

Российский физиологический журнал им. И.М. Сеченова, 2019, T. 105, № 4, стр. 510-519
Блокада транспортеров гамма-аминомасляной кислоты в синапсах головного мозга предохраняет от развития судорог при дыхании кислородом под давлением
А. Н. Москвин 1, Т. Ф. Платонова 1, С. Ю. Жиляев 1, О. С. Алексеева 1, *, Е. Р. Никитина 1, И. Т. Демченко 1
1 Институт эволюционной физиологии и биохимии им. И.М. Сеченова РАН
Санкт-Петербург, Россия
* E-mail: osa72@inbox.ru
Поступила в редакцию 12.12.2018
После доработки 24.02.2019
Принята к публикации 25.02.2019
Аннотация
Дыхание гипербарическим кислородом (ГБО2) вызывает угнетение синтеза гамма-аминомасляной кислоты (ГАМК) в мозге, что приводит к ослаблению тормозной ГАМК-ергической нейропередачи и развитию судорожного синдрома по типу эпилептического припадка. В настоящей работе проверялась гипотеза о том, что ингибирование ГАМК-транспортеров может компенсировать недостаток синтеза тормозного медиатора, усилить ГАМК-ергическую передачу и ослабить или предотвратить развитие кислородных судорог. У бодрствующих крыс в барокамере под давлением кислорода 5 АТА (атмосфер абсолютных) анализировалось развитие судорог после предварительного введения животным в желудочек мозга препаратов, ингибирующих селективно нейрональные (NO-711) и неселективно нейрональные и глиальные ГАМК-транспортеры (nipecotic acid). В отдельной группе крыс ГАМК измерялась в стриатуме с помощью микродиализа, сопряженного с жидкостной хроматографией. Установлено, что ингибирование нейрональных и глиальных ГАМК-транспортеров повышает уровень ГАМК в мозге и ослабляет развитие кислородных судорог. Более эффективный антисудорожный эффект наблюдается после введения в мозговой желудочек неселективного ингибитора ГАМК-транспортеров. Полученные данные свидетельствуют о том, что блокирование функций нейрональных и глиальных ГАМК-транспортеров повышает уровень ГАМК в мозге и ослабляет развитие судорожного синдрома в ГБО2. Антисудорожные эффекты использованных ингибиторов, по-видимому, обусловлены усилением ГАМК-опосредованной синаптической и внесинаптической нейропередач при гипербарической гипероксии. Ингибирование ГАМК-транспортеров может быть перспективным направлением для разработки эффективных методов предотвращения кислородных судорог.
Гипербарический кислород (ГБО2), широко используемый в лечебной медицине и при подводных погружениях, под давлением свыше 2 АТА (атмосфер абсолютных) оказывает острое токсическое действие на ЦНС, проявляющееся в виде генерализованных тонических и клонических судорог по типу эпилептического припадка [1, 2]. Нейротоксический эффект ГБО2 отчетливо демонстрируется также в виде эпилептиформных паттернов на электроэнцефалограмме (ЭЭГ) и повышенной активности симпатической нервной системы [3, 4]. Все эти физиологические реакции свидетельствуют о гиперактивном состоянии головного мозга во время развития кислородных судорог [5]. Принято считать, что причиной появления судорог в ГБО2 является нарушение баланса между возбуждающей и тормозной медиаторными системами мозга за счет угнетения ГАМК-ергической передачи [6, 7]. Данное заключение базируется на исследованиях, показавших уменьшение содержания ГАМК в мозге животных после их экспозиции в ГБО2 [8–12] или снижения количества тормозного медиатора, измеряемого непосредственно в мозге крыс во время развития кислородных судорог с помощью микродиализа, сопряженного с высокоэффективной жидкостной хроматографией [13, 14]. Как было установлено, причиной уменьшения внутримозговой ГАМК в ГБО2 является понижение активности глутаматдекарбоксилазы (ГДК), единственного фермента, катализирующего синтез тормозного медиатора в нервных клетках. Недавние исследования показали, что механизм инактивации ГДК состоит в S-нитрозилировании белка-фермента по остаткам цистеина с помощью оксида азота (NO) и его производных [15]. ГБО2 значительно усиливает генерацию активных форм кислорода и азота (АФКА), таких, как супероксиданионы [6, 16] и оксид азота [13, 17], поэтому вероятность S-нитрозилирования ГДК очень высока, что ведет к посттрансляционной модификации ее структуры и снижению ферментативной активности [18]. В связи с этим возникает вопрос, можно ли предотвратить снижение синтеза ГАМК в ГБО2 или повысить содержание медиатора в синаптической щели и межклеточном пространстве и таким образом противодействовать развитию кислородных судорог. Если