

Российский физиологический журнал им. И.М. Сеченова, 2019, T. 105, № 4, стр. 416-436
Роль гуморальных факторов в дистантном прекондиционировании сердца
Е. С. Прокудина 1, Л. Н. Маслов 1, *, С. Ю. Цибульников 1, Н. Сингх 2, В. С. Клим 1, А. А. Скрябина 1
1 Научно-исследовательский институт кардиологии, Томский национальный
исследовательский медицинский центр РАН
Томск, Россия
2 Пенджабский университет Патиалы
Патиала, Пенджаб, Индия
* E-mail: maslov@cardio-tomsk.ru
Поступила в редакцию 18.09.2018
После доработки 01.02.2019
Принята к публикации 03.02.2019
Аннотация
Гуморальный путь реализации кардиопротекторного эффекта дистантного ишемического прекондиционирования (ДИПре) продемонстрирован на различных моделях ишемии-реперфузии: верхних и нижних конечностей, печени, а также окклюзии/реперфузии мезентериальных и почечных артерий. В качестве гуморальных факторов ДИПре рассматриваются: гидрофобные пептиды, опиоидные пептиды, аденозин, простаноиды, эндованилоиды, эндоканнабиноиды, кальцитонин ген-родственный пептид, лейкотриены, адреномедуллин и микроРНК. Норадреналин, аденозин, которые также принимают участие в реализации кардиопротекторного эффекта ДИПре, рассматриваются как нейрогуморальные факторы защитного действия дистантного ишемического прекондиционирования. Знания о гуморальных факторах кардиопротекции могут быть использованы для разработки методов повышения устойчивости миокарда к ишемическому-реперфузионному повреждению.
Дистантным ишемическим прекондиционированием (ДИПре) сердца принято называть повышение толерантности сердца к действию длительной ишемии-реперфузии после кратковременной ишемии-реперфузии дистантного органа [1–5] . Различают раннюю фазу, когда защитный эффект проявляется сразу же после ДИПре, и позднюю фазу, когда кардиопротекторный эффект формируется через 24 ч после ДИПре [6–8] . В механизме реализации защитного действия ДИПре принято выделять два пути: гуморальный и нейрогенный [9–11] . Преимущественная активация того или иного пути реализации кардиопротекторного эффекта ДИПре, по мнению D.A. Liem и соавт. [12] , зависит от разновидности ишемического прекондиционирования, то есть от того, какой именно орган/ткань будет подвергаться транзиторной ишемии-реперфузии. Например, защитное действие дистантного прекондиционирования миокарда посредством моделирования ишемии-реперфузии почки или кишечника достигается преимущественно за счет активации афферентных нервных путей [13, 14] , тогда как кардиопротекторный эффект, наблюдающийся после транзиторной ишемии скелетной мускулатуры, возможен благодаря активации как гуморального, так и нейрогенного пути [15] . Однако нередко бывает сложно четко разграничить гуморальный либо нервный путь реализации дистантного ишемического прекондиционирования, в таком случае уместнее говорить о нейрогуморальных факторах формирования ДИПре. Тем не менее, поиск гуморальных компонентов формирования и реализации ДИПре, исследование их взаимодействия позволят использовать эти данные в клинической практике для повышения толерантности сердца к ишемическому-реперфузионному повреждению [3] , а также для разработки новых фармакологических агентов для профилактики ишемических и реперфузионных повреждений сердца.
