспорт вовлечены в процессы развития арборизации дендритов в КП коры мозжечка [88]. Мутации в митохондриальной протеазе AFG3L2, входящей в протеом митохондрий, вызывают СЦА28 [89].
Существует два основных пути попадания кальция в цитоплазму КП. В обоих случаях необходимо присутствие глутамата, возбуждающего нейромедиатора. Первый путь представляет собой вход кальция из внеклеточного пространства через VDCC каналы [90]. Активация данных каналов происходит из-за деполяризации мембраны, которая в свою очередь вызывается положительной модуляцией рецептора α-амино-3-гидрокси-5-метил-4-изоксазолпропионовой кислоты (AMPA) молекулами глутамата, которые высвобождаются из пресинаптических терминалей ПВ и ЛВ в синаптичесую щель. Активация VDCC-каналов приводит к вбросу ионов кальция внутрь клетки, что в свою очередь ведет к активации кальций-зависимых калиевых каналов, а также имеет фундаментальное значение для формирования дендритных потенциалов, обусловленных кальциевыми каналами [91]. Отсутствие NMDA-рецепторов, а также кальций-проницаемых АМРА-рецепторов в КП является исключительным случаем среди ГАМКергических нейронов [92]. Второй путь представляет собой активацию метаботропного рецептора глутамата (mGluR) [93], что приводит к выбросу кальция из ЭПР через активированный IP3R-рецептор, данный кальциевый выход называется IP3-индуцируемый кальциевый выход (IICR). Активация mGluR или другого рецептора, сопряженного с G-белком, приводит к активации фосфолипазы С (PLC) на клеточной мембране, что катализирует гидролиз фосфатидилинозитол 4,5-бисфосфата (PIP2) на диацилглицерол (DAG) и IP3, которые являются вторичными мессенджерами и вовлечены в процессы передачи сигнала и липидной сигнализации в клетках различного типа. DAG остается связанным с мембраной, в то время как растворимый IP3 высвобождается в цитозоль. Далее, IP3 связывается с IP3R, приводя к IICR, таким образом происходит увеличение уровня внутриклеточного кальция, что запускает каскад сигнальных изменений и влияет на активность различных белков и ферментов [80].
Активация mGluR1 приводит к DAG-опосредованной положительной модуляции канонического рецептора временного потенциала 3 (TRPC3), представляющего собой неселективный катионный канал, участвующий в формировании медленных возбуждающих постсинаптических токов (ВПСТ). Потеря TRPC3-опосредованных медленных ВПСТ приводила к мозжечковой атаксии, вызванной дисрегуляцией mGluR [94]. Было показано, что транскрипционный фактор RORα вовлечен в экспрессию mGluR1 [95], а также осуществляет апрегуляцию экспрессии многих генов, связанных с кальций-зависимыми сигнальными путями, включая ген SLC1A6, кодирующий транспортер возбуждающих аминокислот 4 (EAAT4), ITPR1, кодирующий IP3R1, а также – ген PCP2/L7, кодирующий белок PcP2/L7 [96]. Для достижения данной апрегуляции, фактор RORα взаимодействует с белком атаксин-1, однако мутантный атаксин-1 с удлиненным полиглутаминовым трактом не способен сформировать транскрипционный комплекс с RORα, поэтому в случае трансгенных СЦА1 мышей наблюдается нарушение экспрессии важных генов регуляции баланса кальция, что ведет к нейродегенерации [97].
Изучение транскриптома мозжечка атаксин-2 КО мышей, а также мышей линии SCA2-42KI выявило, что у данных мышей наблюдаются схожие нарушения регуляции нескольких факторов баланса кальция вместе со многими факторами процессинга РНК, биоэнергетических процессов, клеточной адгезии и роста, а также липидной сигнализации [45]. В частности, наблюдалось уменьшение уровня мРНК генов, кодирующих важные белки, регулирующие баланс кальция, такие как сарко/ЭПР кальциевая АТФаза (SERCA), IP3-фосфатаза (5PP), IP3R, а также – транскрипционный фактор RORα [45].
В экспериментах на планарных липидных бислоях при помощи записи электрофизиологической активности от одиночного канала IP3R1, коэкспрессированного с мутантным атаксином-2 с удлиненным полиглутаминовым трактом, было продемонстрировано, что мутантный атаксин-2 связывается с С-концевым доменом IP3R1 (рис. 1), тогда как атаксин-2 дикого типа не проявляет таких свойств. Присутствие мутантного атаксина-2 значительно увеличивало активацию IP3R молекулами IP3 (рис. 1) [21]. Дальнейшие эксперименты по кальциевому имиджингу, выполненные на первичной культуре КП из мышей-моделей СЦА2 трансгенной линии SCA2-58Q и мышей дикого типа, выявили значительно увеличенный IP3R1-опосредованный IICR в SCA2-58Q КП, но не в КП дикого типа. С целью терапевтического уменьшения IICR посредством ингибирования рианодиновых рецепторов (RyR), еще одного типа внутриклеточных кальциевых каналов, были проведены эксперименты по добавлению препаратов рианодина и дантролена к культурам КП (рис. 1). Применение данных препаратов отменило патологический эффект присутствия мутантного атаксина-2, обратив уровень IICR до уровня дикого типа. Было предположено, что дантролен и рианодин исправили нарушения кальциевого сигналинга посредством уменьшения выброса кальция из ЭПР [21]. Долговременное кормление SCA2-58Q мышей дантроленом значительно улучшало моторную активность мышей и уменьшало дегенерацию КП в данных мышах [21].
Рис. 1.
Кальциевая гипотеза патогенеза СЦА2. Молекулы глутамата, высвобождаемые в синаптическую щель, активируют метаботропный рецептор глутамата (mGluR), приводя к выходу в цитоплазму молекул инозитол – 1,4,5-трисфосфата (IP3) и диацилглицерола (DAG) и к дальнейшей активации рецептора IP3 (IP3R) на мембране эндоплазматического ретикулума (ЭПР), что способствует выбросу кальция из ЭПР в цитоплазму. Данный процесс называют IP3-индуцируемым кальциевым выходом (IICR). Молекулы DAG непосредственно активируют рецептор временного потенциала 3 (TRPC3), осуществляя таким образом дополнительный приток ионов кальция из внеклеточного пространства в цитоплазму. Было показано, что мутантный атаксин-2 (Atxn2mut) с удлиненным полиглутаминовым трактом, в отличие от атаксина-2 дикого типа, ассоциируется с IP3R и увеличивает его чувствительность к IP3. Чрезмерная активация IP3R приводит к нарушению кальциевой сигнализации в клетках Пуркинье (КП) коры мозжечка. Излишнее количество ионов кальция закачивается в митохондрии (Мито) через митохондриальный кальциевый унипортер (MCU), что приводит к разбуханию митохондрий, к последующему выпрямлению крист, разрыву внешней мембраны митохондрий и к дальнейшему выбросу в цитоплазму проапоптотических факторов, таких как цитохром С (Cyt C), таким образом происходит процесс инициации апоптоза и, как следствие, нейродегенерация. Чрезмерно увеличенный IICR может быть подавлен вирус-опосредованной экспрессией фермента IP3-5-фосфатазы (5РР), которая переводит IP3 в неактивную форму IP2. Еще одним способом уменьшить выброс кальция из ЭПР может служить ингибирование рианодиновых рецепторов (RyR) дантроленом (Дан). Кальций-активируемые калиевые каналы малой проводимости (SK каналы) необходимы для поддержания правильной пейсмейкерной активности КП. Активация SK каналов с помощью рилузола, NS13001 или хлорзоксазона (CHZ) увеличивает гиперполяризацию (НР) мембраны КП, таким образом ограничивая работу потенциал-зависимых кальциевых каналов (VDCC) и, в итоге, приводя к уменьшению притока кальция из внеклеточного пространства.
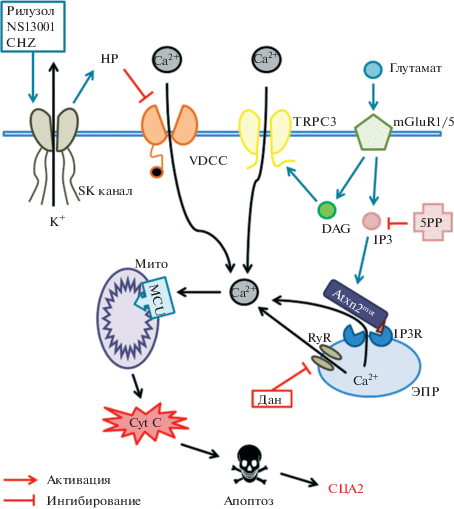
В дальнейших экспериментах с целью уменьшения уровня IP3 в КП мышей трансгенной линии SCA2-58Q с помощью вирус-опосредованной экспрессии 5РР была достигнута хроническая супрессия IICR (рис. 1) [22]. Пэтч-кламп эксперименты на срезах мозжечка выявили улучшение аномальной генерации ПД, характерное для КП стареющих СЦА2 мышей. Оценка моторной координации и последующий нейропатологический анализ показали, что хроническая сверхэкспрессия белка 5РР улучшает нарушения моторных функций и предотвращает смерть КП в SCA2-58Q мышах [22]. Полученные результаты поддерживают гипотезу о важной роли нарушений кальциевой сигнализации в молекулярном патогенезе СЦА2. Таким образом, частичное подавление IICR может иметь терапевтический эффект у СЦА2 пациентов и, возможно, у пациентов с другими типами СЦА [11, 22].
