

Российский физиологический журнал им. И.М. Сеченова, 2019, T. 105, № 12, стр. 1486-1501
Роль аутофагии при инфекциях
И. С. Фрейдлин 1, 2, 3, *, Дж. Т. Маммедова 1, 4, Э. А. Старикова 1, 2
1 Институт экспериментальной медицины
Санкт-Петербург, Россия
2 Первый Санкт-Петербургский государственный медицинский университет
им. академика И.П. Павлова
Санкт-Петербург, Россия
3 Санкт-Петербургский Государственный университет
Санкт-Петербург, Россия
4 Санкт-Петербургский Государственный технологический институт
(Технический университет)
Санкт-Петербург, Россия
* E-mail: irinaf-n@yandex.ru
Поступила в редакцию 06.09.2019
После доработки 20.09.2019
Принята к публикации 20.09.2019
Аннотация
Процесс аутофагии относят к нормальным физиологическим процессам в организме, направленным на регуляцию размеров клеточных популяций за счет поддержания баланса между выживанием и гибелью клеток. Аутофагия позволяет клетке утилизировать отдельные поврежденные молекулы и целые органоиды, выживать и обновляться в условиях стресса или голодания. Аутофагия привлекает особое внимание в связи с гомеостатическими функциями и важной ролью, которую играет этот процесс в антимикробной защите организма. При бактериальных и вирусных инфекциях развитие аутофагии часто индуцируется как способ защиты организма хозяина или механизм стратегии выживания патогена. В последние годы опубликованы результаты экспериментальных исследований, свидетельствующие о важной роли аутофагии в регуляции иммунной защиты организма. Аутофагия в качестве одного из факторов антимикробного иммунитета может способствовать очищению организма от возбудителей и в то же время обеспечивает кросс-презентацию микробных антигенов для индукции противомикробного иммунного ответа. Наряду с этим установлено, что многие патогены в процессе эволюции приобрели факторы вирулентности, способные вмешиваться в процесс аутофагии, извращая ее исходно защитную роль и снижая антимикробную защиту организма.
Процесс аутофагии относят к нормальным физиологическим процессам, направленным на регуляцию размеров клеточных популяций за счет поддержания баланса между выживанием и гибелью клеток. Аутофагия позволяет клетке утилизировать отдельные поврежденные молекулы и целые органоиды, выживать и обновляться в условиях стресса или голодания. Аутофагию в крайней ее форме относят к одному из типов генетически контролируемой и регулируемой формы гибели клеток. Принципиальное отличие аутофагической гибели клеток от других типов программированной клеточной гибели (кроме апоптоза) заключается в том, что ее исходное назначение – защита организма от внутриклеточных молекул, которые при гибели клеток могут высвобождаться в окружающие ткани и вызывать активацию клеток иммунной системы с последующим развитием воспаления. Гибель клеток путем аутофагии носит “щадящий” характер, не сопровождается воспалением.
Аутофагия привлекает особое внимание не только в связи с гомеостатическими функциями, но и важной ролью, которую играет этот процесс во взаимоотношениях организма хозяина и патогена. При бактериальных и вирусных инфекциях развитие аутофагии может выступать и как способ защиты клеток организма хозяина, и как механизм выживания патогена. В последние годы опубликованы результаты экспериментальных исследований, свидетельствующие о важной роли аутофагии в регуляции иммунной защиты организма. Аутофагия в качестве одного из факторов антимикробного иммунитета может способствовать очищению организма от возбудителей и в то же время обеспечивает кросс-презентацию микробных антигенов для индукции противомикробного иммунного ответа. Наряду с этим установлено, что многие патогены в процессе эволюции приобрели факторы вирулентности, способные вмешиваться в процесс аутофагии, извращая ее исходно защитную роль и снижая антимикробную защиту организма. В обзоре основное внимание уделено анализу защитных механизмов аутофагии и стратегии патогенов, направленной на преодоление антимикробной защиты организма при инфекциях.
ОБЩАЯ ХАРАКТЕРИСТИКА АУТОФАГИИ
Аутофагия – ауто-катаболический процесс, направленный на деградацию поврежденных белков и органелл клетки. Этот эволюционно древний механизм позволяет клеточным компонентам самообновляться в условиях стресса, восполнять дефицит метаболитов при голодании и дает возможность клетке выжить и восстановиться после повреждения. Аутофагия на базальном уровне постоянно происходит в клетках, а в условиях стресса, голодания, инфекции наблюдается интенсификация этого процесса [1]. Аутофагия играет важную роль при разных патологических процессах, таких как рак, воспаление, метаболические и нейродегенеративные расстройства, миопатии, бактериальные и вирусные инфекции [2]. Оценка роли аутофагии при разных физиологических и патологических процессах представляется противоречивой, поскольку в некоторых случаях этот процесс играет защитную роль, а в других случаях может способствовать развитию и прогрессированию патологии [3, 4].
Морфологическими признаками аутофагии являются уменьшение количества митохондрий и объема эндоплазматического ретикулума (ER) при одновременном увеличении аппарата Гольджи и формировании в клетке множества вакуолей – аутофагосом (АФ). В ряде случаев усиленная аутофагия может приводить к гибели клеток в результате самопереваривания, с последующим поглощением остатков соседними клетками. Аутофагическая клеточная гибель морфологически отличается от гибели при апоптозе: она проявляется вакуолизацией и деградацией содержимого цитоплазмы при отсутствии конденсации хроматина. При этом, так же как апоптоз, аутофагия является невоспалительной формой клеточной гибели, т. к. в этом случае содержимое цитоплазмы не попадает в окружающую среду. [5].
В настоящее время в зависимости от способа доставки субстрата в АФ выделяют три типа аутофагии – микроаутофагию, макроаутофагию и шаперон-зависимую аутофагию. При микроаутофагии содержимое цитоплазмы попадает в лизосомы в результате формирования инвагинаций мембраны этих органоидов [6]. Шаперон-зависимая аутофагия происходит в результате активного транспорта поврежденных белков цитоплазмы через мембрану лизосомы с помощью белков-шаперонов семейства hsp-70 (heat shock protein 70), вспомогательных белков и LAMP-2 (lysosome-associated membrane protein type 2A) [7]. В результате индукции макроаутофагии происходит рекрутирование специализированных белков ATGs (autophagy-related proteins) с последующей нуклеацией изолирующей мембраны и формированием чашеподобной структуры, получившей название фагофор. В результате постепенной элонгации фагофора происходит ограничение сферического участка цитоплазмы двумя слоями мембраны и формируется АФ [8]. Сформировавшаяся АФ представляет собой ограниченный билипидным слоем участок цитоплазмы, содержащий цитоплазму и разрушенные структуры ER, митохондрий и пероксисом, белковые маркеры рибосом, эндосом и лизосом. Диаметр АФ у млекопитающих составляет 500–1500 нм [3]. При слиянии АФ с лизосомами формируется аутофаголизосома, содержимое которой подвергается деградации под влиянием лизосомальных ферментов. Именно макроаутофагия доминирует в клетках в случае инфекции [8, 9].