инактивация ГДК в нейронах происходит с участием NO, то ослабить или полностью исключить S-нитрозилирование фермента возможно путем блокирования продукции оксида азота. Доказательства этому получены в исследованиях, где неселективное ингибирование NO-синтаз (NOS) ослабляло развитие судорог у крыс [13]. Более того, у мышей, нокаутов по нейрональной NOS, латентный период судорог в ГБО2 достоверно длиннее, чем у обычных животных [19]. Однако подобный способ предотвращения S-нитрозилирования ГДК, а, следовательно, и развития судорог, на практике не пригоден, так как NO вовлекается во многие физиологические процессы и блокирование его синтеза приведет к развитию различных патологий. Другим способом восстановления ГАМК-ергической нейропередачи в ГБО2 может быть увеличение содержания медиатора в синаптической щели и межклеточной среде, что способно компенсировать дефицит его синтеза и обеспечить эффективную тормозную функцию.
После квантового выброса ГАМК в синаптическую щель с последующей активацией постсинаптических рецепторов действие медиатора прекращается путем его удаления ГАМК-транспортерами обратно в пресинаптический нейрон для повторного использования и в глию, где происходит его ферментативное расщепление с участием ГАМК-трансаминазы (ГАМК-Т). Из 4-х типов ГАМК-транспортеров (GAT1, GAT2, GAT3 и BGT1) основными для головного мозга являются GAT1, переносящий ГАМК из синаптической щели преимущественно в нейроны, и GAT3, активно транспортирующий аминокислоту, в основном, в глиальные клетки. Помимо активного транспорта ГАМК из синаптической щели, следует также указать на существование диффузии медиатора в межклеточное пространство, где он может реализовывать внесинаптическую тоническую нейропередачу [20, 21].
Мы предполагаем, что блокирование механизмов удаления нейромедиатора может компенсировать недостаток его синтеза и увеличить содержание ГАМК в синаптической щели и экстраклеточной среде до уровня, достаточного для обеспечения тормозной нейропередачи и, таким образом, ослабить или предотвратить развитие судорожного синдрома в ГБО2. Для проверки этой гипотезы в настоящей работе изучалось развитие судорог у крыс, которым перед экспозицией в ГБО2 в боковой желудочек головного мозга вводились неселективный (nipecotic acid) и селективный (NO-711) ингибиторы ГАМК-транспортеров.
МЕТОДЫ ИССЛЕДОВАНИЯ
Исследования выполнены на крысах линии Sprague Dawley массой 280–340 г, полученных из питомника лабораторных животных “Пущино”. Протокол опытов одобрен Комиссией по использованию животных ИЭФБ РАН в соответствии с Международными рекомендациями (этический кодекс) по проведению медико-биологических исследований с использованием животных (CIOMS, Geneva, 1985). Каждому животному за неделю до опытов под наркозом (нембутал, 50 мг/кг внутрибрюшинно) вводили металлическую канюлю диаметром 0.24 мм в боковой желудочек головного мозга (стереотаксические координаты: Р = –1.0 мм, L = 1.5 мм, D = 3.5 мм). Для укрепления канюли в теменную кость черепа билатерально имплантировали 2 стальных винта. Канюли и металлические винты фиксировались на кости зубопротезным цементом. В день опыта в боковой желудочек мозга вводили один из следующих препаратов: nipecotic acid (NPA), которая является универсальным ингибитором всех ГАМК-транспортеров, за исключением BGT-1, и NO-711, селективного ингибитора ГАМК-транспортера GAT1. Животным контрольной группы вводили искусственный ликвор. Препараты закупали в фирме Sigma Aldrich и перед введением растворяли в искусственном ликворе. За 30 мин до начала опытов животным инъецировали в боковой желудочек мозга препарат в объеме 7 мкл через введенную в канюлю иглу, соединенную с микрошприцем (Hamilton, 10 мкл). Количество вводимого препарата и число использованных животных составляло: искусственный ликвор (контроль, n = 16), NPA – 0.05 мг (n = 16) и NO-711 – 0.02 мг (n = 16). Указанные дозы являются пороговыми величинами, свыше которых наблюдались расстройства двигательной функции у животных, которых мы тестировали на вращающемся стержне (Rotarod test). После введения препаратов крыс помещали в барокамеру объемом 100 литров по одному животному в каждом опыте. Повышение давления кислорода в камере до 5 АТА осуществляли со скоростью 1 АТА/мин. С помощью систем жизнеобеспечения температура в камере поддерживалась в пределах 23–25°С, влажность – около 60%, содержание СО2 не превышало 0.1%.