Недавние эксперименты на мозжечковых срезах SCA2-127Q мышей при помощи комбинированного метода локальной фиксации тока и двухфотонного кальциевого имиджинга показали, что у SCA2-127Q мышей наблюдается значительное повышение уровня соматического кальция наряду с повышенной частотой генерируемых ПД. При этом синаптическая активация mGluR1 посредством электрической стимуляции ПВ выявила значительное увеличение медленных возбуждающих постсинаптических потенциалов (EPSP) и значительно больший внутриклеточный выброс кальция в SCA2-127Q КП по сравнению с КП мышей дикого типа [98]. У данных мышей также наблюдался увеличенный mGluR-сигналинг. Авторы этого исследования пришли к выводу о том, что патология СЦА2 опирается на механизмы положительной обратной связи, включающие в себя взаимодействия между повышением уровня кальция, рецепторами mGluR1 и TRPC3, а также итоговым IICR (рис. 1) [98].
Недавние исследования на нейронах, полученных из иПСК СЦА2 пациентов, показали, что в условиях повышенной концентрации глутамата происходит изменение экспрессии генов глутаматных рецепторов и нарушение сигнализации кальция. При этом добавление к СЦА2- и ПСК-полученным нейронам рилузола, антагониста NMDA-рецептора дизоцилпина, антагониста АМРА-рецептора NBQX или стабилизатора кальция дантролена – блокатора рианодиновых рецепторов, представляющих собой кальциевые каналы ЭПР, уменьшало гибель клеток и улучшало митохондриальную дисфункцию, подтверждая таким образом гипотезу о вовлечении глутаматной эксайтотоксичности в молекулярный патогенез СЦА2 [65].
Изменения в самопроизвольной генерации ПД КП, наблюдаемое в случае различных моделей СЦА2 мышей [22, 58, 99], также может быть объяснено изменениями кальциевой и mGluR-опосредованной сигнализации. Кальций-активируемые калиевые каналы малой проводимости (SK-каналы) отвечают за пейсмейкерную активность КП и, таким образом, пониженная генерация импульсов СЦА2 КП, возможно, происходит по причине постоянно повышенного уровня внутриклеточного кальция [100]. Также было предположено, что постоянно повышенный уровень кальция может приводить к нарушению экспрессии и пост-трансляционной модуляции различных ионных каналов, необходимых для нормального электрофизиологического функционирования КП [100].
Аутофагия
Мутантные белки с удлиненным полиглутаминовым трактом склонны формировать агрегаты и токсические включения, для выведения которых необходимо привлечение механизма аутофагии [15]. Аутофагия является селективной лизосомально-опосредованной деградацией, в процессе которой происходит уничтожение аккумулированных и агрегированных белков, помеченных убиквитином [101]. Среди необходимых для аутофагии белков – белок 1 легкой цепи 3, ассоциированный с микротрубочками (MAP1LC3, ген MAP1LC3), p62 (ген SQSTM1) и связанный с аутофагией белок, содержащий домен FYVE (Alfy, ген WDFY3) [102]. Анализ периферических маркеров аутофагии у пациентов, страдающих заболеваниями полиглутаминового тракта, выявил повышенные уровни экспрессии MAP1LC3B, SQSTM1 и WDFY3 у БХ пациентов, при этом у СЦА2 пациентов был повышен только уровень экспрессии гена WDFY3 [103]. Авторы предположили, что наблюдаемые различия могут быть объяснены допущением о значительно меньшем количестве агрегатов в случае СЦА2 по сравнению с БХ, что также подтверждается низкой частотой встречаемости ядерных включений или их отсутствием в СЦА2 мышах [56].
В экспериментах на фибробластах, полученных от СЦА2 пациентов, и на мышах-моделях СЦА2 было показано, что экспансия полиглутамина в атаксине-2 также приводит к аномальной аутофагии, сопровождаемой повышенной экспрессией белка стауфен 1 (STAU1), представляющего собой белок, связывающий двунитевую РНК, который является необходимым для образования цитоплазматических включений в нейроглию и клеточные культуры, а также вовлечен в регуляцию функций стрессовых гранул (SG) и колокализирующийся с атаксином-2 в SG-подобных структурах [104]. Была выявлено, что повышенный уровень экспрессии STAU1 вызывает нарушения в процессинге РНК мишеней. И хотя необходимо провести более подробные исследования роли атаксина-2 в нарушенной аутофагии, все же было показано, что уменьшение уровня STAU1 приводило к улучшениям моторной координации СЦА2 мышей и, следовательно, может рассматриваться в качестве потенциального терапевтического подхода для лечения СЦА2 [104].
Роль микроглии
Роль мозжечковой микроглии в патогенезе СЦА2 была недавно рассмотрена в исчерпывающей обзорной статье А. Ferro с соавт. [105]. Микроглия является подтипом нейроглии, представляющим собой резидентные макрофаги ЦНС. Данные клетки принимают непосредственное участие в процессах развития и старения; среди их главных функций – формирование иммунного ответа и поддержание гомеостаза, включая утилизацию клеточного “мусора”, фагоцитоз и внеклеточную сигнализацию. Развитие микроглии строго регулируется определенными транскрипционными факторами [106]. В свою очередь, микроглия играет важную роль в развитии мозжечка. Так, она принимает участие в отсечении излишних и нефункциональных синапсов ЛВ-КП во время постнатального развития, что является необходимым для правильной работы проводящих путей мозжечка во взрослой жизни [107]. Исследования на СЦА1 мышах показали, что у данных мышей нарушено развитие ЛВ [108]. Было продемонстрировано, что на ранних этапах своего развития каждая КП иннервирована множеством ЛВ, но со временем только одно ЛВ усиливается и становится доминантным, сохраняя свои синапсы на дендритном древе КП. У SCA1-82Q-S776 мышей перемещение доминантной терминали ЛВ вверх по дендритному древу КП было задержано во времени, и некоторые из терминалей ЛВ оставались на соме КП, в то время как в SCA1-30Q-D776 мышах было нарушено отсечение синапсов, что приводило к гораздо более тяжелым последствиям [108].
Патологическая активация микроглии в мозжечке была обнаружена у СЦА пациентов и на различных мышиных моделях СЦА. В случае нескольких мышиных моделей СЦА1 на ранних стадиях заболевания еще до начала потери нейронов наблюдалось увеличение количества микроглии и TNF-α, при этом данная активация глии четко коррелировала с патологическим развитием заболевания, наводя на мысль о возможном механизме нейродегенерации, основанном на нейровоспалении, вызываемом микроглией [109]. Фармакологическое ингибирование рецептора колоний-стимулирующего фактора 1 (CSF1R), белка, ответственного за выработку, дифференцировку и поддержание функций микроглии, приводило к уменьшению объема мозжечковой микроглии на 69% и к улучшению моторных функций СЦА1 мышей [110].
НЕЙРОФИЗИОЛОГИЯ КП И ИЗМЕНЕНИЯ ФУНКЦИЙ ПРОВОДЯЩИХ ПУТЕЙ МОЗЖЕЧКА ПРИ СЦА2
Мозжечок представляет собой отдел головного мозга, который играет фундаментальную роль в регуляции мышечной активности и контроле моторных функций. Проводящие пути мозжечка являются ключевыми участниками осуществления координации движений. КП считаются главным динамическим элементом мозжечка, поскольку их аксоны формируют единственный выход, идущий от коры мозжечка к ядрам мозжечка и другим глубинным структурам мозга (рис. 2). КП спонтанно генерируют ПД с постоянной частотой [111–118]. Данная тоническая пейсмейкерная активность КП полагается критической для правильного кодирования информации в коре мозжечка [57, 118, 119]. Исследования, проводимые на срезах мозжечка мышей-моделей атаксии, показали, что нейрональная активность стареющих КП в случае различных моделей атаксии нарушена по сравнению с активностью КП мышей дикого типа того же возраста [22, 57, 99, 120–124]. На основе данных результатов была выдвинута идея о том, что начальные симптомы атаксии могут быть вызваны нейрональной дисфункцией, т.е. нарушениями генерации ПД КП, а не клеточной смертью КП. Было предположено, что в случае различных типов атаксий существует общий паттерн электрофизиологической дисфункции КП, а модуляция этих нарушений может иметь потенциальный терапевтический эффект [125].