МОЛЕКУЛЯРНЫЕ МЕХАНИЗМЫ РЕГУЛЯЦИИ АУТОФАГИИ
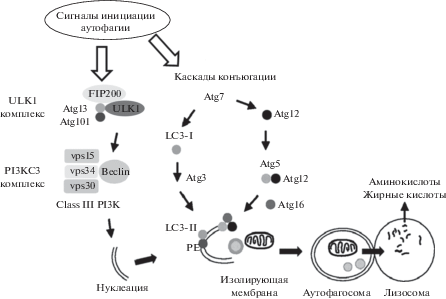
Гены, регулирующие процесс аутофагии принято обозначать Atg (AuTophaGy-related genes). Они кодируют белки, важные для интеграции сигналов аутофагии, а также белки, участвующие в индукции аутофагии, формировании, созревании и рециклинге АФ [10]. Инициация формирования фагофора – сложный процесс, молекулярные механизмы которого до сих пор не до конца изучены. Согласно современным представлениям, при формировании сайтов сборки фагофора происходит активация белкового комплекса ULK1. Сигналами активации аутофагии являются различные стрессорные факторы: дефицит нутриентов, гипоксия, оксидативный стресс, белковые агрегаты и др. Все эти сигналы интегрирует состоящий из ULK1, ATG13, FIP200 и ATG101 белков ULK1-киназный комплекс, который является триггером образования фагофора (рис. 1). Последующее фосфорилирование комплекса PI3KC3, образованного PI3KС3, VPS34, Beclin 1, ATG14, AMBRA 1 и p115 белками, в свою очередь индуцирует локальную продукцию PI3P в характерных структурах ER, получивших название омегасомы. Фосфатидилинозитол 3-киназа (PI3K) является партнером по связыванию с Beclin-1 (продуктом гена Atg6), участвуя в инициации изоляции мембран. PI3P рекрутирует к омегасоме эффекторные белки WIPIs и DFCP1, которые рекрутируют ATG12 ~ ATG5–ATG16L1 комплекс и стимулируют ATG3-опосредованную коньюгацию белков ATG8 (LC3 и GABARAPs) с фосфатидилэтаноламином мембраны фагофора с образованием связанных с мембраной (липидированных) белков. ATG8 не только осуществляют рекрутирование компонентов аутофагического комплекса, но также участвуют в процессах элонгации и слияния мембраны фагофора. В случае селективной аутофагии LC3 участвует в секвестрировании специфически меченых структур клетки для переноса в АФ через LIR транспортный рецептор. Элонгация аутофагосомальной мембраны осуществляется за счет использования разных мембран клетки: плазматической, митохондриальной, эндосомальной, а также мембран аппарата Гольджи. Сиалирование аутофагосомальной мембраны заканчивается формированием двуслойных везикул, которые после созревания сливаются с лизосомой [8]. Слияние вакуолей опосредовано малыми GTP-азами, например, GTP-азой Rab24, которая является одним из маркеров зрелых АФ [11]. Лизосомальные кислые гидролазы осуществляют деградацию содержимого АФ, и утилизированные нутриенты высвобождаются в цитоплазму, чтобы снова использоваться клеткой [8].
Рис. 1.
Процесс аутофагии (Из Yamada and Rajat, 2012 с изменениями). ULK1 – Unc-51 like autophagy activating kinase; FIP200 – 200 kDa FAK family kinase-interacting protein; Atg – autophagy proteins; vsp – vacuolar protein sorting; PIK3C3 – phosphatidylinositol 3-kinase class 3; PE, phosphatidylethanolamine; LC3-I, soluble light chain 3-I; LC3-II, lipid light chain-II.
Регуляция процессов аутофагии осуществляется с участием сигнальных молекул, которые одновременно интегрируют ответ клеток на стрессорные воздействия и регулируют аутофагию. Среди них наиболее полно охарактеризованными в настоящее время являются mTOR (mechanistic target of rapamycin), AMPK (AMP activated protein kinase) и сиртуины (sirtuins).
Сиртуины – это семейство NAD-зависимых (де)ацетилаз, которые являются сенсорами стресса в клетке [12]. Показано, что сиртуин 1 (Sirt1) отвечает за деацетилирование Atg5, Atg7, и LC3 [13], в то время как ацетилтрансфераза p300, напротив, ацетилирует Atg5, Atg7, LC3 и Atg12 [14]. Кроме того, сиртуин 1 деацетилирует транскрипционный фактор forkhead box O3a (FOXO3a) [15]. Ингибирование Akt в результате дефицита ростовых факторов также приводит к активации FOXO3a путем его дефосфорилирования. Последующая транслокация FOXO3a в ядро активирует множество генов, регулирующих аутофагию, таких как ULK2, Beclin 1, VPS34, BNIP3 и BNIP3L, ATG12, ATG4B, LC3 и GABARAPL1 [16]. При голодании диссоциация сиртуин 2 и FOXO1 приводит к ацетилированию последнего, способствуя его взаимодействию с Atg7 и стимуляции аутофагии [17]. AMPK действует как центральный узел, который интегрирует сигналы стресса и инициирует процессы аутофагии. AMPK отслеживает энергетический статус по соотношению AMP : ATP в клетке. Сиртуин 1 и AMPK в условиях депривации нутриентов взаимно усиливают активацию друг друга, формируя петлю обратной положительной связи [18]. Наиболее изученным в настоящее время механизмом индукции аутофагии при участии AMPK является ингибирование mTORC1, путем фосфорилирования TSC2 и Raptor. Кроме того, ингибирование mTORC1 под влиянием AMPK происходит также в случае дефицита ростовых факторов. В ответ на ростовые факторы активированные Akt и ERK1/2 киназы могут фосфорилировать одну или обе субъединицы TSC1/TSC2, и Akt может фосфорилировать Raptor, что в совокупности активирует mTOR [19]. Недавно обнаруженное взаимодействие между AMPK и ULK1 [20] указывает на то, что AMPK может регулировать аутофагию, непосредственно взаимодействуя с этим ключевым компонентом аутофагического комплекса. Индуцированную голоданием аутофагию регулирует также JNK1 (c-Jun N-terminal kinases), которая фосфорилирует Bcl-2, снижая его аффинность с Beclin 1 [21], и Sirt1 и активируя энзиматическую активность [22].
Другим ключевым регулятором аутофагии является TFEB (transcription factor EB) – основной транскрипционный фактор, регулирующий клиренс в клетках, биогенез лизосом и экспрессию лизосомальных ферментов. TFEB контролирует аутофагию за счет регуляции рекрутирования mTORC1 на мембраны лизосом и также регуляции активности этого комплекса за счет усиления экспрессии активирующего mTOR компонента Rag GTP-фазного комплекса RagD по принципу обратной отрицательной связи, что обеспечивает баланс клеточного метаболизма [8].
Разные экзогенные и эндогенные сигналы, такие как депривация аминокислот, двуспиральная вирусная РНК, осмотический стресс, ультрафиолет, гипоксия, оксидативный стресс [23] приводят к активации eIF2a киназ [24, 25]. Фосфорилирование eIF2a подавляет процессы трансляции в клетке, но при этом селективно индуцирует трансляцию транскрипционного фактора ATF4, поддерживающего процессы аутофагии за счет экспрессии LC3 [26].