НЕИДЕНТИФИЦИРОВАННЫЕ ГУМОРАЛЬНЫЕ ФАКТОРЫ
Первые доказательства, указывающие на активацию гуморальных механизмов в реализации защитного действия ДИПре, были опубликованы T. Oxman и соавт. [16] . Исследователи моделировали ишемию (10 мин) и последующую реперфузию (10 мин) нижней конечности крысы с помощью перетягивания эластичного жгута вокруг задней лапы животного, а затем его ослабления. Такое воздействие в 6.5 раз снижало частоту возникновения реперфузионных желудочковых тахикардий во время региональной ишемии (30 мин) и реперфузии (15 мин) изолированного перфузируемого сердца [16] . Реализация кардиопротекторного эффекта ДИПре сопровождалась 1.5-кратным увеличением концентрации норадреналина в перфузионном растворе, оттекающем от изолированного сердца крысы в конце реперфузии [16] . При этом антиаритмический эффект ДИПре не наблюдался после введения крысам симпатолитика резерпина (0.15 мг/кг, внутрибрюшинно за 24 ч до моделирования ДИПре) [16] . Таким образом, норадреналин – один из возможных нейрогуморальных посредников, за счет которого реализуется защитное действие ДИПре на сердце. В исследовании E.W. Dickson и соавт. [17] , выполненном in vivo на кроликах, ишемическое прекондиционирование (ИПре) моделировали с помощью 5 эпизодов ишемии (5 мин) и реперфузии (5 мин) путем окклюзии/реокклюзии огибающей коронарной и почечной артерий. Перекрестная перфузия цельной кровью от кроликов-доноров (с ИПре) кроликам-реципиентам (без ИПре) уменьшала у последних соотношение зоны инфаркта/области риска (ЗИ/ОР) в 4 раза после продолжительной коронароокклюзии (60 мин) и реперфузии (30 мин) [17] . В дальнейшем этим же коллективом было проведено исследование дистантного кардиопротекторного действия ИПре in vitro [18] . На изолированных сердцах-донорах кроликов моделировали ИПре с помощью 3 циклов ишемии (5 мин) и реперфузии (10 мин); за этот период собирали перфузионный раствор, оттекающий от изолированных сердец-доноров (с ИПре). Полученный перфузионный раствор пропускали через интактные сердца-акцепторы (без ИПре), в результате чего повышалась устойчивость их сократительной активности к действию глобальной ишемии (30 мин) и реперфузии (30 мин) [18] . Эти исследования подтверждают наличие гуморальных факторов, вырабатываемых в сердце одного животного после моделирования его ИПре, способных оказывать свое защитное действие на миокард другого животного дистантно. К сожалению, в рамках данных исследований авторы не проводили идентификацию этих гуморальных факторов, однако они предполагают, что такими посредниками могут быть аденозин и/или норадреналин [18] .
В своем исследовании M. Bartekova и соавт. [19, 20] изучали участие низкомолекулярных белков в кардиопротекторном действии ДИПре, которое моделировали путем ишемии и реперфузии печени крысы в опытах in vitro и in vivo. Из перфузионного раствора, оттекающего от изолированной печени крысы после моделирования ее ишемии (30 или 60 мин) и реперфузии (120 мин), методом преципитации сульфатом аммония выделяли низкомолекулярную фракцию белков [19] . Перфузия изолированных сердец крыс раствором, содержащим выделенную фракцию белков, повышала их устойчивость к действию глобальной ишемии (20 мин) и реперфузии (20 мин): это проявлялось снижением частоты реперфузионных аритмий, улучшением постишемического восстановления сократительной функции миокарда, а также увеличением коронарного протока сердца [19] . В то же время, авторы исследования наблюдали взаимосвязь между восстановлением сократимости сердца после перфузии изолированных сердец раствором, содержащим выделенную низкомолекулярную фракцию белков, и увеличением содержания в миокарде АТФ [19] . В следующей работе исследователи моделировали глобальную ишемию печени крысы in situ путем окклюзии портальной вены и печеночной артерии (20 мин) и последующую реперфузию (30 мин) печени [20] . Кардиопротекторный эффект ДИПре оценивался на сердце, изолированном после ишемии печени [20] . ДИПре способствовало лучшему восстановлению сократительной активности миокарда после моделирования глобальной ишемии (20 мин) и реперфузии (30 мин) изолированного сердца крысы [20] . Кроме того, с помощью электрофореза белков в полиакриламидном геле удалось установить, что в плазме крови животных после моделирования ишемии печени наблюдается значительное увеличение количества белков низкомолекулярной фракции [20] . Авторы не уточняют природу этих белков, и, тем не менее, на основе результатов исследований in vivo и in vitro предполагают их роль как одного из гуморальных компонентов реализации кардиопротекторного эффекта ДИПре [19, 20] .
В исследовании F.C. Serejo и соавт. [21] было продемонстрировано дистантное кардиопротекторное действие классического ИПре сердца крысы (три цикла по 5 мин ишемии и 5 мин реперфузии). Перфузионный раствор, оттекающий от изолированного сердца крысы-донора (с моделированным ИПре), оказывал инфаркт-лимитирующий и инотропный эффекты на изолированное сердце крысы-реципиента (без ИПре) после глобальной ишемии (30 мин) и реперфузии (60 мин) [21] . Затем перфузионный раствор, собранный от крыс-доноров, был проанализирован на наличие и механизм действия гуморальных факторов, опосредующих кардиопротекцию ИПре. Во-первых, защитное действие перфузата на сердце крысы-реципиента не проявлялось в условиях блокады протеинкиназы С хелеритрином (100 мкМ, перфузия изолированного сердца за 10 мин до моделирования ИПре). Это указывает на то, что механизм кардиопротекции ИПре при участии исследуемых гуморальных факторов реализуется в условиях активации протеинкиназы С [21] . Во-вторых, дополнительный физико-химический анализ исследуемого перфузионного раствора позволил заключить, что кардиопротекторным эффектом обладает только термолабильная гидрофобная фракция белков с молекулярной массой более 3.5 кДа, механизм действия которой также связан с активацией протеинкиназы С [21] .