В период ГБО2 экспозиции проводили видеорегистрацию поведения животных в барокамере. Экспозиция продолжалась до появления генерализованных клонических или тонических судорог, а при их отсутствии, максимально 60 мин. Время декомпрессии составляло 8 мин.
В другой серии опытов измеряли содержание внеклеточной ГАМК в стриатуме крыс после предварительного введения в него NPA с помощью микродиализа. Эту работу проводили в рамках ранее выполненных исследований [7], но результаты измерений ГАМК после введения указанного препарата еще не публиковались. Наркотизированным животным (уретан + хлоралоза) в стриатум (координаты: А = +1.0 мм, LМ ±2.5 мм, D = 5.8 мм) вводили микродиализные канюли (CMA/11, CMA/Microdialysis AB, Швеция). Во время ГБО2 экспозиции канюли перфузировали искусственным ликвором со скоростью 1.0 мкл/мин, а пробы диализата автоматически отбирали каждые 15 мин (CMA 142 Microfraction Collector, AB, Швеция). Измерения ГАМК в пробах проводили после опыта с помощью высокопроизводительной жидкостной хроматографии (HPLC) с электрохимической детекцией ГАМК в диализате (ESA model 5100A). Содержание ГАМК в пробах диализата определяли в мкмоль/л по калибровочным стандартам. Параметры ГБО2 экспозиции были такими же, как и в первой части исследований. Всего в этих опытах использовано 9 животных, у которых микродиализные канюли перфузировали раствором NPA (0.07 мМ) в течение 30 мин до компрессии кислородом, а во время ГБО2 экспозиции под давлением 5 АТА искусственным ликвором. ГАМК в стриатуме измеряли при дыхании воздухом (контроль), после ингибирования ГАМК-транспортеров и в барокамере при дыхании кислородом под давлением 5 АТА на фоне блокирования транспортеров тормозного нейромедиатора.
Для статистического анализа использовали результаты измерений латентного периода (в минутах) появления разных типов моторных судорожных реакций. В тех случаях, когда у животных не наблюдалось никаких судорожных проявлений за все время ГБО2 экспозиции за латентный период принимали время окончания эксперимента, равное 60 мин. Данные анализировали с помощью дисперсионного анализа SigmaPlot 13.0 (Systat Software, Inc., San Jose, CA). Однофакторный дисперсионный анализ использовали для сравнения латентных периодов судорожных реакций при введении искусственного ликвора и препаратов. Двухфакторный дисперсионный анализ использовали для определения эффектов ГБО2 на содержание ГАМК. Для выявления достоверности использовали парный t-критерий. Все данные представлены как М ± SD, при этом в качестве статистически значимых принимали значения Р < 0.05.
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЙ
Крысы контрольной группы в ходе ГБО2 экспозиции демонстрировали различные паттерны моторной судорожной активности и определенную последовательность появления, в которой мы выделили 4 стадии. Первая стадия начиналась после 10–15 мин ГБО2 с появления интенсивного груминга, легкого потряхивания головы и передних лап, переходящего в кратковременные встряхивания всего тела, типа эффекта “мокрой собаки” длительностью 1–3 с. На стадии 2 наблюдались повторяющиеся локальные сокращения мышц мордочки, головы и передних конечностей, а также всего тела. Миоклонии продолжались от 5 до 15 с и могли повторяться через несколько минут. На стадии 3 у животных появлялись ритмические сокращения мышц всего тела продолжительностью от 10 до 25 с, при этом животные могли вставать на задние лапы, пятиться назад, сильно бить хвостом. На стадии 4 у животных наблюдались генерализованные клонические или тонические конвульсии. Судорожный припадок сопровождался тахикардией, гипервентиляцией и другими признаками расстройств вегетативной нервной системы.