Очевидно, существует координация между регуляцией процессов апоптоза и аутофагии на генетическом уровне. Многие сигналы апоптоза индуцируют аутофагию, а сигналы угнетения апоптоза ингибируют аутофагию. Гены, контролирующие митохондриальную регуляцию апоптоза, взаимодействуют с генами, регулирующими аутофагию [1, 3]. Показано, что bcl-2 ген, кодирующий анти-апоптотический белок Bcl-2, необходим для выживания лейкемических клеток человека HL-60, а его подавление (нокдаун гена) индуцирует аутофагию. Снижение экспрессии Bcl-2 усиливает аутофагию каспаза-независимым путем: в клетках формируются многочисленные АФ при интактных ядрах и интактных митохондриях без вовлечения активации каспаз [27]. Bcl-2 ингибирует аутофагию, напрямую взаимодействуя с белком Beclin 1, который является компонентом сигнального комплекса PI3KC3, регулирующего формирование фагофора. Для проявления аутофагической активности Beclin 1 должен диссоциировать из комплекса с Bcl-2 [28].
КЛЕТОЧНО-МОЛЕКУЛЯРНЫЕ МЕХАНИЗМЫ РЕГУЛЯЦИИ АУТОФАГИИ ПРИ ИНФЕКЦИИ
В отличие от гомеостатического процесса аутофагии, направленного на переработку внутриклеточных молекул и органоидов, процесс элиминации внутриклеточных патогенов в ходе развития иммунного ответа получил название ксенофагия [29]. Ксенофагия представляет собой защитный механизм, существенно ограничивающий внутриклеточный рост бактерий, среди которых Salmonella enterica серовар typhimurium [30], Mycobacterium tuberculosis [31, 32], Listeria monocytogenes [33] и стрептококки группы A [29, 34]. Ксенофагия препятствует внутриклеточному размножению бактерий, отграничивая их от цитозоля, богатого питательными веществами, и транспортируя их в бактерицидные аутолизосомы [35]. Процесс ксенофагии также используется клеткой для уничтожения широкого спектра внутриклеточных патогенов, включая Shigella flexneri, Mycobacterium tuberculosis, альфавирусы, вирус простого герпеса, и инвазирующих внеклеточных бактерий, например, Streptococcus гр. А [1].
Ксенофагия может быть инициирована в процессе адгезии или фагоцитоза патогена клетками организма-хозяина [36, 37]. Фагосомы, содержащие бактерии, могут непосредственно рекрутировать LC3 на свою поверхность, этот процесс получил название LC3-ассоцированный фагоцитоз LAP (LC3-associated phagocytosis) [29]. Один из центральных вопросов ксенофагии касается специфичности этого процесса, а именно, каким образом аутофагический аппарат распознает патогены в цитозоле и направляет их для деградации в АФ. В настоящее время установлено, что выбор мишени для аутофагии осуществляется с помощью универсального аппарата распознавания, мечения и деградации. В случае проникновения в цитоплазму, микробы подвергаются убиквитинилированию специфическими убиквитин лигазами и распознаются секвестосома-подобными рецепторами SLRs (Sequestosome-1-like receptors) – p62/SQSTM1, NDP52, оптиневрином, NBR1 и TAX1BP1 [38, 39]. Эти адапторные белки связывают убиквитинилированные бактерии через LC3 с зарождающимся фагофором или белками, ассоциированными с гаммааминомасляным рецептором. Активация и рекрутирование к фагосоме белков комплекса ULK1 инициирует формирование фагофора, изолирующего микробную клетку [36, 37, 40]. В любом случае, результатом LAP и канонической аутофагии становится заключение бактерий в компартмент, декорированный LC3 с целью дальнейшей их деградации после слияния с лизосомами [29, 41, 42].
Вызванные инфицированием клеток организма-хозяина голодание, энергетический и оксидативный стресс, дефекты сборки белков в ER, гипоксия, повреждение митохондрий – все эти процессы приводят к активации аутофагии [43]. Отдельные микробные продукты, консервативные структуры патогенных микроорганизмов (PAMPs), молекулы, ассоциированные с повреждением клеток (DAMPs), и провоспалительные цитокины тоже являются сигналами усиления аутофагии. Распознающие PAMPs рецепторы – TLRs способны распознавать множество микробных продуктов как на поверхности клетки, так и внутри. Было показано, что Poly (I : C), LPS и ssRNA, которые являются лигандами для TLR3, TLR4 и TLR7 соответственно, индуцируют формирование АФ через активацию MyD88-зависимого внутриклеточного сигнального пути [44]. Индукцию аутофагии в ходе инфекции регулирует IFN-γ (через активацию GTPases). M.G. Gutierrez с соавт. [31] показали, что IFN-γ опосредованно через Irgm1 (immunity-related p47 guanosine triphosphatases) мимикрирует эффект рапамицина или голодания, что выражается в ингибировании mTOR сигнального пути и усиливает аутофагию при микобактериальной инфекции [44]. Транскрипционный фактор NF-kB, некоторые другие киназы (IKK комплекс, TAK1) этого сигнального пути связывают иммунные сигналы с сигналами, регулирующими процессы аутофагии [45]. Субъединица NF-kB p105 может напрямую взаимодействовать с некоторыми белками (например, Beclin 1), участвующими в процессе аутофагии [20]. Предполагают, что при стрессе в клетках формируется “мета-инфламмасома” – специализированный белковый комплекс, образованный PKR, eIF2a, JNK, IRS, IKK сигнальными молекулами. В рамках “мета-инфламмасомы” PKR фосфорилирует eIF2a, активирует провоспалительные киназы IKKb и JNK1 и фосфорилирует IRS1, что в совокупности приводит к индукции аутофагии [24]. Оксидативный стресс, который наблюдается в клетках в ходе инфекции, может индуцировать аутофагию посредством множества механизмов. Экзогенная перекись водорода может активировать PERK (Protein kinase R-like ER kinase), локализованную на мембранах ER, стимулируя фосфорилирование проаутофагической сигнальной молекулы eIF2a [46]. Непосредственное окисление протеазы Atg4 при повышенной продукции перекиси водорода способствует формированию протеолитически зрелого LC3 [47].
Активные формы кислорода (АФК), которые при инфекции продуцируются активированными фагоцитами для элиминации патогенов, ингибируют mTOR прямо [46] или опосредованно через активацию PARP1 и цитоплазматического пула ATM с последующей активацией LKB1 и AMPK1 [48, 49]. При повышении продукции АФК происходит активация MAP-киназ, включая JNK1, которая также может усиливать процессы аутофагии [50]. Кроме того, повреждения ДНК, вызванные АФК, также стимулируют экспрессию проаутофагических p53-индуцированных генов. p53 играет двойственную роль в индукции процессов аутофагии при стрессе: как транскрипционный фактор он активирует индукторы аутофагии DRAM1 и Sestrin2 [51], а в случае цитоплазматической локализации p53 действует как ингибитор аутофагии [52, 53].
СТРАТЕГИЯ ПАТОГЕНОВ В РЕГУЛЯЦИИ АУТОФАГИИ ПРИ ИНФЕКЦИЯХ
Инвазия патогенных микроорганизмов, как правило, начинается с их взаимодействия со специфическими рецепторами на клетках организма-хозяина и эндоцитоза, но дальнейшая их судьба может складываться по-разному. Некоторые (сальмонеллы, микобактерии и легионеллы) окружаются мембранами для формирования репликативных компартментов, другие (шигеллы, листерии и стрептококки группы А (GAS)) высвобождаются из эндоцитозных вакуолей и реплицируются в богатом питательными веществами цитозоле [38].