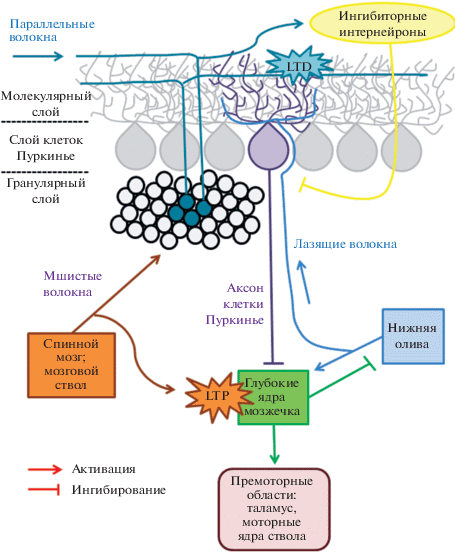
Рис. 2.
Схематическое представление проводящих путей мозжечка. Клетки Пуркинье (КП) образуют слой КП в коре мозжечка, а их аксоны формируют единственный выход, идущий от коры мозжечка к глубоким ядрам мозжечка. КП получают тормозные и возбуждающие импульсы через многочисленные синапсы, образованные на их дендритном древе, которое лежит в молекулярном слое коры мозжечка. Лазящие волокна (ЛВ, обозначены на схеме голубым цветом) берут свое начало в нейронах нижней оливы, восходят по мозжечку и формируют от 250 до 1500 синапсов на дендритном древе одиночной КП, в уникальном соотношении 1 : 1. Параллельные волокна (ПВ, обозначены еловым цветом) представляют собой аксоны гранулярных клеток, располагающихся в гранулярном слое коры мозжечка, и формируют по несколько синапсов с дистальной частью дендритного древа одиночной КП. Дендритное древо каждой отдельно взятой КП формирует огромное количество ПВ-КП синапсов, поскольку с каждой КП контактирует около 200 000 ПВ. Мшистые волокна (МВ, обозначены оранжевым цветом) берут свое начало в нейронах спинного мозга и мозгового ствола и переносят информацию от периферии и коры головного мозга к КП через аксоны гранулярных клеток, формирующие ПВ. Еловая и оранжевая звездочки изображают предполагаемые сайты ЛВ-опосредованных, ассоциативных форм синаптической пластичности, необходимых для ассоциативного моторного обучения (долговременная депрессия (LTD) синапсов ПВ и долговременная потенциация (LTP) синапсов МВ соответственно). Данные формы пластичности скорее всего нарушены в СЦА2. Адаптировано из [8, 100].
Нарушение электрофизиологических функций КП было впервые обнаружено у мышей трансгенной линии SCA2-127Q: для КП этих мышей было характерно прогрессирующее зависимое от возраста уменьшение частоты генерации ПД, соответствующее наблюдаемому прогрессирующему ухудшению моторных функций, при этом начало данных нарушений наблюдалось еще до начала дегенерации КП [62]. Далее, на мышах трансгенной линии SCA2-58Q также было показано значительное уменьшение частоты генерации ПД КП с возрастом, однако в добавок к этому наблюдалось значительное уменьшение точности генерации ПД и наличие аномальных пачечных паттернов активности [22, 57].
Недавние исследования, проводимые на мышах трансгенной линии SCA2-127Q, выявили, что уменьшение частоты генерации ПД в случае SCA2-127Q КП наблюдается одновременно с уменьшением уровня экспрессии потенциал-зависимых калиевых каналов Kv3.3 и кальций-активируемых калиевых каналов большой проводимости (BK channels), что также приводило к недавно обнаруженной следовой гиперполяризации (AHP) мембраны с медленной кинетикой и уменьшенной амплитудой [126]. У КП мышей трансгенной линии SCA2-127Q также наблюдалось нарушение генерации спонтанной тонической активности, а именно: наблюдалось значительное снижение регулярности генерации спайков [126]. Значение тока, необходимого для того, чтобы SCA2-127Q КП перешли в состояние деполяризационного блока регулярных спайков, было значительно ниже по сравнению со значениями, необходимыми для КП мышей дикого типа [126]. Известно, что мутации в генах различных ионных каналов приводят к патологии СЦА. В случае атаксий полиглутаминового тракта нарушения функций ионных каналов наблюдаются в связи с изменениями в их транскрипционных уровнях [127]. Недавно проведенное панельное исследование на пациентах с аутосомно-доминантными формами прогрессирующей мозжечковой атаксии выявило наличие у данных пациентов точечных мутаций в генах субъединицы альфа 1 А VDCC (CACNA1A), а также в гене, вызывающем наследственную спастическую параплегию (SPG7) [128]. Мутации других ионных каналов также могут вызывать атаксию и фенотип двигательных расстройств. В частности, новая мутация в альфа-субъединице потенциалзависимого натриевого канала VIII типа (SCN8A) была обнаружена недавно на мышах с хроническим расстройством координации движения, для которых характерно появление тремора на ранних этапах развития, проявление дистонии во взрослом возрасте, а также – укороченная продолжительность жизни и нарушение моторных функций. КП данных мышей также проявляли недостаток спонтанной и индуцированной электрофизиологической активности [129].
Несмотря на то, что при СЦА в первую очередь наблюдается поражение КП, нарушения проводящих путей мозжечка также наблюдаются при данной патологии. Нормальное функционирование возбуждающих входов на КП, таких как ЛВ и ПВ, и их синаптической пластичности необходимо для нормальной работы мозжечка и осуществления двигательной активности (рис. 2) [8]. Известно, что дисфункция КП и волокон мозжечка приводит к проявлению фенотипа атаксии еще до потери КП, при этом ЛВ контролирует нормальную работу КП [8]. Своевременное ингибирование КП в схеме мозжечка является необходимым для формирования памяти в мозжечке [130]. Существуют экспериментальные доказательства того, что ЛВ принимают участие в процессах моторного обучения и в функциях контроля координации движений, а также могут играть немаловажную роль в развитии мозжечка [131]. Анализ патологии синапса ЛВ-КП в мозжечке пациентов, страдающих различными дегенеративными заболеваниями движений, выявил специфические патологические изменения ЛВ среди данных заболеваний, скорее всего отображая таким образом различные механизмы двигательных патологий [132].
КП генерируют два типа спайков: простые спайки (ПС) и сложные спайки (СС). Данные два типа спайков возникают благодаря двум главным типам афферентных волокон мозжечка, мшистым волокнам (МВ) и ЛВ (рис. 2). МВ берут свое начало в нейронах спинного мозга и мозгового ствола и переносят информацию от периферии и коры мозга к КП через аксоны гранулярных клеток, формирующие ПВ. ЛВ берут свое начало в ядре нижней оливы (НО) и посылают информацию от коры головного мозга к КП, формируя многочисленные синаптические контакты с проксимальными дендритами КП. ЛВ переносят возбуждение, благодаря которому КП генерируют СС: начальный высокоамплитудный ПД, за которым следуют высокочастотные пачки потенциалов меньшей амплитуды (их также называют спайклетами). В тоже время КП генерирует ПС в ответ на возбуждающие потенциалы, приносимые ПВ [114, 133, 134]. Фармакологическая стимуляция оливо-мозжечкового пути гармалином, алкалоидом, который является специфическим активатором нейронов нижней оливы [135], выявила in vivo нарушения паттерна электрофизиологической активности и формы СС КП мышей трансгенной линии SCA2-58Q по сравнению с КП мышей дикого типа того же возраста [59]. Нарушения синапса ЛВ-КП ухудшались с возрастом. Было предположено, что патологические изменения в синапсе ЛВ-КП являются одной из причин симптомов атаксии в случае СЦА2 и, возможно, других типов СЦА [59].