В 2008 г. было показано [22] , что диализат плазмы крови, полученный от кроликов-доноров, у которых моделировали ДИПре (4 цикла ишемии и реперфузии задней конечности), способен улучшать реперфузионные показатели сократительной активности миокарда (давление, развиваемое левым желудочком и конечное диастолическое давление) после кардиоплегической остановки изолированного сердца кролика-реципиента, то есть после воздействия ишемии-реперфузии. Перфузия изолированных сердец кроликов-реципиентов диализатом плазмы крови кролика-донора (с ДИПре) также повышала устойчивость миокардиальных митохондрий к действию кардиоплегии, что проявлялось увеличением скорости дыхания митохондрий в состоянии 3 (после добавления АДФ в среду инкубации), а также сохранением целостности внешней мембраны митохондрий и уменьшением высвобождения цитохрома С из этих органелл [22] . Группа G. Heusch показала, что перфузия изолированного сердца крысы раствором, содержащим плазму крови свиней после ДИПре, способствует уменьшению соотношения ЗИ/ОР, увеличивает дыхание митохондрий в состоянии 3, усиливает синтез АТФ и уменьшает продукцию активных форм кислорода этими органеллами [1] . В 2014 г. C.H. Leung и соавт. [23] показали, что защитное действие ДИПре на митохондрии миокарда может быть связано с активацией аденозиновых рецепторов, расположенных в сердце. Другому коллективу физиологов [24] удалось продемонстрировать, что механизм кардиопротекторного действия ДИПре связан с активацией АТФ-зависимых К-каналов (КАТФ-каналов) и не зависит от активации афферентных нервов, иннервирующих сердце. Эксперимент заключался в проведении трансплантации сердца от свиней-доноров свиньям-реципиентам, у которых заранее моделировали ДИПре путем перетягивания эластичного жгута вокруг задней конечности – 4 цикла ишемии (5 мин) и реперфузии (5 мин) [24] . После процедуры трансплантации у животного-реципиента моделировали окклюзию левой восходящей коронарной артерии (30 мин) и реперфузию (2 ч). ДИПре уменьшало соотношение ЗИ/ОР на 57% [24] . Инфаркт-лимитирующий эффект ДИПре не проявлялся в условиях блокады КАТФ-каналов глибенкламидом (0.5 мг/кг внутривенно за 10 мин до моделирования ДИПре) у прекондиционированных животных-реципиентов [24] .
Работа M. Shimizu и соавт. [25] также была посвящена поиску гуморальных факторов, участвующих в механизме кардиопротекции при ДИПре. Согласно данному исследованию [25] , кардиопротекторное действие ДИПре [4 цикла ишемии (5 мин) и реперфузии (5 мин) нижней конечности кролика] обусловлено циркуляцией в плазме крови гидрофобных низкомолекулярных соединений (менее 15 кДа), цитопротекторный и инфаркт-лимитирующий эффект которых не проявлялся в условиях неселективной блокады опиоидных рецепторов налоксоном (100 мкМ) [25] . В 2017 г. L. Maciel и соавт. [26] попытались определить с помощью ультрафильтрации молекулярную массу гуморального фактора. Они собирали коронарный перфузат, оттекающий от прекондиционированного сердца, и использовали его для перфузии других сердец, которые подвергали ишемии-реперфузии. Выяснилось, что инфаркт-лимитирующий эффект оказывают гидрофобные пептиды с молекулярной массой 4–12 кДа. Их кардиопротекторный эффект был связан с активацией протеинкиназы С и открытием КАТФ-каналов [26]. Один из этих пептидов был идентифицирован, он оказался heat shock protein (HSP-10).