Ксенофагия в качестве одного из факторов антибактериального иммунитета представляет для многих бактерий реальную угрозу гибели внутри клеток. Поэтому большинство патогенов приобрело способность избегать аутофагии, ингибировать или извращать процессы аутофагии. Патогенные микроорганизмы могут вмешиваться в процесс аутофагии на всех его стадиях: формирования АФ, захвата патогенов АФ, выхода патогенов из АФ в цитозоль, слияния АФ с лизосомами. Некоторые внутриклеточно паразитирующие бактерии используют АФ в качестве защитных ниш для внутриклеточного выживания и репликации [54]. Другие вызывают нарушения формирования АФ и реплицируются внутри структур, имеющих характеристики АФ: двойную мембрану, маркеры ER. Для таких бактерий немедленное после проникновения в клетку попадание в АФ является условием их выживания за счет избегания лизосомальной деградации. Такие патогены продуцируют эффекторные молекулы, вызывающие отсроченное созревание АФ [11]. Кроме того, облигатно внутриклеточные паразиты способны использовать ресурсы нутриентов клеток хозяина для своей репликации.
При стрептококковых инфекциях в нефагоцитирующих эпителиальных и эндотелиальных клетках были обнаружены АФ-подобные вакуоли, проявляющие способность к слиянию с лизосомами [55]. Колокализация Streptococcus pyogenes с маркерами АФ LC3 и LAMP-1 позволяет прогнозировать их последующую гибель при слиянии АФ с лизосомами. В клетках с дефектами аутофагии GAS выживали и размножались [56]. В кератиноцитах 80% захваченных GAS погибали в течение 4 ч за счет аутофагической активности. Через 24 ч 50% инфицированных клеток погибали апоптозом, т.е. аутофагический киллинг бактерий не защищал клетки, но способствовал элиминации возбудителя [57].
Многие патогены приобрели разные стратегии избегания или эксплуатации механизмов аутофагии для создания оптимальных условий своего внутриклеточного выживания и репликации. Причем стратегия одного и того же патогена может меняться на разных стадиях развития инфекции. Например, сразу после инвазии патоген избегает аутофагии, а в дальнейшем использует этот процесс в своих целях [58]. Способность высоко вирулентного штамма S. pyogenes серотипа M1T1 избегать аутофагии и эффективно реплицироваться в цитоплазме инфицированных клеток объясняется наличием у данного патогена механизмов, предотвращающих его распознавание аутофагическим аппаратом клетки. Бактерии этого штамма продуцируют стрептококковую цистеинпротеазу (SpeB), которая разрушает адапторные протеины p62, NDP52, NBR1, ответственные за распознавание убиквитинилированных бактерий. Дефектный по SpeB мутант не был способен избегать распознавания и подвергался разрушению путем ксенофагии [59]. В эмбриональных фибробластах мышей, нокаутированных по atg5 (atg5–/–), стрептококки выживали, размножались и выходили из клеток. В отличие от этого, в клетках дикого типа выходящие в цитозоль стрептококки попадали внутрь АФ и подвергались лизосомальному гидролизу [57].
Секретируемый S. pyogenes порообразующий токсин стрептолизин (SLO) участвует в индукции ксенофагии, обеспечивая выход бактерий из эндосом в цитозоль. SLO-дефицитные мутанты стрептококка не могут покинуть эндосомы. Кроме того, под действием SLO-опосредованной транслокации ко-токсина NAD-гликогидролазы ингибируется слияние стрептококк-содержащих АФ с лизосомами, что защищает стрептококк от ксенофагической гибели [60]. Повышенная экспрессия SLO снижает эффективность pH-зависимого киллинга, т.е. способствует выживанию GAS в клетках хозяина.
Staphylococcus aureus тоже секретирует порообразующий токсин α-гемолизин, который активирует ксенофагию, а будучи захвачен АФ, S. aureus ингибирует ее созревание и слияние с лизосомами. Находящийся в цитозоле S. aureus запускает Atg5- независимый путь аутофагии с потреблением нутриентов и репликацией, в то же время избегая ксенофагии [61].
Как многие другие внутриклеточно паразитирующие бактерии, Listeria monocytogenes обладает способностью реплицироваться в цитозоле клеток хозяина. L. monocytogenes выходят из фагосом благодаря секреции холестерин-зависимого порообразующего цитолизина листериолизина О. Находящаяся в цитозоле L. monocytogenes избегает распознавания рецепторами аутофагии за счет секреции двух факторов вирулентности ActA и InlK, которые усиливают мобилизацию актина и других белков хозяина для формирования защитной оболочки, маскирующей бактерию. Таким образом, L. monocytogenes избегает убиквитинилирования и аутофагического распознавания. Репликация L. monocytogenes в цитоплазме клетки сопровождается истощением запаса аминокислот, ингибированием mTOR-пути и активацией аутофагии. Другая внутриклеточно паразитирующая бактерия Francisella tularensis маскируется за счет продукции поверхностного полисахарида (О-антигена), который препятствует распознаванию бактерий в цитозоле и их ксенофагии [44].
Многие кишечные бактерии секретируют белки, которые вмешиваются в регуляцию формирования АФ. Примером является Shigella flexneri, секретирующая фактор вирулентности – поверхностный белок VirG (IcsA), который является лигандом одного из белков аутофагии – ATG5 и участвует в индукции аутофагии. Однако Sh. flexneri дополнительно секретирует эффекторную молекулу IcsB, способную конкурентно связываться с поверхностным белком VirG и ингибировать рекрутирование бактерий в фагофоры [62]. Через несколько минут после инвазии в макрофаги Sh. flexneri могут лизировать фагоцитарную вакуоль и выходить в цитозоль для репликации, избегая ксенофагии. Однако в цитозоле бактерии подвергаются убиквитинилированию с последующим распознаванием и захватом АФ [38]. Поведение Sh. flexneri при инфицировании макрофагов имеет еще одну особенность: бактерии индуцируют активацию каспазы-1, что ведет к активации инфламмасом и клеточной гибели путем NLRC4-зависимого пироптоза. При этом и каспаза-1 и NLRC4 ингибируют формирование АФ в макрофагах, инфицированных Sh. flexneri, что ведет к угнетению ксенофагии [44].
Ингибирование аутофагии является важным механизмом вирулентности, который используется Salmonella typhimurium при инфицировании клеток хозяина. S. typhimurium избегает ксенофагической гибели и демонстрирует высокую степень адаптации к внутриклеточному существованию. Захваченные макрофагами S. typhimurium предотвращают аутофагию, активируя основной репрессор аутофагии mTOR. S. typhimurium секретирует в цитоплазму клетки хозяина более 30 эффекторных белков, активирующих AKT/mTOR-сигнальный путь и супрессирующих аутофагию [42].
Кишечные бактерии Sh. flexneri и S. typhimurium вызывают клеточный стресс, связанный с голоданием и подавлением активации AKT/mTOR. Однако в данном случае ингибирование процессов аутофагии носит кратковременных характер, т. к. эти патогены выработали механизмы реактивации mTORС1 за счет лизосомальной деградации AMPK, LKB1 и Sirt1 с последующим ингибированием аутофагии [44].