ПОТЕНЦИАЛЬНЫЕ ТЕРАПЕВТИЧЕСКИЕ ПОДХОДЫ ДЛЯ ЛЕЧЕНИЯ СЦА2
Одним из наиболее многообещающих терапевтических подходов для лечения СЦА2 на настоящий момент является антисенс-терапия, поскольку данная методика показала обнадеживающие результаты в клинических испытаниях на БХ пациентах [136]. В случае антисенс-терапии последовательность олигонуклеотидов, комплементарная таргетной мРНК, используется для того, чтобы подавить экспрессию этой мРНК и, таким образом, уменьшить уровень белка интереса [137]. В недавних исследованиях in vitro был проведен скрининг 152-х антисенс олигонуклеотидов (ASO), спроектированных in silico в качестве мишеней для человеческого атаксина-2, в результате чего олигонуклеотид ASO7 был выявлен в качестве лучшего кандидата для антисенс-терапии СЦА2 [17]. Последующие эксперименты на трансгенных мышах показали, что инъекции ASO7 в желудочки головного мозга подопытных мышей приводили к уменьшению экспрессии человеческого мутантного атаксина-2 в мозжечке мышей в среднем на 75%, при этом у подопытных мутантных мышей значительно уличшались моторные функции, генерация спайков восстанавливалась до уровня мышей дикого типа, а также происходила нормализация белковых уровней нескольких СЦА2-связанных белков, экспрессируемых в КП, включая белки Rgs8, Pcp2, Pcp4, Homer3 и Cep76 [17].
Другим многообещающим подходом в терапии СЦА2 является использование мезенхимальных стволовых клеток (МСК). МСК представляют собой мультипотентные стромальные фибробласт-подобные клетки, которые способны дифференцироваться в клетки различных типов, а также проявляют некоторые иммуно-модулирующие функции, например, выброс нейротрофических факторов [138]. Исследования на трансгенных СЦА2 мышах показали, что внутривенные инъекции человеческих МСК приводили к значительному улучшению моторных функций СЦА2 мышей и предотвращали дегенерацию КП в коре мозжечка [139]. Клинические испытания I/IIа фазы на шести СЦА3 пациентах подтвердили безопасность и хорошую переносимость внутривенных инъекций МСК, полученных от здоровых доноров, также наблюдалось незначительное улучшение моторных функций, однако для того, чтобы делать релевантные выводы необходимо проведение клинических испытаний с большим количеством пациентов [19].
Последним из известных клинических испытаний на СЦА пациентах является рандомизированное, плацебо-контролируемое исследование, основанное на двойном слепом методе, посвященное оценке действия рилузола, небольшой молекулы, которая способна связывать и аллостерически модулировать активность SK каналов [140]. Проведенное исследование выявило значительное улучшение показателей координации движения и не имело каких-либо серьезных побочных эффектов [20].
ЗАКЛЮЧЕНИЕ
На настоящий момент СЦА2 все еще остается неизлечимым прогрессирующим нейродегенеративным заболеванием, однако многочисленные исследования физиологических, биохимических и функциональных характеристик мозга в условиях патологии СЦА2 продолжаются и в данный момент с тем, чтобы найти новые терапевтические мишени для лечения СЦА2 на ранних стадиях и предотвратить развитие дальнейших симптомов. Современные подходы в исследовании молекулярных механизмов заболевания позволили научному сообществу выделить стратегические направления потенциальной терапии и помогли в создании многообещающих терапевтических подходов.
Список литературы
Ashizawa T., Oz G., Paulson H. L. Spinocerebellar ataxias: Prospects and challenges for therapy development. Nat. Rev. Neurol. 14(10): 590–605. 2018.
Magana J.J., Velazquez-Perez L., Cisneros B. Spinocerebellar ataxia type 2: Clinical presentation, molecular mechanisms, and therapeutic perspectives. Mol. Neurobiol. 47(1): 90–104. 2013.
Paulson H.L., Shakkottai V.G., Clark H.B., Orr H.T. Polyglutamine spinocerebellar ataxias – from genes to potential treatments. Nat. Rev. Neurosci. 18(10): 613–626. 2017.
Scoles D.R., Pulst S.M. Spinocerebellar Ataxia Type 2. Adv. Exp. Med. Biol. 1049: 175–195. 2018.
Buijsen R.A.M., Toonen L.J.A., Gardiner S.L., van Roon-Mom W.M.C. Genetics, mechanisms, and therapeutic progress in polyglutamine spinocerebellar ataxias. Neurotherapeutics. 2019.
Satterfield T.F., Pallanck L.J. Ataxin-2 and its Drosophila homolog, ATX2, physically assemble with polyribosomes. Hum. Mol. Genet. 15(16): 2523–32. 2006.
Alves-Cruzeiro J.M., Mendonca L., Pereira de Almeida L., Nobrega C. Motor Dysfunctions and neuropathology in mouse models of spinocerebellar ataxia type 2: A Comprehensive Review. Front Neurosci. 10: 572. 2016.
Smeets C.J., Verbeek D.S. Climbing fibers in spinocerebellar ataxia: A mechanism for the loss of motor control. Neurobiol. Dis. 88: 96–106. 2016.
Takeuchi T., Nagai Y. Protein Misfolding and aggregation as a therapeutic target for polyglutamine diseases. Brain Sci. 7(10). 2017.
Massey T.H., Jones L. The central role of DNA damage and repair in CAG repeat diseases. Dis. Model Mech. 11(1). 2018.
Egorova P., Popugaeva E., Bezprozvanny I. Disturbed calcium signaling in spinocerebellar ataxias and Alzheimer’s disease. Semin. Cell Dev. Biol. 40: 127–133. 2015.
Egorova P.A., Bezprozvanny I.B. Inositol 1,4,5-trisphosphate receptors and neurodegenerative disorders. FEBS J. 285(19): 3547–3565. 2018.
Hisatsune C., Hamada K., Mikoshiba K. Ca(2+) signaling and spinocerebellar ataxia. Biochim. Biophys. Acta Mol. Cell Res. 11: 1733–1744. 2018.
Mark M.D., Schwitalla J.C., Groemmke M., Herlitze S. Keeping our calcium in balance to maintain our balance. Biochem. Biophys. Res. Commun. 483(4): 1040–1050. 2017.
Ashkenazi A., Bento C.F., Ricketts T., Vicinanza M., Siddiqi F., Pavel M., Squitieri F., Hardenberg M.C., Imarisio S., Menzies F.M., Rubinsztein D.C. Polyglutamine tracts regulate autophagy. Autophagy. 13(9): 1613–1614. 2017.
Yau W.Y., O’Connor E., Sullivan R., Akijian L., Wood N.W. DNA repair in trinucleotide repeat ataxias. FEBS J. 285(19): 3669–3682. 2018.
Scoles D.R., Meera P., Schneider M.D., Paul S., Dansithong W., Figueroa K.P., Hung G., Rigo F., Bennett C.F., Otis T.S., Pulst S.M. Antisense oligonucleotide therapy for spinocerebellar ataxia type 2. Nature. 544(7650): 362–366. 2017.
Teive H.A.G., Camargo C.H.F., Munhoz R.P. Antisense Oligonucleotide Therapy for Spinocerebellar Ataxias: Good News for Terrible Diseases. Mov. Disord. Clin. Pract. 5(4): 400–403. 2018.
Tsai Y.A., Liu R.S., Lirng J.F., Yang B.H., Chang C.H., Wang Y.C., Wu Y.S., Ho J.H., Lee O.K., Soong B.W. Treatment of spinocerebellar ataxia with mesenchymal stem cells: A Phase I/IIa Clinical Study. Cell Transplant. 26(3): 503–512. 2017.
Romano S., Coarelli G., Marcotulli C., Leonardi L., Piccolo F., Spadaro M., Frontali M., Ferraldeschi M., Vulpiani M. C., Ponzelli F., Salvetti M., Orzi F., Petrucci A., Vanacore N., Casali C., Ristori G. Riluzole in patients with hereditary cerebellar ataxia: A randomised, double-blind, placebo-controlled trial. Lancet Neurol. 14(10): 985–991. 2015.
Liu J., Tang T.S., Tu H., Nelson O., Herndon E., Huynh D.P., Pulst S.M., Bezprozvanny I. Deranged calcium signaling and neurodegeneration in spinocerebellar ataxia type 2. J. Neurosci. 29(29): 9148–9162. 2009.
Kasumu A.W., Liang X., Egorova P., Vorontsova D., Bezprozvanny I. Chronic suppression of inositol 1,4,5-triphosphate receptor-mediated calcium signaling in cerebellar purkinje cells alleviates pathological phenotype in spinocerebellar ataxia 2 mice. J. Neurosci. 32(37): 12786–12796. 2012.
Bushart D.D., Chopra R., Singh V., Murphy G.G., Wulff H., Shakkottai V. G. Targeting potassium channels to treat cerebellar ataxia. Ann. Clin. Transl. Neurol. 5(3): 297–314. 2018.
Gispert S., Twells R., Orozco G., Brice A., Weber J., Heredero L., Scheufler K., Riley B., Allotey R., Nothers C. Chromosomal assignment of the second locus for autosomal dominant cerebellar ataxia (SCA2) to chromosome 12q23-24.1. Nat. Genet. 4(3): 295–299. 1993.