Выше речь шла о ранней фазе ДИПре, но существует еще и поздняя фаза, которая формируется через 24 ч после ДИПре [7] . В 2017 г. появились сведения о том, что гуморальный фактор(ы) участвует в поздней фазе ДИПре [27]. У крыс моделировали ДИПре (крысы-доноры), через 48 ч их плазму трансфузировали крысам- реципиентам, у которых воспроизводили коронароокклюзию и реперфузию через час и 24 ч после переливания. В качестве контроля использовали плазму непрекондиционированных крыс. Плазма крови прекондиционированных и непрекондиционированных крыс оказывала инфаркт-лимитирующий эффект, но плазма крови прекондиционированных крыс действовала сильнее, если оценку размера инфаркта производили через 24 ч после трансфузии. Трансфузия плазмы крови прекондиционированных крыс через 24 ч после переливания вызывала повышение уровня фосфорилированных киназ Akt и ERK1/2 в миокарде крыс реципиентов, но не влияла на фосфорилирование транскрипционного фактора STAT [28]. Принято считать, что существуют два сигнальных пути, которые обеспечивают повышение толерантности сердца к ишемии-реперфузии: (1) RISK (Reperfusion-Induced Salvage Kinase), который включает киназы PI3, Akt, MEK1/2, ERK1/2; (2) SAFE (Survivor Activating Factor Enhancement), который включает киназу JAK и транскрипционный фактор STAT [29, 30]. Авторы заключили, что гуморальный фактор активирует RISK [28]. Позднее тот же коллектив авторов показал, что трансфузия плазмы прекондиционированных крыс может не только ограничивать размер инфаркта, но и предупреждать возникновение желудочковой фибрилляции при коронароокклюзии и реперфузии [31].
Клинические данные также свидетельствуют о существовании гуморального фактора(ов) [32–34]. Так, было обнаружено увеличение уровня интерлейкина-1α в крови кардиохирургических пациентов после ДИПре [32]. В раствор, которым перфузировали изолированное сердце крысы, добавляли 0.5% плазмы людей, которых подвергали ДИПре [33]. Оказалось, что плазма крови молодых добровольцев способствует уменьшению соотношения ЗИ/ОР на 34%. Плазма крови пожилых добровольцев не влияла на размер инфаркта [33]. В 2018 г. J. Ney и соавт. [34] попытались обнаружить гуморальный фактор у кардиохирургических больных с ДИПре. Оказалось, что ДИПре вызывает повышение уровня macrophage migration inhibitory factor (MIF) и stromal cell-derived factor 1 (CXCL12) в сыворотке крови. Однако ДИПре не оказывало кардиопротекторного эффекта. Следовательно, CXCL12 и MIF не могут рассматриваться в качестве гуморальных факторов.
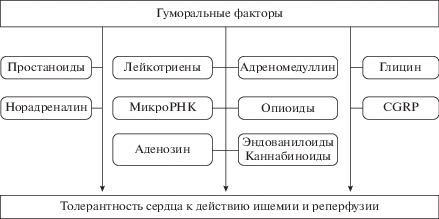
Таким образом, гуморальный путь реализации кардиопротекции при ДИПре был продемонстрирован на различных моделях прекондиционирования. Дальнейшие исследования механизма реализации ДИПре сердца дают основания предполагать, что в качестве гуморальных компонентов его формирования и реализации могут выступать такие соединения, как гидрофобные пептиды [26], эндогенные опиоиды (опиоидные пептиды) [35–40], производные арахидоновой кислоты (простаноиды) [41], агонисты каннабиноидных рецепторов [42] и эндогенные ванилоиды [43], кальцитонин ген-родственный пептид [44, 45], а также аденозин [12, 46–49], лейкотриены [50], норадреналин [51], адреномедуллин [52], глицин [53] и микроРНК [54–58].
ОПИОИДЫ
Первые доказательства участия опиоидных пептидов в реализации кардиопротекторного эффекта ДИПре были приведены в работе E.W. Dickson и соавт. [35]. Перфузия изолированных сердец кроликов-реципиентов раствором, собранным после моделирования ИПре на изолированных сердцах кроликов-доноров, уменьшала соотношение ЗИ/ОР левого желудочка в ответ на глобальную ишемию (40 мин) и последующую реперфузию (120 мин) [35]. Добавление в перфузионный раствор кроликов-реципиентов неселективного антагониста опиоидных рецепторов налоксона (в конечной концентрации 2 мкМ) устраняло инфаркт-лимитирующий эффект ИПре [35]. Гуморальные факторы, продуцируемые миокардом во время моделирования ИПре, оказывали антинекротическое действие не только на сам миокард, но и на другие органы, например, на стенку тонкого кишечника [37]. Защитное действие такой формы прекондиционирования сердца в отношении кишечника не проявлялось после блокады опиоидных рецепторов (ОР) налоксоном (5 мкМ) или блокады КАТФ-каналов глибенкламидом (5 мкМ) [37]. Кардиопротекторное действие ДИПре путем окклюзии/реперфузии мезентериальной артерии не проявлялось в условиях блокады кардиальных ОР налоксоном (10 мг/кг) [59]. По данным исследования R.Q. Xie и соавт. [60] антиапоптотическое действие на миокард ДИПре, которое моделировали с помощью окклюзии (15 мин) и реперфузии (10 мин) бедренной артерии у свиней, также связано с активацией опиоидных рецепторов. Использование ганглиоблокатора гексаметония у животных перед моделированием ДИПре не снижало проявлений антиапоптозного эффекта прекондиционирования, что в данном случае исключает участие нейрогенного механизма реализации кардиопротекторного действия [60].