У крыс в ГБО2, которым вводились изучаемые препараты, стадийное развитие судорожного синдрома сохранялось (табл. 1). Тестируемые препараты задерживали появление каждой стадии судорожного синдрома по отношению к контрольным значениям. Наибольший антисудорожный эффект вызывала NPA – неселективный ингибитор ГАМК-транспортеров, при введении которого 3-я и 4-я стадии не развивались. При введении NO-711 временные пороги появления всех стадий развития судорожного синдрома были короче, чем при введении неселективного ингибитора.
Таблица 1.
Временные пороги стадийного развития судорожного синдрома у крыс в ГБО2 при блокировании ГАМК-транспортеров в головном мозге
| Препараты | Количество животных | Стадия 1 (мин) |
Стадия 2 (мин) |
Стадия 3 (мин) |
Стадия 4 (мин) |
|---|---|---|---|---|---|
| Контроль | 16 | 19.5 ± 6.8 | 24 ± 7.3 | 27 ± 6.9 | 35.3 ± 6.1 |
| NO-711 | 16 | 31.3 ± 7.6 | 39.2 ± 8.6* | 43.7 ± 7.5* | 56.2 ± 2.5* |
| NPA | 16 | 46.9 ± 9* # | 58.5 ± 1.5* # | 60* | 59.9 ± 0.2* |
Общее состояние животных после часовой экспозиции в барокамере оценивалось по балльной шкале: животным без видимых двигательных нарушений присваивалось 4 балла, с нарушениями координации движений – 3 балла, продолжающих судорожить – 2 балла и погибшим в барокамере или в течение одного часа после декомпрессии – 1 балл. Анализ показал лучшее состояние у крыс с введенной NPA (3.8 ± 0.2 балла), по сравнению с контрольной группой (2.1 ± 0.4 балла, p < 0.05) или с группой животных, которым вводился NO-711, селективный блокатор нейронального захвата ГАМК (3.0 ± 0.4 балла, p < 0.05).
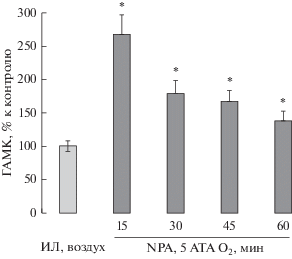
Концентрация ГАМК в стриатуме наркотизированных крыс при дыхании воздухом составляла 0.067 ± 0.028 мкмоль/л. Внутримозговое введение NPA значительно повышало уровень ГАМК в стриатуме крыс при дыхании воздухом (рис. 1). В ГБО2 содержание медиатора снижалось, но оставалось выше контрольного уровня до конца гипероксической экспозиции (рис. 2).
Рис. 1.
Содержание ГАМК в стриатуме головного мозга крыс при дыхании атмосферным воздухом после ингибирования ГАМК-транспортеров. По оси абсцисс: ИЛ – перфузия микродиализной канюли искусственным ликвором (n = 9); NPA – перфузия раствором нипекотиновой кислоты (n = 9). По оси ординат: концентрация ГАМК, мкмоль/л. **P < 0.01 по сравнению с ИЛ.

Рис. 2.