Fernandez M., McClain M.E., Martinez R.A., Snow K., Lipe H., Ravits J., Bird T.D., La Spada A.R. Late-onset SCA2: 33 CAG repeats are sufficient to cause disease. Neurology. 55(4): 569–572. 2000.
Pulst S.M. The complex structure of ATXN2 genetic variation. Neurol. Genet. 4(6): e299. 2018.
Almaguer-Mederos L.E., Mesa J.M.L., Gonzalez-Zaldivar Y., Almaguer-Gotay D., Cuello-Almarales D., Aguilera-Rodriguez R., Falcon N.S., Gispert S., Auburger G., Velazquez-Perez L. Factors associated with ATXN2 CAG/CAA repeat intergenerational instability in Spinocerebellar ataxia type 2. Clin. Genet. 94(3–4): 346–350. 2018.
Sena L.S., Castilhos R.M., Mattos E.P., Furtado G.V., Pedroso J.L., Barsottini O., de Amorim M.M.P., Godeiro C., Pereira M.L.S., Jardim L.B. Selective forces related to spinocerebellar ataxia type 2. Cerebellum. 18(2): 188–194. 2019.
van de Loo S., Eich F., Nonis D., Auburger G., Nowock J. Ataxin-2 associates with rough endoplasmic reticulum. Exp. Neurol. 215(1): 110–118. 2009.
Kiehl T.R., Nechiporuk A., Figueroa K.P., Keating M.T., Huynh D.P., Pulst S.M. Generation and characterization of Sca2 (ataxin-2) knockout mice. Biochem. Biophys. Res. Commun. 339(1): 17–24. 2006.
Lastres-Becker I., Brodesser S., Lutjohann D., Azizov M., Buchmann J., Hintermann E., Sandhoff K., Schurmann A., Nowock J., Auburger G. Insulin receptor and lipid metabolism pathology in ataxin-2 knock-out mice. Hum. Mol. Genet. 17(10): 1465–1481. 2008.
Pfeffer M., Gispert S., Auburger G., Wicht H., Korf H.W. Impact of Ataxin-2 knock out on circadian locomotor behavior and PER immunoreaction in the SCN of mice. Chronobiol. Int. 34(1): 129–137. 2017.
Lim C., Allada R. ATAXIN-2 activates PERIOD translation to sustain circadian rhythms in Drosophila. Science. 340(6134): 875–879. 2013.
Zhang Y., Ling J., Yuan C., Dubruille R., Emery P. A role for Drosophila ATX2 in activation of PER translation and circadian behavior. Science. 340(6134): 879–882. 2013.
Seidel K., Siswanto S., Fredrich M., Bouzrou M., den Dunnen W.F.A., Ozerden I., Korf H.W., Melegh B., de Vries J.J., Brunt E.R., Auburger G., Rub U. On the distribution of intranuclear and cytoplasmic aggregates in the brainstem of patients with spinocerebellar ataxia type 2 and 3. Brain Pathol. 27(3): 345–355. 2017.
Lim N.S., Kozlov G., Chang T.C., Groover O., Siddiqui N., Volpon L., De Crescenzo G., Shyu A.B., Gehring K. Comparative peptide binding studies of the PABC domains from the ubiquitin-protein isopeptide ligase HYD and poly(A)-binding protein. Implications for HYD function. J. Biol. Chem. 281(20): 14376-14382. 2006.
Lastres-Becker I., Nonis D., Eich F., Klinkenberg M., Gorospe M., Kotter P., Klein F.A., Kedersha N., Auburger G. Mammalian ataxin-2 modulates translation control at the pre-initiation complex via PI3K/mTOR and is induced by starvation. Biochim. Biophys. Acta. 1862(9): 1558–1569. 2016.
Nonhoff U., Ralser M., Welzel F., Piccini I., Balzereit D., Yaspo M.L., Lehrach H., Krobitsch S. Ataxin-2 interacts with the DEAD/H-box RNA helicase DDX6 and interferes with P-bodies and stress granules. Mol. Biol. Cell. 18(4): 1385–1396. 2007.
Bakthavachalu B., Huelsmeier J., Sudhakaran I.P., Hillebrand J., Singh A., Petrauskas A., Thiagarajan D., Sankaranarayanan M., Mizoue L., Anderson E.N., Pandey U.B., Ross E., Vij-ayRaghavan K., Parker R., Ramaswami M. RNP-Granule Assembly via Ataxin-2 Disordered Domains Is Required for Long-Term Memory and Neurodegeneration. Neuron. 98(4): 754–766 e4. 2018.
Ostrowski L.A., Hall A.C., Szafranski K.J., Oshidari R., Abraham K.J., Chan J.N.Y., Krustev C., Zhang K., Wang A., Liu Y., Guo R., Mekhail K. Conserved Pbp1/Ataxin-2 regulates retrotransposon activity and connects polyglutamine expansion-driven protein aggregation to lifespan-controlling rDNA repeats. Commun. Biol. 1: 187. 2018.
Sen N.E., Drost J., Gispert S., Torres-Odio S., Damrath E., Klinkenberg M., Hamzeiy H., Akdal G., Gulluoglu H., Basak A.N., Auburger G. Search for SCA2 blood RNA biomarkers highlights Ataxin-2 as strong modifier of the mitochondrial factor PINK1 levels. Neurobiol. Dis. 96: 115–126. 2016.
Li P.P., Sun X., Xia G., Arbez N., Paul S., Zhu S., Peng H.B., Ross C.A., Koeppen A.H., Margolis R.L., Pulst S.M., Ashizawa T., Rudnicki D.D. ATXN2-AS, a gene antisense to ATXN2, is associated with spinocerebellar ataxia type 2 and amyotrophic lateral sclerosis. Ann. Neurol. 80(4): 600–615. 2016.
Fittschen M., Lastres-Becker I., Halbach M.V., Damrath E., Gispert S., Azizov M., Walter M., Muller S., Auburger G. Genetic ablation of ataxin-2 increases several global translation factors in their transcript abundance but decreases translation rate. Neurogenetics. 16(3): 181–192. 2015.
Meierhofer D., Halbach M., Sen N.E., Gispert S., Auburger G. Ataxin-2 (Atxn2)-Knock-Out Mice Show Branched Chain Amino Acids and Fatty Acids Pathway Alterations. Mol. Cell Proteomics. 15(5): 1728–39. 2016.
Halbach M.V., Gispert S., Stehning T., Damrath E., Walter M., Auburger G. Atxn2 Knockout and CAG42-Knock-in Cerebellum Shows Similarly Dysregulated Expression in Calcium Homeostasis Pathway. Cerebellum. 16(1): 68-81. 2017.
Louis E.D., Kuo S.H., Tate W.J., Kelly G.C., Gutierrez J., Cortes E.P., Vonsattel J.G., Faust P.L. Heterotopic Purkinje cells: a Comparative postmortem study of essential tremor and spinocerebellar ataxias 1, 2, 3, and 6. Cerebellum. 17(2): 104–110. 2018.
Nibbeling E.A.R., Duarri A., Verschuuren-Bemelmans C.C., Fokkens M.R., Karjalainen J. M., Smeets C., de Boer-Bergsma J.J., van der Vries G., Dooijes D., Bampi G. B., van Diemen C., Brunt E., Ippel E., Kremer B., Vlak M., Adir N., Wijmenga C., van de Warrenburg B.P.C., Franke L., Sinke R.J., Verbeek D.S. Exome sequencing and network analysis identifies shared mechanisms underlying spinocerebellar ataxia. Brain. 140(11): 2860–2878. 2017.
Aboulhoda B.E., Hassan S.S. Effect of prenatal tramadol on postnatal cerebellar development: Role of oxidative stress. J. Chem. Neuroanat. 94: 102–118. 2018.
Squadrone S., Brizio P., Mancini C., Abete M.C., Brusco A. Altered homeostasis of trace elements in the blood of SCA2 patients. J. Trace Elem. Med. Biol. 47: 111–114. 2018.
Guevara-Garcia M., Gil-del Valle L., Velasquez-Perez L., Garcia-Rodriguez J.C. Oxidative stress as a cofactor in spinocerebellar ataxia type 2. Redox Rep. 17(2): 84–89. 2012.
Almaguer-Gotay D., Almaguer-Mederos L.E., Aguilera-Rodriguez R., Rodriguez-Labrada R., Cuello-Almarales D., Estupinan-Dominguez A., Velazquez-Perez L.C., Gonzalez-Zaldivar Y., Vazquez-Mojena Y. Spinocerebellar Ataxia Type 2 Is Associated with the Extracellular Loss of Superoxide Dismutase but Not Catalase Activity. Front Neurol. 8: 276. 2017.