Таким образом, в механизме реализации защитного действия ДИПре определенно важную роль играет активация опиоидных рецепторов эндогенными агонистами. Дальнейшие исследования в этой области были направлены на установление субтипов опиоидных рецепторов, а также их эндогенных агонистов, которые принимают участие в механизме кардиопротекторного действия ДИПре.
В исследовании [39], выполненном на модели коронароокклюзии (30 мин) и реперфузии (30 мин) сердца крысы in vivo, сообщается об участии δ1-ОР в реализации инфаркт-лимитирующего эффекта ДИПре, которое осуществляли путем окклюзии/реокклюзии почечной артерии. Защитное действие ДИПре устраняла селективная блокада δ1-ОР препаратом BNTX (3 мг/кг) [39]. Китайским физиологам удалось выявить роль κ-ОР, а также участие эндогенного агониста опиоидных рецепторов динорфина в механизме кардиопротекторного действия ДИПре [40]. Выраженность инфаркт-лимитирующего и антинекротического эффектов ДИПре, которое моделировали с помощью трех циклов окклюзии (5 мин) и реперфузии (5 мин) бедренной артерии у крыс, была сопоставима с таковыми при использовании селективных агонистов κ-ОР синтетического (U-50.488H, 10 мг/кг внутривенно за 30 мин до коронароокклюзии) и эндогенного (динорфин, 20 нг/кг внутривенно за 30 мин до коронароокклюзии) происхождения [40]. Столь высокая биологическая активность динорфина (20 нг/кг) не может не удивлять читателя, потому что опиоидные пептиды обычно ограничивают размер инфаркта в дозе несколько десятков и сотен микрограмм на кг, а U-50.488H оказывает кардиопротекторный эффект в дозе 1 мг/кг [61]. Кардиопротекторный эффект указанных воздействий не проявлялся в условиях селективной блокады κ-опиоидных рецепторов нор-биналторфимином (10 мг/кг, внутривенно за 10 мин до ДИПре или инъекции U-50.488H) [40]. У крыс, спустя 2 ч после моделирования ДИПре, также было обнаружено 3-кратное увеличение концентрации динорфина в плазме крови [40]. Следует отметить, что физиологи не зафиксировали увеличения содержания в плазме крови у крыс после ДИПре мет-энкефалина (эндогенного δ-ОР-агониста), а также продемонстрировали, что селективная блокада δ-ОР не влияла на проявление кардиопротекции при ДИПре [40]. Отличие полученных данных [40] от результатов предыдущего исследования [39], вероятно, обусловлено использованием различных моделей ДИПре.
В качестве одного из потенциальных эндогенных агонистов опиоидных рецепторов, повышающих толерантность миокарда к ишемии/реперфузии, Е.W. Dickson и соавт. [36] рассматривают пептид Met5-enkephalin-Arg6-Phe7 (MEAP). Основываясь на результатах предыдущих исследований, посвященных оценке участия ОР в реализации протекторного действия ИПре [35, 37], физиологи продолжили поиск эндогенных лигандов ОР, способных действовать как гуморальные факторы кардиопротекции. Так, было продемонстрировано, что внутримышечная инъекция MEAP (25 мг) за 24 ч до моделирования глобальной ишемии (25 мин) и реперфузии (120 мин) изолированного сердца кролика способна уменьшать соотношение ЗИ/ОР в 2.6 раза [36]. Способность MEAP оказывать кардиопротекторный эффект дистантно позволяют предполагать участие данного пептида в качестве одного из гуморальных факторов реализации ДИПре.