Изменения ГАМК в стриатуме головного мозга крыс при дыхании кислородом под давлением 5 АТА после ингибирования ГАМК-транспортеров. По оси абсцисс: ИЛ – перфузия микродиализной канюли искусственным ликвором (n = 9); NPA – перфузия раствором нипекотиновой кислоты (n = 9). Цифры – время (мин) измерения ГАМК при 5 АТА О2. По оси ординат: изменения ГАМК, в % к контролю (ИЛ), *P < 0.05 по сравнению с ИЛ.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
В настоящей работе получено несколько новых данных, касающихся механизмов развития судорог и способов их предотвращения при дыхании кислородом под давлением. Во-первых, определен паттерн локальных миоклоний у крыс в ГБО2, который является маркером продромального периода развития кислородных судорог. Во-вторых, установлено, что ингибирование нейрональных и глиальных ГАМК- транспортеров повышает уровень тормозного медиатора в головном мозге. В-третьих, блокирование функции ГАМК-транспортеров предохраняет от развития судорог в ГБО2.
У крыс, свободно передвигающихся в барокамере под давлением кислорода 5 АТА, в определенной последовательности развивался судорожный синдром, начиная с локальных миоклоний и до генерализованных конвульсий. Такие судорожные реакции известны давно, и латентное время появления клонических или тонических судорог обычно используется для оценки нейротоксического действия ГБО2. В данной работе при 60-минутной экспозиции в ГБО2 мы не наблюдали генерализованных судорог у крыс, которым вводились препараты, поэтому для сравнительной оценки антисудорожной эффективности препаратов был использован другой показатель. Как оказалось, у животных контрольной и опытных групп в ГБО2 сначала появлялись одиночные сокращения мышц головы, шеи и передних конечностей, которые всегда предшествовали генерализованным конвульсиям. Эти локальные миоклонии и были использованы в качестве продромального маркера появления генерализованных конвульсиий. Мы рассчитывали усредненное латентное время появления определенной группы локальных миоклоний и использовали эту величину для оценки эффективности противосудорожных препаратов. Применение этого способа позволило установить, что два ингибитора ГАМК-транспортеров задерживали развитие кислородных судорог у крыс в барокамере по сравнению с животными, которым в мозговой желудочек вводился искусственный ликвор. Латентные периоды появления каждой из 4-х стадий судорожного синдрома в опыте достоверно отличались от контрольных значений. Среди двух использованных препаратов более эффективным антисудорожным действием обладал неселективный ингибитор ГАМК-транспортеров.
Мы полагаем, что основным механизмом противосудорожного действия NO-711 и NPA в ГБО2 является повышение ГАМК в синаптическом пространстве и межклеточной среде. Правомерность этого предположения подтверждается результатами, полученными в настоящей работе. Во-первых, задержка в развитии кислородных судорог была значительно больше при использовании NPA по сравнению с селективным ингибитором NO-711. NPA является блокатором всех переносчиков ГАМК, поэтому при использовании этого ингибитора уровень медиатора в синапсе гипотетически может быть выше, чем при блокировании только нейрональных транспортеров. Во-вторых, ингибирование ГАМК-транспортеров в стриатуме крыс с помощью микродиализной доставки NPA более чем в 3 раза повышало содержание ГАМК в межклеточном пространстве (рис. 1). Кроме того, после предварительного ингибирования ГАМК-транспортеров уровень медиатора в ГБО2 оставался повышенным по отношению к контрольным значениям, тогда как в наших ранних исследованиях содержание ГАМК в стриатуме крыс с интактными транспортерами понижалось в ГБО2 на 37 ± 7.4% [7]. Метод внутримозгового микродиализа, использованный в данной работе, позволяет измерять содержание ГАМК в межклеточной жидкости вокруг мембраны канюли. До 50–70% уровня межклеточной ГАМК формируется из медиатора, осуществляющего синаптическую нейропередачу [22], и какая-то часть из ГАМК, выделяемой из глиальных клеток [23]. Следовательно, ингибирование ГАМК-транспортеров может повышать уровень тормозного медиатора как в синаптической щели, так и в межклеточной среде, а это предполагает, что нейропередача в ГБО2 восстанавливается благодаря усилению фазной синаптической и тонической внесинаптической ГАМК-нейропередач.