Almaguer-Mederos L.E., Almaguer-Gotay D., Aguilera-Rodriguez R., Gonzalez-Zaldivar Y., Cuello-Almarales D., Laffita-Mesa J., Vazquez-Mojena Y., Zayas-Feria P., Rodriguez-Labrada R., Velazquez-Perez L., MacLeod P. Association of glutathione S-transferase omega polymorphism and spinocerebellar ataxia type 2. J. Neurol. Sci. 372: 324-328. 2017.
Monte T.L., Pereira F.S., Reckziegel E.D.R., Augustin M.C., Locks-Coelho L.D., Santos A.S.P., Pedroso J.L., Barsottini O., Vargas F.R., Saraiva-Pereira M.L., Jardim L.B. Neurological phenotypes in spinocerebellar ataxia type 2: Role of mitochondrial polymorphism A10398G and other risk factors. Parkinsonism Relat. Disord. 42: 54–60. 2017.
Hamzeiy H., Savas D., Tunca C., Sen N.E., Gundogdu Eken A., Sahbaz I., Calini D., Tiloca C., Ticozzi N., Ratti A., Silani V., Basak A.N. Elevated Global DNA Methylation Is Not Exclusive to Amyotrophic Lateral Sclerosis and Is Also Observed in Spinocerebellar Ataxia Types 1 and 2. Neurodegener. Dis. 18(1): 38–48. 2018.
Wilke C., Bender F., Hayer S.N., Brockmann K., Schols L., Kuhle J., Synofzik M. Serum neurofilament light is increased in multiple system atrophy of cerebellar type and in repeat-expansion spinocerebellar ataxias: A pilot study. J. Neurol. 265(7): 1618–1624. 2018.
Huynh D.P., Figueroa K., Hoang N., Pulst S.M. Nuclear localization or inclusion body formation of ataxin-2 are not necessary for SCA2 pathogenesis in mouse or human. Nat. Genet. 26(1): 44–50. 2000.
Kasumu A.W., Hougaard C., Rode F., Jacobsen T.A., Sabatier J.M., Eriksen B.L., Strobaek D., Liang X., Egorova P., Vorontsova D., Christophersen P., Ronn L.C., Bezprozvanny I. Selective positive modulator of calcium-activated potassium channels exerts beneficial effects in a mouse model of spinocerebellar ataxia type 2. Chem. Biol. 19(10): 1340–1353. 2012.
Egorova P.A., Zakharova O.A., Vlasova O.L., Bezprozvanny I.B. In vivo analysis of cerebellar Purkinje cell activity in SCA2 transgenic mouse model. J. Neurophysiol. 115(6): 2840–2851. 2016.
Egorova P.A., Gavrilova A.V., Bezprozvanny I.B. In Vivo Analysis of the Climbing Fiber-Purkinje Cell Circuit in SCA2-58Q Transgenic Mouse Model. Cerebellum. 17(5): 590–600. 2018.
Aguiar J., Fernandez J., Aguilar A., Mendoza Y., Vazquez M., Suarez J., Berlanga J., Cruz S., Guillen G., Herrera L., Velazquez L., Santos N., Merino N. Ubiquitous expression of human SCA2 gene under the regulation of the SCA2 self promoter cause specific Purkinje cell degeneration in transgenic mice. Neurosci. Lett. 392(3): 202–206. 2006.
Damrath E., Heck M.V., Gispert S., Azizov M., Nowock J., Seifried C., Rub U., Walter M., Auburger G. ATXN2-CAG42 sequesters PABPC1 into insolubility and induces FBXW8 in cerebellum of old ataxic knock-in mice. PLoS Genet. 8(8): e1002920. 2012.
Hansen S. T., Meera P., Otis T.S., Pulst S. M. Changes in Purkinje cell firing and gene expression precede behavioral pathology in a mouse model of SCA2. Hum. Mol. Genet. 22(2): 271–283. 2013.
Pflieger L.T., Dansithong W., Paul S., Scoles D.R., Figueroa K.P., Meera P., Otis T.S., Facelli J.C., Pulst S.M. Gene co-expression network analysis for identifying modules and functionally enriched pathways in SCA2. Hum. Mol. Genet. 26(16): 3069–3080. 2017.
Dansithong W., Paul S., Figueroa K.P., Rinehart M.D., Wiest S., Pflieger L.T., Scoles D.R., Pulst S.M. Ataxin-2 regulates RGS8 translation in a new BAC-SCA2 transgenic mouse model. PLoS Genet. 11(4): e1005182. 2015.
Chuang C.Y., Yang C.C., Soong B.W., Yu C.Y., Chen S.H., Huang H.P., Kuo H.C. Modeling spinocerebellar ataxias 2 and 3 with iPSCs reveals a role for glutamate in disease pathology. Sci. Rep. 9(1): 1166. 2019.
Marthaler A.G., Schmid B., Tubsuwan A., Poulsen U.B., Hyttel P., Nielsen T.T., Nielsen J.E., Holst B. Generation of spinocerebellar ataxia type 2 patient-derived iPSC line H196. Stem. Cell. Res. 16(1): 199–201. 2016.
Maguire J.A., Gagne A.L., Gonzalez-Alegre P., Davidson B.L., Shakkottai V., Gadue P., French D.L. Generation of Spinocerebellar Ataxia Type 2 induced pluripotent stem cell lines, CHOPi002-A and CHOPi003-A, from patients with abnormal CAG repeats in the coding region of the ATXN2 gene. Stem. Cell. Res. 34: 101361. 2019.
Todd T.W., Lim J. Aggregation formation in the polyglutamine diseases: Protection at a cost? Mol. Cells. 36(3): 185–194. 2013.
Koyano S., Yagishita S., Kuroiwa Y., Tanaka F., Uchihara T. Neuropathological staging of spinocerebellar ataxia type 2 by semiquantitative 1C2-positive neuron typing. Nuclear translocation of cytoplasmic 1C2 underlies disease progression of spinocerebellar ataxia type 2. Brain Pathol. 24(6): 599–606. 2014.
Ueda M., Li S., Itoh M., Hayakawa-Yano Y., Wang M.X., Hayakawa M., Hasebe-Matsubara R., Ohta K., Ohta E., Mizuno A., Hida Y., Matsumoto M., Chen H., Nakagawa T. Polyglutamine expansion disturbs the endoplasmic reticulum formation, leading to caspase-7 activation through Bax. Biochem. Biophys. Res. Commun. 443(4): 1232–1238. 2014.
Cornelius N., Wardman J. H., Hargreaves I.P., Neergheen V., Bie A. S., Tumer Z., Nielsen J.E., Nielsen T.T. Evidence of oxidative stress and mitochondrial dysfunction in spinocerebellar ataxia type 2 (SCA2) patient fibroblasts: Effect of coenzyme Q10 supplementation on these parameters. Mitochondrion. 34: 103–114. 2017.
Lo R.Y., Figueroa K.P., Pulst S.M., Lin C.Y., Perlman S., Wilmot G., Gomez C., Schmahmann J., Paulson H., Shakkottai V.G., Ying S., Zesiewicz T., Bushara K., Geschwind M., Xia G., Subramony S.H., Ashizawa T., Kuo S.H. Coenzyme Q10 and spinocerebellar ataxias. Mov. Disord. 30(2): 214–220. 2015.
Brown A.S., Meera P., Altindag B., Chopra R., Perkins E.M., Paul S., Scoles D.R., Tarapore E., Magri J., Huang H., Jackson M., Shakkottai V.G., Otis T.S., Pulst S.M., Atwood S.X., Oro A.E. MTSS1/Src family kinase dysregulation underlies multiple inherited ataxias. Proc. Natl. Acad. Sci. U. S. A. 115(52): E12407–E12416. 2018.
Verbeek D.S., Goedhart J., Bruinsma L., Sinke R.J., Reits E.A. PKC gamma mutations in spinocerebellar ataxia type 14 affect C1 domain accessibility and kinase activity leading to aberrant MAPK signaling. J. Cell Sci. 121(Pt 14): 2339–2349. 2008.
Shimobayashi E., Kapfhammer J.P. Calcium Signaling, PKC Gamma, IP3R1 and CAR8 Link Spinocerebellar Ataxias and Purkinje Cell Dendritic Development. Curr. Neuropharmacol. 16(2): 151–159. 2018.
Chopra R., Wasserman A.H., Pulst S.M., De Zeeuw C.I., Shakkottai V.G. Protein kinase C activity is a protective modifier of Purkinje neuron degeneration in cerebellar ataxia. Hum. Mol. Genet. 27(8): 1396–1410. 2018.
Meffert M.K., Chang J.M., Wiltgen B.J., Fanselow M.S., Baltimore D. NF-kappa B functions in synaptic signaling and behavior. Nat. Neurosci. 6(10): 1072–1078. 2003.