В исследовании H. Surendra и соавт. [38] было продемонстрировано, что обработка изолированных кардиомиоцитов кролика диализатом плазмы крови, полученным после моделирования ДИПре с помощью 4 циклов ишемии (5 мин) и реперфузии (5 мин) нижней конечности животного, в 1.5 раза снижала % некротической гибели кардиомиоцитов после их аноксии (45 мин) и реоксигенации (60 мин). Селективная блокада δ-ОР налтриндолом (10 нМ) или κ-ОР блокатором GNTI (1 нМ) перед моделированием аноксии/реоксигенации изолированных кардиомиоцитов устраняла цитопротекторное действие диализата плазмы крови, полученной от прекондиционированных кроликов [38]. Определение содержания в исследуемом диализате эндогенных агонистов ОР показало отсутствие Met5-энкефалина (агонист δ-ОР) и динорфина В (агонист κ-ОР), а также лишь небольшие количества MEAP (31 нМ), концентрация агонистов аденозиновых рецепторов – аденозина (8 нМ) и инозина (456 нМ) – также была невелика [38]. Стоит отметить, что имитация феномена прекондиционирования путем инкубации изолированных кардиомиоцитов перед аноксией/реоксигенацией с экзогенными Met5-энкефалином (100 мкМ) или динорфином (100 мкМ) не оказывала цитопротекторного действия в случае блокады аденозиновых А-рецепторов 8-(р-сульфофенил)теофиллин (8-SPT, 100 мкМ) [38]. Кроме того, селективная блокада аденозиновых А1-рецепторов препаратом 8-cyclopentyl-1,diproylxanthine (DPCPX, 10 мкМ) устраняла цитопротекторное действие инкубации изолированных кардиомиоцитов кролика с диализатом плазмы крови от прекондиционированных животных, т.е. защитное действие ДИПре также опосредуется участием аденозиновых А1-рецепторов [38]. Эти данные свидетельствуют о функциональном взаимодействии (трансактивации) опиоидных и аденозиновых рецепторов при их активации эндогенными лигандами в условиях моделирования ДИПре [62].
На примере описанных выше исследований можно заключить, что эндогенные опиоидные пептиды принимают участие в качестве гуморальных факторов в реализации кардиопротекторного действия ДИПре. Важную роль в этом механизме играет активация опиоидных δ- и κ-опиоидных рецепторов. Окончательное решение об участии конкретных эндогенных лигандов ОР в гуморальном механизме ДИПре принять сложно, так как участие того либо иного опиоида в ДИПре зависит от используемой модели прекондиционирования, а также от выбранного объекта исследования.
АДЕНОЗИН
Существует немало доказательств участия аденозиновых рецепторов в реализации кардиопротекторного действия ДИПре [12, 46–49]. Впервые об этом сообщается в работе T.J. Pell и соавт. [47]. ДИПре сердца моделировали на кроликах путем окклюзии (10 мин) и реперфузии (10 мин) левой почечной артерии [47]. Было продемонстрировано, что ДИПре способствует 2-кратному уменьшению ЗИ/ОР после моделирования коронароокклюзии (30 мин) и реперфузии (2 ч) сердца кролика in vivo [47]. Для оценки участия аденозина в реализации защитного эффекта ДИПре был использован неселективный антагонист аденозиновых рецепторов 8-SPT (7.5 мг/кг). Внутривенное введение 8-SPT кроликам за 10 мин до моделирования ДИПре устраняло способность последнего ограничивать размер инфаркта [47]. Таким образом, авторами исследования было показано, что аденозиновые рецепторы участвуют в реализации инфаркт-лимитирующего эффекта ДИПре.
В работе A. Takaoka и соавт. [49] выполняли окклюзию (10 мин) почечной артерии и реперфузию (20 мин) с последующим моделированием 40-минутной ишемии миокарда in vivo у кроликов. Классическое ИПре моделировали с помощью окклюзии коронарной артерии (5 мин) и реперфузии (20 мин) перед последующей 40-минутной коронароокклюзией. Как дистантное, так и классическое ИПре значительно снижали соотношение ЗИ/ОР у кроликов [49]. Кроме того, оценка содержания фосфокреатинина и АТФ в миокарде методом ЯМР-спектроскопии показала, что дистантное и классическое ИПре предупреждают реперфузионное снижение данных показателей у кроликов [49]. Неселективная блокада аденозиновых рецепторов 8-SPT (10 мг/кг) за 2 мин до коронароокклюзии у животных из групп с дистантным и классическим ИПре устраняла как проявление инфаркт-лимитирующего действия прекондиционирования, так и защитный эффект, направленный на сохранение энергетического метаболизма миокарда [49]. Эти данные подтверждают, что ДИПре так же, как и классическое ИПре, предупреждает нарушения энергетического метаболизма в миокарде во время ишемии и реперфузии, а также ограничивает размер инфаркта миокарда при участии аденозиновых рецепторов.