Одним из важных аспектов рассматриваемой проблемы является определение путей и звеньев ГАМК-ергической передачи, которые угнетаются в ГБО2, вызывая судороги. Выраженное противосудорожное действие использованных препаратов косвенно свидетельствует о том, что ГБО2 не подавляет функцию ГАМК-транспортеров. В противном случае, вводимые в мозг препараты не вызывали бы такого выраженного антисудорожного эффекта. Вместе с тем, действие ГБО2 на ГАМК-транспортеры все-таки остается неясным, так как проведенное нами компьютерное секвенирование GAT1 показало наличие в его структуре цистеина с тиоловыми группами, являющимися потенциальной мишенью для S-нитрозилирования (данные не представлены). Гипербарический кислород также не меняет активности ГАМК-трансаминазы [15], хотя ингибирование этого фермента с помощью вигабатрина ослабляет развитие кислородных судорог у крыс [24], свиней [25] и мышей [26].
Основным механизмом подавления ГАМК-ергической передачи в ГБО2 является снижение ферментативной активности глутаматдекарбоксилазы и соответственно снижения синтеза тормозного медиатора. Такой механизм вовлечения ГАМК- опосредованной нейропередачи в нейротоксическое действие гипербарического кислорода был высказан еще в 60-х годах прошлого века [8, 9], и понадобилось более 50 лет, чтобы понять молекулярную природу инактивации глутаматдекарбоксилазы при экстремальной гипероксии. Глутаматдекарбоксилаза присутствует в головном мозге в виде двух изоформ, ГДК65 и ГДК67, участвующих в синтезе тормозного медиатора, используя глутамат в качестве субстрата. Как оказалось, в ГБО2 инактивируется изоформа ГДК65, локализованная в терминалях аксонов нервных клеток, а механизмом инактивации является ее S-нитрозилирование, приводящее к изменению структуры и функции белка [15, 18]. В исследованиях установлено, что ГБО2 не меняет ферментативной активности изоформы ГДК67, локализованной преимущественно в телах нейронов, и ее S-нитрозилирования в гипербарической гипероксии не выявлено [15]. Отсюда следует, что снижение синтеза тормозного нейромедиатора в ГБО2 происходит преимущественно в терминалях аксонов ГАМК-ергических нейронов вследствие подавления ферментативной активности ГДК65. Логично предположить, что пониженная продукция медиатора приведет к уменьшению его выброса в синаптическую щель и, следовательно, к ослаблению ГАМК-опосредованного фазного торможения. Вместе с тем, наличие связи между содержанием медиатора в синаптической щели, в прилежащих к ней глиальных клетках и в межклеточной среде позволяет предположить, что внесинаптическая ГАМК-ергическая нейропередача также может угнетаться в ГБО2. Данное предположение базируется на известных данных о наличии в мозге тонической внесинаптической ГАМК-опосредованной нейропередачи через ГАМКА-рецепторы, локализованные на теле нейронов [21]. Имеющиеся данные пока не позволяют ответить на вопрос, какая из двух ГАМК-ергических нейропередач, синаптическая или внесинаптическая, премущественно угнетается в условиях гипероксической экспозиции. Найденное в данной работе у крыс повышение межклеточного ГАМК в стриатуме, а у других авторов в гиппокампе [27] при ингибировании ГАМК-транспортеров и более эффективное антисудорожное действие NPA по сравнению с NO-711 косвенно свидетельствуют о возможном снижении ГАМК-опосредованной внесинаптической передачи в ГБО2.
Таким образом, дефицит ГАМК-ергической нейропередачи в ГБО2, возникающий за счет угнетения синтеза медиатора, может быть преодолен путем повышения его уровня в синаптическом пространстве и во внеклеточной среде. Тестирование этой гипотезы показало, что введенные в мозговой желудочек ингибиторы ГАМК-транспортеров повышают межклеточный уровень ГАМК в мозге (NPA) и предотвращают развитие кислородных судорог. Следовательно, блокирование механизмов клиренса синаптической ГАМК может полноценно компенсировать снижение синтеза тормозного медиатора в ГБО2, увеличить содержание ГАМК до уровня, достаточного для обеспечения тормозной нейромедиации, и, тем самым, ослабить или предотвратить развитие судорожного синдрома.
ИСТОЧНИКИ ФИНАНСИРОВАНИЯ
Работа выполнена при поддержке Программы фундаментальных исследований президиума РАН № 18 “Биомедицинские технологии: инновационные разработки” (проект № 0132-2018-0011) и, частично, в рамках государственного задания ИЭФБ РАН (№ 007-00096-18, рег. № АААА-А18-118012290142-9).