Ferro A., Qu W., Lukowicz A., Svedberg D., Johnson A., Cvetanovic M. Inhibition of NF-kappaB signaling in IKKbetaF/F;LysM Cre mice causes motor deficits but does not alter pathogenesis of Spinocerebellar ataxia type 1. PLoS One. 13(7): e0200013. 2018.
Li Y.X., Sibon O.C.M., Dijkers P.F. Inhibition of NF-kappaB in astrocytes is sufficient to delay neurodegeneration induced by proteotoxicity in neurons. J. Neuroinflammation. 15(1): 261. 2018.
Alberts B. Molecular biology of the cell. 4th ed. New York. Garland Science. 2002.
Huang M., Verbeek D.S. Why do so many genetic insults lead to Purkinje Cell degeneration and spinocerebellar ataxia? Neurosci. Lett. 688: 49–57. 2019.
Schwaller B., Meyer M., Schiffmann S. 'New' functions for 'old' proteins: The role of the calcium-binding proteins calbindin D-28k, calretinin and parvalbumin, in cerebellar physiology. Studies with knockout mice. Cerebellum. 1(4): 241–258. 2002.
Kreiner L., Christel C.J., Benveniste M., Schwaller B., Lee A. Compensatory regulation of Cav2.1 Ca2+ channels in cerebellar Purkinje neurons lacking parvalbumin and calbindin D-28k. J. Neurophysiol. 103(1): 371–381. 2010.
Kano M., Nakayama H., Hashimoto K., Kitamura K., Sakimura K., Watanabe M. Calcium-dependent regulation of climbing fibre synapse elimination during postnatal cerebellar development. J. Physiol. 591(Pt 13): 3151–3158. 2013.
Barnes J.A., Ebner B.A., Duvick L.A., Gao W., Chen G., Orr H.T., Ebner T.J. Abnormalities in the climbing fiber-Purkinje cell circuitry contribute to neuronal dysfunction in ATXN1[82Q] mice. J. Neurosci. 31(36): 12778–12789. 2011.
Long C., Grueter C.E., Song K., Qin S., Qi X., Kong Y.M., Shelton J.M., Richardson J.A., Zhang C.L., Bassel-Duby R., Olson E.N. Ataxia and Purkinje cell degeneration in mice lacking the C-AMTA1 transcription factor. Proc. Natl. Acad. Sci. U. S. A. 111(31): 11521–11526. 2014.
Kawaguchi S.Y., Hirano T. Gating of long-term depression by Ca2+/calmodulin-dependent protein kinase II through enhanced cGMP signalling in cerebellar Purkinje cells. J. Physiol. 591(Pt 7): 1707–1730. 2013.
Fukumitsu K., Hatsukano T., Yoshimura A., Heuser J., Fujishima K., Kengaku M. Mitochondrial fission protein Drp1 regulates mitochondrial transport and dendritic arborization in cerebellar Purkinje cells. Mol. Cell Neurosci. 71: 56–65. 2016.
Di Bella D., Lazzaro F., Brusco A., Plumari M., Battaglia G., Pastore A., Finardi A., Cagnoli C., Tempia F., Frontali M., Veneziano L., Sacco T., Boda E., Brussino A., Bonn F., Castellotti B., Baratta S., Mariotti C., Gellera C., Fracasso V., Magri S., Langer T., Plevani P., Di Donato S., Muzi-Falconi M., Taroni F. Mutations in the mitochondrial protease gene AFG3L2 cause dominant hereditary ataxia SCA28. Nat. Genet. 42(4): 313–321. 2010.
Kandel E.R. Principles of Neural Science Ed. Sydor A.L.H. United States of America. The McGraw-Hill Companies. 2013.
Indriati D.W., Kamasawa N., Matsui K., Meredith A.L., Watanabe M., Shigemoto R. Quantitative localization of Cav2.1 (P/Q-type) voltage-dependent calcium channels in Purkinje cells: Somatodendritic gradient and distinct somatic coclustering with calcium-activated potassium channels. J. Neurosci. 33(8): 3668–3678. 2013.
Piochon C., Levenes C., Ohtsuki G., Hansel C. Purkinje cell NMDA receptors assume a key role in synaptic gain control in the mature cerebellum. J. Neurosci. 30(45): 15330–15335. 2010.
Crupi R., Impellizzeri D., Cuzzocrea S. Role of Metabotropic Glutamate Receptors in Neurological Disorders. Front Mol Neurosci. 12: 20. 2019.
Hirai H., Kano M. Type 1 metabotropic glutamate receptor and its signaling molecules as therapeutic targets for the treatment of cerebellar disorders. Curr. Opin. Pharmacol. 38: 51–58. 2018.
Serra H.G., Duvick L., Zu T., Carlson K., Stevens S., Jorgensen N., Lysholm A., Burright E., Zoghbi H.Y., Clark H.B., Andresen J.M., Orr H.T. RORalpha-mediated Purkinje cell development determines disease severity in adult SCA1 mice. Cell. 127(4): 697–708. 2006.
Gold D.A., Baek S.H., Schork N.J., Rose D.W., Larsen D.D., Sachs B.D., Rosenfeld M.G., Hamilton B.A. RORalpha coordinates reciprocal signaling in cerebellar development through sonic hedgehog and calcium-dependent pathways. Neuron. 40(6): 1119–1131. 2003.
Orr H.T. SCA1-phosphorylation, a regulator of Ataxin-1 function and pathogenesis. Prog. Neurobiol. 99(3): 179–185. 2012.
Meera P., Pulst S., Otis T. A positive feedback loop linking enhanced mGluR function and basal calcium in spinocerebellar ataxia type 2. Elife. 6. 2017.
Collins A.J., Foley R.N., Herzog C., Chavers B., Gilbertson D., Ishani A., Johansen K., Kasiske B., Kutner N., Liu J., St Peter W., Ding S., Guo H., Kats A., Lamb K., Li S., Roberts T., Skeans M., Snyder J., Solid C., Thompson B., Weinhandl E., Xiong H., Yusuf A., Zaun D., Arko C., Chen S.C., Daniels F., Ebben J., Frazier E., Hanzlik C., Johnson R., Sheets D., Wang X., Forrest B., Constantini E., Everson S., Eggers P., Agodoa L. US Renal Data System 2012 Ann. Data Report. Am. J. Kidney Dis. 61(1 Suppl 1): A7, e1-476. 2013.
Meera P., Pulst S.M., Otis T.S. Cellular and circuit mechanisms underlying spinocerebellar ataxias. J. Physiol. 594(16): 4653–4660. 2016.
Grumati P., Dikic I. Ubiquitin signaling and autophagy. J. Biol. Chem. 293(15): 5404–5413. 2018.
Jatana N., Ascher D.B., Pires D.E.V., Gokhale R.S., Thukral L. Human LC3 and GABARAP subfamily members achieve functional specificity via specific structural modulations. Autophagy. 14: 1–17. 2019.
Puorro G., Marsili A., Sapone F., Pane C., De Rosa A., Peluso S., De Michele G., Filla A., Sacca F. Peripheral markers of autophagy in polyglutamine diseases. Neurol. Sci. 39(1): 149–152. 2018.
Paul S., Dansithong W., Figueroa K.P., Scoles D.R., Pulst S.M. Staufen1 links RNA stress granules and autophagy in a model of neurodegeneration. Nat. Commun. 9(1): 3648. 2018.
Ferro A., Sheeler C., Rosa J.G., Cvetanovic M. Role of Microglia in Ataxias. J. Mol. Biol. 2019.
Thion M.S., Low D., Silvin A., Chen J., Grisel P., Schulte-Schrepping J., Blecher R., Ulas T., Squarzoni P., Hoeffel G., Coulpier F., Siopi E., David F.S., Scholz C., Shihui F., Lum J., Amoyo A.A., Larbi A., Poidinger M., Buttgereit A., Lledo P.M., Greter M., Chan J.K.Y., Amit I., Beyer M., Schultze J.L., Schlitzer A., Pettersson S., Ginhoux F., Garel S. Microbiome Influences Prenatal and Adult Microglia in a Sex-Specific Manner. Cell. 172(3): 500–516 e16. 2018.
Nakayama H., Abe M., Morimoto C., Iida T., Okabe S., Sakimura K., Hashimoto K. Microglia permit climbing fiber elimination by promoting GABAergic inhibition in the developing cerebellum. Nat. Commun. 9(1): 2830. 2018.
Ebner B.A., Ingram M.A., Barnes J.A., Duvick L.A., Frisch J.L., Clark H.B., Zoghbi H.Y., Ebner T.J., Orr H.T. Purkinje cell ataxin-1 modulates climbing fiber synaptic input in developing and adult mouse cerebellum. J. Neurosci. 33(13): 5806–5820. 2013.