О взаимосвязи активации аденозиновых рецепторов со стимуляцией афферентных нервных волокон в условиях моделирования ДИПре сердца у крыс сообщается в исследовании D.A. Liem и соавт. [12] . ДИПре моделировали путем окклюзии верхней брыжеечной артерии (15 мин) и реперфузии (10 мин). Такое воздействие в 1.4 раза уменьшало соотношение ЗИ/ОР после коронароокклюзии (60 мин) и реперфузии (120 мин) сердца in vivo у крыс. Неселективная блокада аденозиновых рецепторов 8-SPT (50 мг/кг внутривенно) полностью устраняла инфаркт-лимитирующее действие ДИПре, в то же время селективная блокада А1-рецепторов 8-SPT (10 мг/кг внутривенно) или А3-рецепторов MRS-1191 (3.3 мг/кг внутривенно) снижала проявление кардиопротекторного эффекта ДИПре только на 65%, но не устраняла полностью [12] . Стимуляция аденозиновых рецепторов путем инфузии аденозина (10 мкг/мин) в верхнюю мезентериальную артерию оказывала кардиопротекторный эффект, сопоставимый с таковым при ДИПре, который не проявлялся при использовании ганглиоблокатора гексаметония (20 мг/кг внутривенно) [12] . Использование гексаметония на 5 мин реперфузии брыжеечной артерии не влияло на способность ДИПре уменьшать размер инфаркта миокарда, тогда как блокада всех аденозиновых рецепторов 8-SPT (50 мг/кг внутривенно) устраняла защитное действие ДИПре на сердце крысы. Использование гексаметония перед моделированием ДИПре также устраняла защитное действие прекондиционирования [12] . Аналогичным действием обладала блокада миокардиальных аденозиновых рецепторов: неселективный антагонист А-рецепторов 8-SPT (50 мг/кг внутривенно) устранял инфаркт-лимитирующий эффект ДИПре в то время как селективная блокада А1-рецепторов 8-SPT (10 мг/кг внутривенно) или А3-рецепторов MRS-1191 (3.3 мг/кг внутривенно) снижала его проявление до 61 ± 2% и 53 ± 3% соответственно [12] . В совокупности эти данные указывают на то, что локальная активация аденозиновых рецепторов во время окклюзии верхней брыжеечной артерии стимулирует афферентные нервные волокна в мезентериальной области во время последующей реперфузии. Это событие, в свою очередь, приводит к активации миокардиальных аденозиновых рецепторов, что и опосредует реализацию кардиопротекторного действия ДИПре [12] . Следовательно, аденозин выступает как фактор нейрогуморальной регуляции механизма кардиопротекции при ДИПре.
Об участии аденозина как фактора нейрогуморальной регуляции ДИПре сообщается в работе J.H. Dong и соавт. [46]. Дистантное прекондиционирование они воспроизводили путем ишемии/реперфузии нижней конечности крысы. ДИПре повышало устойчивость миокарда к действию коронароокклюзии (30 мин) и реперфузии (120 мин). Селективная блокада аденозиновых А1-рецепторов с помощью антагониста 8-cyclopentyl-1,diproylxanthine (DPCPX, 32 нМ/кг) частично устраняла кардиопротекторный эффект ДИПре [46]. В 2004 г. G. Schulte и соавт. показали [48], что отсроченное (через 24 ч) ДИПре воспроизводили с помощью транзиторной ишемии головного мозга мыши путем двусторонней перевязки внутренних сонных артерий. Концентрация эндогенного аденозина в крови мышей после ДИПре возрастала в 2.5 раза спустя 30 мин после перевязки сонных артерий [48]. Через 24 ч после подобного ДИПре изолировали сердце и подвергали его ишемии и реперфузии. Оказалось, что ДИПре повышает толерантность сердца к ишемии-реперфузии. Кардиопротекторный эффект ДИПре не проявлялся у мышей, нокаутированных по гену аденозиновых А1-рецепторов (A1R–/–). Следует отметить, что период полужизни аденозина в цельной крови сравнительно невелик и составляет менее 10 с [63], очевидно поэтому аденозин оказывает инфаркт-лимитирующий эффект, если его инфузируют в течение трех часов [64], что ставит под сомнение его роль как гуморального фактора прекондиционирования. Возможно, что аденозин принимает участие в повышении толерантности сердца под действием гуморального фактора, выступая в роли посредника между гуморальным фактором и кардиомиоцитами. Инфаркт-лимитирующий эффект аденозина связывают с активацией киназ (MEK1/2, ERK1/2, Akt, тирозинкиназы, протеинкиназа С) и КАТФ-каналов [5] .