Манипуляция ксенофагии путем ингибирования созревания АФ подробно изучена на примере Mycobacterium tuberculosis. Главная черта возбудителя туберкулеза – способность выживать внутри альвеолярных макрофагов. Белок, повышающий внутриклеточное выживание M. tuberculosis, – EIS ингибирует аутофагию, активируя путь AKT/mTOR с усилением продукции IL-10. M. tuberculosis использует и другие механизмы нарушения эффективности ксенофагии: препятствует созреванию АФ, ингибирует ацидификацию содержимого фагосом [44]. Один из факторов вирулентности M. tuberculosis Esat-6 играет существенную роль при блокировании созревания АФ в макрофагах и дендритных клетках, а секретируемая M. tuberculosis N-ацетилтрансфераза подавляет LAP, препятствуя мобилизации NADPH- оксидазы в фагосомы [58].
Legionella pneumophila реплицируется в альвеолярных макрофагах, используя стратегию блокирования слияния фагосом с лизосомами. Попадание в АФ помогает патогену избежать немедленной внутриклеточной гибели, после чего бактерия вызывает отсроченное созревание АФ, чтобы иметь достаточно времени для репликации внутри АФ. В дальнейшем развитии инфекции L. pneumophila ингибирует аутофагию путем секреции RavZ – цистеинпротеазы, которая нарушает коньюгацию фосфатидилэтаноламина с LC3 (Atg8) и формирование АФ. Эффекторный белок L. pneumophila RavZ был идентифицирован как фактор, блокирующий возможность распознавания фагофорами убиквитинилированных бактерий [63].
Escherichia coli, Mycobacterium marinum, Chlamydia trachomatis, Yersinia pestis активно индуцируют аутофагию, эффективно распознаются клетками хозяина и захватываются АФ, но избегают гибели путем блокирования слияния АФ с лизосомами [38].
Yersinia pseudotuberculosis использует аутофагические вакуоли как ниши для собственной репликации. В этом случае стимуляция аутофагии способствует репликации бактерий, а ингибирование аутофагии препятствует их выживанию. F. tularensis индуцирует аутофагию, чтобы использовать аминокислоты организма хозяина для построения бактериальных белков [29].
Многие патогенные микроорганизмы экспрессируют аргининдеиминазу, которая осуществляет гидролиз аргинина с образованием аммиака и цитрулина [64, 65]. Аргинин является единственным субстратом iNOS – фермента, осуществляющего реакцию синтеза NO в клетках млекопитающих. Продуцируемая бактериями аргининдеиминаза может подавлять бактерицидные эффекты аутофагии благодаря снижению синтеза NO [66], а также защелачивания среды фаголизосомы за счет продукции аммиака [64, 67]. Поэтому аргининдеиминаза может играть важную роль для выживания патогена в аутофаголизосомах. С другой стороны, активность аргининдеиминазы может приводить к истощению аргинина в клетке с последующим ингибированием сигнального пути mTOR и активацией аутофагии. Значение этого фермента для избегания патогенными микроорганизмами гибели при ксенофагии остается неизученной.
Поскольку репликация внутри клеток организма хозяина является необходимой стадией развития вирусной инфекции, эти патогены так же, как бактерии, выработали эффективные стратегии, позволяющие избегать аутофагии или эксплуатировать этот процесс в своих целях. При инфекциях, вызванных разными ДНК- и РНК-содержащими вирусами, в инфицированных клетках повышается количество АФ. Сама вирусная инфекция часто вызывает стресс в клетках, который проявляется усиленной продукцией АФК с индукцией аутофагии. В инфицированных HSV клетках развивается полноценный аутофагический ответ. Вирус герпеса (HSV-1) способен связывать Beclin-1 и ингибировать формирование АФ. Другие вирусы: HIV-1, вирус гриппа А, вирус коксаки B3, полиовирус ингибируют аутофагию, блокируя созревание или деградацию АФ. Многие вирусы выработали стратегии прямого или опосредованного нарушения аутофагии, чтобы использовать ее на разных стадиях вирусного жизненного цикла. К провирусным эффектам аутофагии относятся: предоставление аутофагией мембранной платформы для репликации вируса, сборки и выхода из клетки, предоставление ресурсов энергии для вирусной репликации. Аутофагия усиливает репликацию HSV за счет ингибирования иммунного ответа. Путь аутофагии пересекается с биосинтезом белков вируса HIV-1 и регулирует вирусную нагрузку в макрофагах [68]. Деградация вируса кори при аутофагии способствует презентации вирусных пептидов и развитию адаптивного протективного иммунитета [2].
Из приведенных ранее примеров видно, что заражение клеток организма патогенными микроорганизмами может иметь разные исходы: (i) механизмы ксенофагии успешно секвестрируют и разрушают патогенные микроорганизмы, (ii) патогенные микроорганизмы после индукции аутофагии успешно избегают попадания в АФ при выходе из эндосом в цитозоль или ингибируют созревание АФ и слияние их с лизосомами, (iii) некоторые из наиболее успешных патогенов не только преодолевают аутофагию, но и используют возможности аутофагии для своего выживания и репликации в клетках хозяина (рис. 2).

Антибактериальные механизмы аутофагии связаны с уничтожением патогенов с помощью лизосомальных ферментов, продукции NO и АФК, что было показано при GAS-инфицировании клеток макрофагоподобной линии RAW264.7 и мышиных перитонеальных макрофагов [57, 69]. Кроме прямой бактерицидной активности ксенофагия может способствовать изоляции внутриклеточных патогенов в АФ и снижению их диссеминации. Кроме того, аутофагия обеспечивает сохранение питательного статуса клетки в период микробного паразитизма по аналогии с ролью аутофагии при голодании [3]. Защитные функции аутофагии более успешны при инфекциях, вызванных внеклеточно паразитирующими бактериями.
Кроме выполнения функций одного их факторов врожденного антимикробного иммунитета, аутофагия вносит свой вклад в регуляцию адаптивного иммунитета. Классическая парадигма MHC(major histocompatibility complex)-рестриктированной презентации антигенов предполагает, что после деградации в протеосомах внутриклеточные антигены презентируются в составе молекул MHC I класса, в то время как внеклеточные антигены после протеолиза в лизосомах нагружаются в молекулы MHC II класса. Однако аутофагия позволяет осуществлять презентацию фрагментов антигенов внутриклеточно-паразитирующих микробов в составе MHC II класса. При этом содержимое АФ поступает в специальные вакуоли антигенпрезентирующих клеток для последующей презентации в составе молекул MHC II [70]. Этот феномен получил название кросс-презентации. Комплексы антигенных пептидов с молекулами MHC II презентируются CD4+ Т- клеткам, что приводит к их активации с последующей индукцией адаптивного иммунного ответа. Связь между макроаутофагией и процессингом антигенов для презентации в составе молекул МНС впервые была продемонстрирована, когда установили, что значительная часть лигандов MHC II класса (20–30%) является производными внутриклеточных цитозольных и ядерных белков [71]. В дальнейшем эти данные были подтверждены с использованием экспериментов по включению и отключению молекул аутофагического аппарата. Было показано, что ингибирование PI3 киназы и сайленсинг Atg12 нарушает процессинг ядерного антигена 1 вируса Эпштейн–Барр (EBNA1) и неомицин-фосфотрансферазы II (NeoR) [72–74]. Потеря Atg5 нарушает CD4+ T-клеточный ответ и MHCII-рестриктированную селекцию T-клеток в тимусе in vivo [75, 76]. В экспериментах по включению функции аутофагии вирусные антигены были соединены с LC3B для направленного транспорта в аутофагосомы [77–80]. Это приводило к усилению презентации антигена в составе MHC II класса специфическим CD4+ T-клеточным клонам в 20-раз. Предполагают, что макроаутофагия – это не единственный способ доставки внутриклеточных белков для процессинга и презентации в составе молекул МНС II класса. Определенную роль в процессе презентации внутриклеточных антигенов в составе МНС II класса может также играть шаперон-опосредованная аутофагия [81, 82]. Активированные Т лимфоциты могут стимулировать макроаутофагию и слияние АФ с лизосомами с помощью продукции цитокинов: IFNγ, TNF и молекул семейства TNF: TRAIL, CD40L [83]. Таким образом, процессы аутофагии могут способствовать презентации эндогенных антигенов в составе молекул MHC II класса и влиять на развитие адаптивного иммунитета. Эффективность участия аутофагии в регуляции антимикробного адаптивного иммунитета в значительной степени зависит от особенностей возбудителя и его стратегии в отношении аутофагии.