Cvetanovic M., Ingram M., Orr H., Opal P. Early activation of microglia and astrocytes in mouse models of spinocerebellar ataxia type 1. Neuroscience. 289: 289–299. 2015.
Qu W., Johnson A., Kim J.H., Lukowicz A., Svedberg D., Cvetanovic M. Inhibition of colony-stimulating factor 1 receptor early in disease ameliorates motor deficits in SCA1 mice. J. Neuroinflammation. 14(1): 107. 2017.
Llinas R., Sugimori M. Electrophysiological properties of in vitro Purkinje cell dendrites in mammalian cerebellar slices. J. Physiol. 305: 197–213. 1980.
Llinas R., Sugimori M. Electrophysiological properties of in vitro Purkinje cell somata in mammalian cerebellar slices. J. Physiol. 305: 171–195. 1980.
Raman I.M., Bean B.P. Resurgent sodium current and action potential formation in dissociated cerebellar Purkinje neurons. J. Neurosci. 17(12): 4517–4526. 1997.
Raman I.M., Bean B.P. Ionic currents underlying spontaneous action potentials in isolated cerebellar Purkinje neurons. J. Neurosci. 19(5): 1663–1674. 1999.
Nam S.C., Hockberger P.E. Analysis of spontaneous electrical activity in cerebellar Purkinje cells acutely isolated from postnatal rats. J. Neurobiol. 33(1): 18–32. 1997.
Womack M., Khodakhah K. Active contribution of dendrites to the tonic and trimodal patterns of activity in cerebellar Purkinje neurons. J. Neurosci. 22(24): 10603–10612. 2002.
Smith S.L., Otis T.S. Persistent changes in spontaneous firing of Purkinje neurons triggered by the nitric oxide signaling cascade. J. Neurosci. 23(2): 367–372. 2003.
De Zeeuw C.I., Hoebeek F.E., Bosman L.W., Schonewille M., Witter L., Koekkoek S.K. Spatiotemporal firing patterns in the cerebellum. Nat. Rev. Neurosci. 12(6): 327–344. 2011.
Hoebeek F.E., Stahl J.S., van Alphen A.M., Schonewille M., Luo C., Rutteman M., van den Maagdenberg A.M., Molenaar P.C., Goossens H.H., Frens M.A., De Zeeuw C.I. Increased noise level of purkinje cell activities minimizes impact of their modulation during sensorimotor control. Neuron. 45(6): 953–965. 2005.
Alvina K., Khodakhah K. The therapeutic mode of action of 4-aminopyridine in cerebellar ataxia. J. Neurosci. 30(21): 7258–7568. 2010.
Dell'Orco J.M., Wasserman A.H., Chopra R., Ingram M.A., Hu Y.S., Singh V., Wulff H., Opal P., Orr H.T., Shakkottai V.G. Neuronal atrophy early in degenerative ataxia is a compensatory mechanism to regulate membrane excitability. J. Neurosci. 35(32): 11292–11307. 2015.
Mark M.D., Krause M., Boele H.J., Kruse W., Pollok S., Kuner T., Dalkara D., Koekkoek S., De Zeeuw C.I., Herlitze S. Spinocerebellar ataxia type 6 protein aggregates cause deficits in motor learning and cerebellar plasticity. J. Neurosci. 35(23): 8882–8895. 2015.
Shakkottai V.G., do Carmo Costa M., Dell’Orco J.M., Sankaranarayanan A., Wulff H., Paulson H.L. Early changes in cerebellar physiology accompany motor dysfunction in the polyglutamine disease spinocerebellar ataxia type 3. J. Neurosci. 31(36): 13002–13014. 2011.
Walter J.T., Alvina K., Womack M.D., Chevez C., Khodakhah K. Decreases in the precision of Purkinje cell pacemaking cause cerebellar dysfunction and ataxia. Nat. Neurosci. 9(3): 389–397. 2006.
Chopra R., Shakkottai V.G. Translating cerebellar Purkinje neuron physiology to progress in dominantly inherited ataxia. Future Neurol. 9(2): 187–196. 2014.
Dell'Orco J.M., Pulst S.M., Shakkottai V. G. Potassium channel dysfunction underlies Purkinje neuron spiking abnormalities in spinocerebellar ataxia type 2. Hum. Mol. Genet. 26(20): 3935–3945. 2017.
Bushart D.D., Shakkottai V.G. Ion channel dysfunction in cerebellar ataxia. Neurosci. Lett. 688: 41-48. 2019.
Coutelier M., Coarelli G., Monin M.L., Konop J., Davoine C.S., Tesson C., Valter R., Anheim M., Behin A., Castelnovo G., Charles P., David A., Ewenczyk C., Fradin M., Goizet C., Hannequin D., Labauge P., Riant F., Sarda P., Sznajer Y., Tison F., Ullmann U., Van Maldergem L., Mochel F., Brice A., Stevanin G., Durr A. A panel study on patients with dominant cerebellar ataxia highlights the frequency of channelopathies. Brain. 140(6): 1579–1594. 2017.
Jones J.M., Dionne L., Dell’Orco J., Parent R., Krueger J.N., Cheng X., Dib-Hajj S.D., Bunton-Stasyshyn R.K., Sharkey L.M., Dowling J.J., Murphy G.G., Shakkottai V.G., Shrager P., Meisler M.H. Single amino acid deletion in transmembrane segment D4S6 of sodium channel Scn8a (Nav1.6) in a mouse mutant with a chronic movement disorder. Neurobiol. Dis. 89: 36–45. 2016.
Lee K.H., Mathews P.J., Reeves A.M., Choe K.Y., Jami S.A., Serrano R.E., Otis T.S. Circuit mechanisms underlying motor memory formation in the cerebellum. Neuron. 86(2): 529–540. 2015.
Lang E.J., Apps R., Bengtsson F., Cerminara N.L., De Zeeuw C.I., Ebner T.J., Heck D.H., Jaeger D., Jorntell H., Kawato M., Otis T.S., Ozyildirim O., Popa L.S., Reeves A.M., Schweighofer N., Sugihara I., Xiao J. The Roles of the Olivocerebellar Pathway in Motor Learning and Motor Control. A Consensus Paper. Cerebellum. 16(1): 230–252. 2017.
Kuo S.H., Lin C.Y., Wang J., Sims P.A., Pan M.K., Liou J.Y., Lee D., Tate W.J., Kelly G.C., Louis E.D., Faust P.L. Climbing fiber-Purkinje cell synaptic pathology in tremor and cerebellar degenerative diseases. Acta Neuropathol. 133(1): 121–138. 2017.
Burroughs A., Wise A.K., Xiao J., Houghton C., Tang T., Suh C.Y., Lang E.J., Apps R., Cerminara N.L. The dynamic relationship between cerebellar Purkinje cell simple spikes and the spikelet number of complex spikes. J. Physiol. 595(1): 283–299. 2017.
Davie J.T., Clark B.A., Hausser M. The origin of the complex spike in cerebellar Purkinje cells. J. Neurosci. 28(30): 7599–7609. 2008.
Karelina T.V., Grigor’ian R.A. Effect of harmaline of the complex spike waveform and depression time in cerebellar Purkinje cell discharge in rat postnatal ontogenesis. Zh. Evol. Biokhim. Fiziol. 46(3): 218–224. 2010.
van Roon-Mom W.M.C., Roos R.A.C., de Bot S.T. Dose-dependent lowering of mutant huntingtin using antisense oligonucleotides in huntington disease patients. Nucleic. Acid Ther. 28(2): 59–62. 2018.
Rinaldi C., Wood M.J.A. Antisense oligonucleotides: The next frontier for treatment of neurological disorders. Nat. Rev. Neurol. 14(1): 9–21. 2018.
Volkman R., Offen D. Concise Review: Mesenchymal Stem cells in neurodegenerative diseases. Stem Cells. 35(8): 1867–1880. 2017.
Chang Y.K., Chen M.H., Chiang Y.H., Chen Y.F., Ma W.H., Tseng C.Y., Soong B.W., Ho J.H., Lee O.K. Mesenchymal stem cell transplantation ameliorates motor function deterioration of spinocerebellar ataxia by rescuing cerebellar Purkinje cells. J. Biomed. Sci. 18: 54. 2011.
Cho L.T., Alexandrou A.J., Torella R., Knafels J., Hobbs J., Taylor T., Loucif A., Konopacka A., Bell S., Stevens E.B., Pandit J., Horst R., Withka J.M., Pryde D.C., Liu S., Young G.T. An Intracellular Allosteric Modulator Binding Pocket in SK2 Ion Channels Is Shared by Multiple Chemotypes. Structure. 26(4): 533–544 e3. 2018.
Дополнительные материалы отсутствуют.
Инструменты
Российский физиологический журнал им. И.М. Сеченова