Список литературы
Зальцман Г.Л. Стадии развития кислородной эпилепсии и функциональное состояние нервной системы. В кн.: Гипербарические эпилепсия и наркоз. Л. Наука. С. 129–136. 1968. [Zaltsman G.L. Stages of formation of oxygen epilepsy and the functional state of the centres of the nervous system. In book: Hyperbaric epilepsy and narcosis. Ed. Zaltsman G.L. Leningrad. Nauka. Р. 129–136. 1968. (In Russ)].
Balentine J.D. Pathology of oxygen toxicity. New York. Academic. 1982.
Арсеньева В.И., Селивра А.И. Биоэлектрическая активность периферического отдела симпатической нервной системы в процессе кислородной эпилепсии. В кн.: Гипербарические эпилепсия и наркоз. Л. Наука. С. 70–78. 1968. [Arsenjeva V.I., Selivra A.I. Bioelectric activity of the peripheral part of sympathetic nervous system in the course of formation of oxygen epilepsy. In book: Hyperbaric epilepsy and narcosis. Ed. Saltsman G.L. Leningrad. Nauka. Р. 70–78. 1968. (In Russ)].
Селивра А.И. Гипербарическая оксигенация. Физиологические механизмы реакций центральной нервной системы на гипероксию. Л. Наука. 1983. [Selivra A.I. Hyperbaricheskaya oksigenaciya. Fiziologicheskie mehanizmy reakcii centralnoi nervnoi sistemy na hyperoksiyu. [Hyperbaric oxygenation. Physiological mechanisms of central nervous system responses to hyperoxia.] Leningrad. Nauka. 1983].
Dean J.B., Mulkey D.K., Henderson R.A., Potter S.J., Putnam R.W. Hyperoxia, reactive oxygen species, and hyperventilation: Oxygen sensitivity of brain stem neurons. J. Appl. Physiol. 96: 784–791. 2004.
Dean J.B., Mulkey D.K., Garcia A.J., Putnam R.W., Henderson R.A. Neuronal sensitivity to hyperoxia, hypercapnia, and inert gases at hyperbaric pressures. J. Appl. Physiol. 95: 883–909. 2003.
Demchenko I.T., Piantadosi C.A. Nitric oxide amplifies the excitatory to inhibitory neurotransmitter imbalance accelerating oxygen seizures. Undersea Hyperb. Med. 33(3): 169–174. 2006.
Щербакова Г.В. Активность глютаматдекарбоксилазы и содержание γ-аминомасляной кислоты в мозге крыс при разных функциональных состояниях, вызванных повышенным давлением кислорода. Докл. АН СССР. 146(5): 1213–1215. 1962. [Shcherbakova G.V. Glutamate decarboxylase activity and γ-aminobutyric acid content in rat brain at different functional states caused by high oxygen pressure. Dokl. AN USSR. 146(5): 1213–1215. 1962. (In Russ)].
Wood J.D., Watson W.J. Protective action of gamma-aminobutyric acid against oxygen toxicity. Nature. 195: 296. 1962.
Кричевская А.А., Шугалей В.С., Щербина Л.А., Ермоленко Г.Г. Содержание γ-аминомасляной кислоты и активность глютаматдекарбоксилазы в мозге крыс при гипербарической оксигенации и защитном действии мочевины. Вопр. мед. хим. 20(3): 294–298. 1974. [Krichevskaya A.A., Shugaley V.S., Shcherbina L.A., Ermolenko G.G. The content of γ-aminobutyric acid and the activity of glutamate decarboxylase in the brain of rats under hyperbaric oxygen and the protective action of urea. Vopr. med. chem. 20(3): 294–298. 1974. (In Russ)].
Singh A.K., Banister E.W. Effect of 6-hydroxydopamine on brain and blood catecholamine, ammonia, and amino acid metabolism in rats subjected to high pressure oxygen induced convulsions. Can. J.Physiol. Pharmacol. 56(2): 334–336. 1978.
Mialon P., Gibey R., Bigot J.C., Barthelemy L. Changes in striatal and cortical amino acid and ammonia levels of rat brain after one hyperbaric oxygen-induced seizures. Aviat. Space Environ. Med. 63(4): 287–291. 1992.