ЗАКЛЮЧЕНИЕ
Аутофагия выполняет в организме гомеостатические функции, обеспечивая деградацию поврежденных внутриклеточных структур, восполнение дефицита питательных веществ и выживание клеток. Осуществляя селективную деградацию нежелательного цитоплазматического материала, аутофагия служит важным противовоспалительным механизмом. Аутофагию усиливают различные стрессорные сигналы, включая осмотический шок, пищевое и сывороточное голодание, а также инфекция. В последнее время появляется все больше данных, которые позволяют рассматривать аутофагию как ключевой механизм врожденного иммунитета, направленный на уничтожение внутриклеточных патогенов и призванный контролировать инфекцию. Однако некоторые наиболее успешные микроорганизмы выработали разнообразные стратегии, позволяющие им обходить механизмы аутофагии или даже использовать этот процесс для создания репликативных ниш в различных клетках организма-хозяина и для получения нутриентов. Кроме того, аутофагия может способствовать выживанию бактерий за счет своего противовоспалительного действия. Поэтому патогены могут не только ингибировать аутофагию, но и усиливать этот процесс для подавления защитных реакций иммунитета. В связи с этим при инфекциях стратегия возбудителей в отношении регуляции аутофагии может быть разнонаправленной: с одной стороны, бактерии пытаются избежать губительного действия ксенофагии, с другой стороны, поддерживают аутофагию как гомеостатический процесс. На разных этапах развития инфекции патогенные микроорганизмы могут инициировать аутофагию для избегания защитного действия иммунной системы хозяина, или, напротив, блокировать ее, чтобы иметь возможность длительно персистировать в зараженных клетках. Аутофагия играет важную роль в ходе развития адаптивного иммунного ответа при инфекции, усиливая кросс-презентацию антигенов внутриклеточно паразитирующих микробов. Результаты дальнейшего изучения тонких механизмов регуляции аутофагии при инфекции могут послужить основой разработки новых лекарственных препаратов, повышающих антимикробную защиту организма.
Список литературы
Levine B., Kroemer G. Autophagy in the pathogenesis of disease. Cell. 132: 27–42. 2008.
Jiang P., Mizushima N. Autophagy and human diseases. Cell Res. 24: 69. 2014.
Levine B., Klionsky D.J. Development by self-digestion: Molecular mechanisms and biological functions of autophagy. Dev. Cell. 6: 463–477. 2004.
Giampieri F., Afrin S., Forbes-Hernandez T.Y., Gasparrini M., Cianciosi D., Reboredo-Rodriguez P., Varela-Lopez A., Quiles J.L., Battino M. Autophagy in Human Health and Disease: Novel Therapeutic Opportunities. Antioxid. Redox Signal. 30(4): 577–634. 2019.
Labbé K., Saleh M. Cell death in the host response to infection. Cell Death Differ. 15(9): 1339–1349. 2008.
de Duve C., Wattiaux R. The lysosome. Annu. Rev. Physiol. 28: 435–492. 1966
Del Roso A., Vittorini S., Cavallini G., Donati A., Gori Z., Masini M., Pollera M., Bergamini E. Ageing-related changes in the in vivo function of rat liver macroautophagy and proteolysis. Exp. Gerontol. 38: 519–527. 2003.
Dikic I., Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 19(6): 349–364. 2018.
Degenhardt K., Mathew R., Beaudoin B., Bray K., Anderson D., Chen G., Mukherjee C., Shi Y., Gelinas C., Fan Y., Nelson D.A., Jin S., White E. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 10: 51–64. 2006.
Klionsky D.J., Cregg J.M., Dunn W.A. Jr., Emr S.D., Sakai Y., Sandoval I.V., Sibirny A., Subramani S., Thumm M., Veenhuis M., Ohsumi Y. A unified nomenclature for yeast autophagy-related genes. Dev. Cell. 5(4): 539–545. 2003.
Kirkegaard K., Taylor M.P., Jackson W.T. Cellular autophagy: surrender, avoidance and subversion by microorganisms. Nat. Rev. Microbiol. 2(4): 301–314. 2004.
Haigis M.C., Sinclair D.A. Mammalian sirtuins: biological insights and disease relevance. Annu. Rev. Pathol. 5: 253–295. 2010.
Lee I.H., Cao L., Mostoslavsky R., Lombard D.B., Liu J., Bruns N.E., Tsokos M., Alt F.W., Finkel T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Natl. Acad. Sci. U.S.A. 105: 3374–3379. 2008.
Lee I.H., Finkel T. Regulation of autophagy by the p300 acetyltransferase. J. Biol. Chem. 284(10): 6322–6328. 2009.
Kume S., Uzu T., Horiike K., Chin-Kanasaki M., Isshiki K., Araki S., Sugimoto T., Haneda M., Kashiwagi A., Koya D. Calorie restriction enhances cell adaptation to hypoxia through Sirt1-dependent mitochondrial autophagy in mouse aged kidney. J. Clin. Invest. 120(4): 1043–1055. 2010.
Mammucari C., Milan G., Romanello V., Masiero E., Rudolf R., Del Piccolo P., Burden S.J., Di Lisi R., Sandri C., Zhao J., Goldberg A.L., Schiaffino S., Sandri M. FoxO3 controls autophagy in skeletal muscle in vivo. Cell. Metab. 6(6): 458–471. 2007.
Zhao Y., Wang L., Yang J., Zhang P., Ma K., Zhou J., Liao W., Zhu W.G. Anti-neoplastic activity of the cytosolic FoxO1 results from autophagic cell death. Autophagy. 6(7): 988–990. 2010.
Ruderman N.B., Xu X.J., Nelson L., Cacicedo J.M., Saha A.K., Lan F., Ido Y. AMPK and SIRT1: a long-standing partnership? Am. J. Physiol. Endocrinol. Metab. 298(4): E751–E760. 2010.
Neufeld T.P. TOR-dependent control of autophagy: Biting the hand that feeds. Curr. Opin. Cell Biol. 22(2): 157–168. 2010.
Behrends C., Sowa M.E., Gygi S.P., Harper J.W. Network organization of the human autophagy system. Nature. 466(7302): 68–76. 2010.
Wei Y., Sinha S., Levine B. Dual role of JNK1-mediated phosphorylation of Bcl-2 in autophagy and apoptosis regulation. Autophagy. 4(7): 949–951. 2008.
Nasrin N., Kaushik V.K., Fortier E., Wall D., Pearson K.J., de Cabo R., Bordone L. JNK1 phosphorylates SIRT1 and promotes its enzymatic activity. PLoS One. 4(12): e8414. 2009.
Harding H.P., Zhang Y., Zeng H., Novoa I., Lu P.D., Calfon M., Sadri N., Yun C., Popko B., Paules R., Stojdl D.F., Bell J.C., Hettmann T., Leiden J.M., Ron D. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell. 11: 619–633. 2003.
Nakamura T., Furuhashi M., Li P., Cao H., Tuncman G., Sonenberg N., Gorgun C.Z., Hotamisligil G.S. Double-stranded RNA-dependent protein kinase links pathogen sensing with stress and metabolic homeostasis. Cell. 140: 338–348. 2010.
He C., Klionsky D.J. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 43: 67–93. 2009.
Milani M., Rzymski T., Mellor H.R., Pike L., Bottini A., Generali D., Harris A.L. The role of ATF4 stabilization and autophagy in resistance of breast cancer cells treated with Bortezomib. Cancer Res. 69: 4415–4423. 2009.
Saeki K., Yuo A., Okuma E., Yazaki Y., Susin S.A., Kroemer G., Takaku F. Bcl-2 down-regulation causes autophagy in a caspase-independent manner in human leukemic HL60 cells. Cell Death Differ. 7: 1263–1269. 2000.
Maiuri M.C., Criollo A., Kroemer G. Crosstalk between apoptosis and autophagy within the Beclin 1 interactome. EMBO J. 29(3): 515–516. 2010.
Escoll P., Rolando M., Buchrieser C. Modulation of Host Autophagy during Bacterial Infection: Sabotaging Host Munitions for Pathogen Nutrition. Front. Immunol. 7: 81. 2016.
Birmingham C.L., Smith A.C., Bakowski M.A., Yoshimori T., Brumell J.H. Autophagy controls Salmonella infection in response to damage to the Salmonella-containing vacuole. J. Biol. Chem. 281: 11374–11383. 2006.
Gutierrez M.G., Master S.S., Singh S.B., Taylor G.A., Colombo M.I., Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 119: 753–766. 2004.
Zhao Z., Fux B., Goodwin M., Dunay I.R., Strong D., Miller B.C., Cadwell K., Delgado M.A., Ponpuak M., Green K.G., Schmidt R.E., Mizushima N., Deretic V., Sibley L.D., Virgin H.W. Autophagosome-independent essential function for the autophagy protein Atg5 in cellular immunity to intracellular pathogens. Cell Host Microbe. 4: 458–469. 2008.
Py B.F., Lipinski M.M., Yuan J. Autophagy limits Listeria monocytogenes intracellular growth in the early phase of primary infection. Autophagy. 3: 117–125. 2007.
Nakagawa I., Amano A., Mizushima N., Yamamoto A., Yamaguchi H., Kamimoto T., Nara A., Funao J., Nakata M., Tsuda K., Hamada S., Yoshimori T. Autophagy defends cells against invading Group A Streptococcus. Science. 306: 1037–1040. 2004.
Joubert P.E., Meiffren G., Grégoire I.P., Pontini G., Richetta C., Flacher M., Azocar O., Vidalain P.O., Vidal M., Lotteau V., Codogno P., Rabourdin-Combe C., Faure M. Autophagy induction by the pathogen receptor CD46. Cell Host Microbe. 6(4): 354–366. 2009.
Travassos L.H., Carneiro L.A.M., Ramjeet M., Hussey S., Kim Y.G., Magalhães J.G., Yuan L., Soares F., Chea E., Le Bourhis L., Boneca I.G., Allaoui A., Jones N.L., Nuñez G., Girardin S.E., Philpott D.J. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat. Immunol. 11: 55–62. 2010.
Sanjuan M.A., Dillon C.P., Tait S.W.G., Moshiach S., Dorsey F., Connell S., Komatsu M., Tanaka K., Cleveland J.L., Withoff S., Green D.R. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature. 450: 1253–1257. 2007.
Casanova J.E. Bacterial Autophagy: Offense and Defense at the Host-Pathogen Interface. Cell Mol. Gastroenterol. Hepatol. 4(2): 237–243. 2017.
Gatica D., Lahiri V., Klionsky, D.J. Cargo recognition and degradation by selective autophagy. Nat. Cell Biol. 20(3): 233–242. 2018.
Kageyama S., Omori H., Saitoh T., Sone T., Guan J.-L., Akira S., Imamoto F., Noda T., Yoshimori T. The LC3 recruitment mechanism is separate from Atg9L1-dependent membrane formation in the autophagic response against Salmonella. Mol. Biol. Cell. 22: 2290–2300. 2011.
Huang J., Brumell J.H. Bacteria-autophagy interplay: a battle for survival. Nat. Rev. Microbiol. 12: 101–114. 2014.
Sharma V., Verma S., Seranova E., Sarkar S., Kumar D. Selective Autophagy and Xenophagy in Infection and Disease. Front Cell Dev. Biol. 6: 147. 2018.
Sumpter R.Jr., Levine B. Autophagy and innate immunity: triggering, targeting and tuning. Semin. Cell. Dev. Biol. 21: 699–711. 2010.
Siqueira M.D.S., Ribeiro R.M., Travassos L.H. Autophagy and Its Interaction With Intracellular Bacterial Pathogens. Front Immunol. 9: 935. 2018.
Baud V., Karin M. Is NF-kappaB a good target for cancer therapy? Hopes and pitfalls. Nat. Rev. Drug Discov. 8(1): 33–40. 2009.
Liu L., Wise D.R., Diehl J.A., Simon M.C. Hypoxic reactive oxygen species regulate the integrated stress response and cell survival. J. Biol. Chem. 283: 31153–31162. 2008.
Scherz-Shouval R., Shvets E., Fass E., Shorer H., Gil L., Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 26: 1749–1760. 2007.
Alexander A., Cai S.L., Kim J., Nanez A., Sahin M., MacLean,K.H., Inoki K., Guan K.L., Shen J., Person M.D., Kusewitt D., Mills G.B., Kastan M.B., Walker C.L. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc. Natl. Acad. Sci. U.S.A. 107: 4153–4158. 2010.
Huang Q., Wu Y.T., Tan H.L., Ong C.N., Shen H.M. A novel function of poly(ADP-ribose) polymerase-1 in modulation of autophagy and necrosis under oxidative stress. Cell Death Differ. 16: 264–277. 2009.
Wong C.H., Iskandar K.B., Yadav S.K., Hirpara J.L., Loh T., Pervaiz S. Simultaneous induction of non-canonical autophagy and apoptosis in cancer cells by ROS-dependent ERK and JNK activation. PLoS ONE. 5: e9996. 2010.
Bensaad K., Cheung E.C., Vousden K.H. Modulation of intracellular ROS levels by TIGAR controls autophagy. EMBO J. 28: 3015–3026. 2009.
Green D.R., Kroemer G. Cytoplasmic functions of the tumour suppressor p53. Nature. 458: 1127–1130. 2009.