Demchenko I.T., Boso A.E., Whorton A.R., Piantadosi C.A. Nitric oxide production is enhanced in rat brain before oxygen-induced convulsions. Brain Res. 917(2): 253–261. 2001.
Zhang S., Takeda Y., Hagioka S., Takata K., Aoe H., Nakatsuka H., Yokoyama M., Morita K. Measurement of GABA and glutamate in vivo levels with high sensitivity and frequency. Brain Res. Protoc. 14(2): 61–66. 2005.
Gasier H.G., Demchenko I.T., Tatro L.G., Piantadosi C.A. S-nitrosylation of GAD65 is implicated in decreased GAD activity and oxygen-induced seizures. Neurosci. Lett. 653: 283–287. 2017.
D’Agostino D.P., Putnam R.W., Dean J.B. Superoxide (O2_) production in CA1 neurons of rat hippocampal slices exposed to graded levels of oxygen. J. Neurophysiol. 98: 1030–1041. 2007.
Thom S.R., Bhopale V., Fisher D. et al. Stimulation of nitric oxide synthase in cerebral cortex due to elevated partial pressures of oxygen: An oxidative stress response. J. Neurobiol. 51: 85–100. 2002.
Gould N., Doulias P.T., Tenopoulou M., Raju K., Ischiropoulos H. Regulation of protein function and signaling by reversible cysteine S-nitrosylation. J. Biol. Chem. 288(37): 26473–26479. 2013.
Atochin D.N., Demchenko I.T., Astern J, Boso A.E., Piantadosi C.A., Huang P.L. Contributions of endothelial and neuronal nitric oxide synthases to cerebrovascular responses to hyperoxia. J. Cereb. Blood Flow Metab. 23: 1219–1226. 2003.
Richerson G.B., Wu Y. Dynamic equilibrium of neurotransmitter transporter. Not Just for Reuptake Anymore. J. Neurophysiol. 90: 1363–1374. 2003.
Семьянов А.В. Диффузная внесинаптическая нейропередача посредством глутамата и ГАМК. Ж. Высш. Нерв. Деят. им. И.П. Павлова. 54(1): 66–82. 2004. [Semyanov A.V. Glutamate and GABA-mediated extrasynaptic diffuse signaling in the hippocampus. Zh. Vyssh. Nerv. Deiat. I P Pavlova. 54(1): 66–82. 2003. (In Russ)].
Miranda van der Zeyden, Oldenziel W.H., Rea K., Cremers T.I., Westerink B.H. Microdialysis of GABA and glutamate: Analysis, interpretation and comparison with microsensors. Pharmacol., Biochem. and Behav. 90: 135–147. 2008.
Del Arco A., Segovia G., Fuxe R., Mora F. Changes in dialysate concentrations of glutamate and GABA in the brain: An index of volume transmission mediated actions? J. Neurochem. 85: 23–33. 2003.
Tzuk-Shina T., Bitterman N., Harel D. The effect of vigabatrin on central nervous system oxygen toxicity in rats. Eur. J. Pharmacol. 202(2): 171–175. 1991.
Hall A.A., Young C., Bodo M., Mahon R.T. Vigabatrin prevents seizure in swine subjected to hyperbaric hyperoxia. J. Appl. Physiol. 115(6): 861–867. 2013
Demchenko I.T., Zhilyaev S.Y., Moskvin A.N., Krivchenko A.I., Piantadosi C.A., Allen B.W. A-ntiepileptic drugs prevent seizures in hyperbaric oxygen: A novel model of epileptiform activity. Brain Res. 1657: 347–354. 2017.
Kersante F., Rowley S., Pavlov I., Gutierrez-Mecinas M., Semyanov A., Reul J., Walker M., Linthorst A. A functional role for both γ-aminobutyric acid (GABA) transporter-1 and GABA transporter-3 in the modulation of extracellular GABA and GABAergic tonic conductances in the rat hippocampus. J. Physiol. 591(10): 2429–2441. 2013.
Дополнительные материалы отсутствуют.
Инструменты
Российский физиологический журнал им. И.М. Сеченова