Kroemer G., Marino G., Levine B. Autophagy and the Integrated Stress Response. Mol. Cell. 40(2): 280–293. 2010.
Seay M., Patel S., Dinesh-Kumar S.P. Autophagy and plant innate immunity. Cell Microbiol. 8(6): 899–906. 2006.
Bento C.F., Renna M., Ghislat G., Puri C., Ashkenazi A., Vicinanza M., Menzies F.M., Rubinsztein D.C. Mammalian Autophagy: How Does It Work? Annu. Rev. Biochem. 85: 685–713. 2016.
Sakurai A., Maruyama F., Funao J., Nozawa T., Aikawa C., Okahashi N., Shintani S., Hamada S., Ooshima T., Nakagawa I. Specific behavior of intracellular Streptococcus pyogenes that has undergone autophagic degradation is associated with bacterial streptolysin O and host small G proteins Rab5 and Rab7. J. Biol. Chem. 285: 22666–22675. 2010.
Nakagawa I., Amano A., Mizushima N., Yamamoto A., Yamaguchi H., Kamimoto T., Nara A., Funao J., Nakata M., Tsuda K., Hamada S., Yoshimori T. Autophagy defends cells against invading group A Streptococcus. Science. 306(5698): 1037–1040. 2004.
Bah A., Vergne I. Macrophage Autophagy and Bacterial Infections. Front Immunol. 8: 1483. 2017.
Barnett T.C., Liebl D., Seymour L.M., Gillen C.M, Lim J.Y., Larock C.N., Davies M.R., Schulz B.L., Nizet V., Teasdale R.D., Walker M.J. The globally disseminated M1T1 clone of group A Streptococcus evades autophagy for intracellular replication. Cell Host Microbe. 14: 675–672. 2013.
O'Seaghdha M., Wessels M.R. Streptolysin O and its co-toxin NAD-glycohydrolase protect group A Streptococcus from Xenophagic killing. PLoS Pathog. 9(6): e1003394. 2013.
Schnaith A., Kashkar H., Leggio S.A., Addicks K., Kronke M., Krut O. Staphylococcus aureus subvert autophagy for induction of caspaseindependent host cell death. J. Biol. Chem. 282: 2695–2706. 2007.
Ogawa M., Yoshimori T., Suzuki T., Sagara H., Mizushima N., Sasakawa C. Escape of intracellular Shigella from autophagy. Science. 307: 727–731. 2005.
Choy A., Dancourt J., Mugo B., O’Connor T.J., Isberg R.R., Melia T.J., Roy C.R. The Legionella effector RavZ inhibits host autophagy through irreversible Atg8 deconjugation. Science. 338(6110): 1072–1076. 2012.
Cunin R., Glansdorff N., Piérard A., Stalon V. Biosynthesis and metabolism of arginine in bacteria. Microbiol. Rev. 50(3): 314–352. 1986.
Gallego P., Planell R., Benach J., Querol E., Perez-Pons J.A., Reverter D. Structural characterization of the enzymes composing the arginine deiminase pathway in Mycoplasma penetrans. PLoS One. 7(10): e47886. 2012.
Starikova E.A., Sokolov A.V., Vlasenko A.Y., Burova L.A., Freidlin I.S., Vasilyev V.B. Biochemical and biological activity of arginine deiminase from Streptococcus pyogenes M22. Biochem. Cell Biol. 94(2): 129–137. 2016.
Cusumano Z.T., Caparon M.G. Citrulline protects Streptococcus pyogenes from acid stress using the arginine deiminase pathway and the F1Fo-ATPase. J. Bacteriol. 197(7): 1288–1296. 2015.
Chiramel A.I., Brady N.R., Bartenschlager R. Divergent Roles of Autophagy in Virus Infection. Cells. 2: 83–104. 2013.
Vural A., Kehrl J.H. Autophagy in macrophages: Impacting inflammation and bacterial infection. Scientifica (Cairo). 2014: 825463. 2014.
Münz C. Enhancing immunity through autophagy. Annu. Rev. Immunol. 27: 423–449. 2009.
Dengjel J., Schoor O., Fischer R., Reich M., Kraus M., Müller M., Kreymborg K., Altenberend F., Brandenburg J., Kalbacher H., Brock R., Driessen C., Rammensee H.G., Stevanovic S. Autophagy promotes MHC class II presentation of peptides from intracellular source proteins. Proc. Natl. Acad. Sci. U. S. A. 102(22): 7922–7927. 2005.
Paludan C., Schmid D., Landthaler M., Vockerodt M., Kube D., Tuschl T., Münz C. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science. 307(5709): 593–596. 2005.
Nimmerjahn F., Milosevic S., Behrends U., Jaffee E.M., Pardoll D.M., Bornkamm G.W., Mautner J. Major histocompatibility complex class II-restricted presentation of a cytosolic antigen by autophagy. Eur. J. Immunol. 33(5): 1250–1259. 2003.
Leung C.S., Haigh T.A., Mackay L.K., Rickinson A.B., Taylor G.S. Nuclear location of an endogenously expressed antigen, EBNA1, restricts access to macroautophagy and the range of CD4 epitope display. Proc. Natl. Acad. Sci. U. S. A. 107(5): 2165–2170. 2010.
Lee H.K., Mattei L.M., Steinberg B.E., Alberts P., Lee Y.H., Chervonsky A., Mizushima N., Grinstein S., Iwasaki A. In vivo requirement for Atg5 in antigen presentation by dendritic cells. Immunity. 32(2): 227–239. 2010.
Nedjic J., Aichinger M., Emmerich J., Mizushima N., Klein L. Autophagy in thymic epithelium shapes the T-cell repertoire and is essential for tolerance. Nature. 455(7211): 396–400. 2008.
Schmid D., Pypaert M., Münz C. Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity. 26(1): 79–92. 2007.
Comber J.D., Robinson T.M., Siciliano N.A., Snook A.E., Eisenlohr L.C. Functional macroautophagy induction by influenza A virus without a contribution to major histocompatibility complex class II-restricted presentation. J. Virol. 85(13): 6453–6463. 2011.
Jin Y., Sun C., Feng L., Li P., Xiao L., Ren Y., Wang D., Li C., Chen L. Regulation of SIV antigen-specific CD4+ T cellular immunity via autophagosome-mediated MHC II molecule-targeting antigen presentation in mice. PLoS One. 9(3): e93143. 2014.
Fonteneau J.F., Brilot F., Münz C., Gannagé M. The Tumor Antigen NY-ESO-1 Mediates Direct Recognition of Melanoma Cells by CD4+ T Cells after Intercellular Antigen Transfer. J. Immunol. 196(1): 64–71. 2016.
Zhou D., Li P., Lin Y., Lott J.M., Hislop A.D., Canaday D.H., Brutkiewicz R.R., Blum J.S. Lamp-2a facilitates MHC class II presentation of cytoplasmic antigens. Immunity. 22(5): 571–581. 2005.
Münz C. Autophagy Beyond Intracellular MHC Class II Antigen Presentation. Trends Immunol. 37(11): 755–763. 2016.
Schmid D., Münz C. Innate and adaptive immunity through autophagy. Immunity. 27(1): 11–21. 2007.
Дополнительные материалы отсутствуют.
Инструменты
Российский физиологический журнал им. И.М. Сеченова