Таким образом, несмотря на очевидные доказательства участия активации аденозиновых рецепторов в механизме кардиопротекции при ДИПре, говорить о самом аденозине как гуморальном факторе, нужно с долей сомнения.
ПРОСТАНОИДЫ
Впервые исследование участия простаноидов в реализации защитного механизма ДИПре обсуждается в работе T. Oxman и соавт. [16] . Прекондиционирование воспроизводили на крысах путем моделирования 10-минутной ишемии задней конечности с помощью перетягивания вокруг нее эластичного жгута и 10-минутной реперфузии с последующей изоляцией сердца. На модели региональной ишемии (30 мин) и реперфузии (15 мин) изолированного перфузируемого сердца крысы исследователи продемонстрировали кардиопротекторный и антиаритмический эффекты ДИПре [16] . Авторы сообщают, что исходная концентрация простациклина (ПГI2) в перфузате, оттекающем от сердца прекондиционированных крыс, в 1.35 раза выше, чем в группе контрольных крыс без моделирования ДИПре [16] . В то же время моделирование региональной ишемии изолированного сердца приводило к значительному снижению содержания простациклина в перфузионном растворе, оттекающем от сердца. Однако к окончанию первой минуты реперфузии изолированного прекондиционированного сердца крысы наблюдалось пиковое увеличение концентрации ПГI2 в перфузионном растворе, которая затем снижалась к концу реперфузии [16] . Схожая динамика высвобождения простациклина из миокарда наблюдалась и в группе контрольных крыс, причем процентные изменения концентрации ПГI2 не отличались от таковых в группе животных с ДИПре. Таким образом, исследователям не удалось выявить взаимосвязь между динамикой высвобождения простациклина в период реперфузии из миокарда крыс после ДИПре и его способностью уменьшать частоту возникновения реперфузионных тахиаритмий [16] .
Исследование R. Sharma и соавт. [41] было посвящено изучению роли тромбоксана А2 в реализации кардиопротекторного эффекта ДИПре. Прекондиционирование моделировали с помощью четырех циклов надувания (5 мин) и сдувания (5 мин) манжеты, расположенной вокруг задней конечности крысы. Такое воздействие способствовало ограничению размера инфаркта, снижению степени некроза миокарда и улучшению постишемического восстановления сократительной активности изолированного сердца крысы в ответ на моделирование глобальной ишемии (30 мин) и реперфузии (120 мин) изолированного сердца крысы [41]. Для оценки участия тромбоксана А2 в реализации кардиопротекторного эффекта ДИПре использовалось внутрибрюшинное введение крысам препаратов озагрел (ингибитор тромобксансинтазы, 15 и 30 мг/кг) и сератродаст (антагонист рецепторов тромбоксана А2, 15 и 30 мг/кг) за 30 мин до моделирования ДИПре. Ингибирование тромбоксансинтазы, а также блокада рецепторов тромобоксана А2 устраняли проявления защитных эффектов ДИПре, что свидетельствует об участии тромбоксана А2 в реализации кардиопротекторного эффекта дистантного ишемического прекондиционирования [41]. Следует отметить, что роль тромбоксана А2 в ДИПре пока не подтверждена другими исследователями. Вместе с тем, время его полужизни (half-life) в крови составляет 30–47 с [65], поэтому он может претендовать на роль гуморального фактора дистанционного прекондиционирования.
Таким образом, об участии простаноидов в реализации кардиопротекторного эффекта ДИПре еще накоплено мало данных, и требуются дальнейшие исследования, однако на основании результатов исследований роли простаноидов в реализации классического ИПре [66] можно предполагать, что данная группа биологически активных веществ может принимать участие в реализации кардиопротекторного действия дистантного ишемического прекондиционирования.
КАННАБИНОИДЫ И ЭНДОВАНИЛОИДЫ
Об участии эндогенных каннабиноидов и каннабиноидных рецепторов в механизме защитного действия ДИПре сообщается в исследовании A.R. Hajrasouliha и соавт. [42]. Прекондиционирование сердца моделировали у крыс с помощью окклюзии мезентериальной артерии (15 мин) и последующей реперфузией (15 мин) [42]. Блокада СВ2-рецепторов препаратом AM630 (1 мг/кг) за 15 мин до окклюзии мезентериальных артерий устраняла инфаркт-лимитирующий и антиаритмический эффекты ДИПре в ответ на моделирование коронароокклюзии (30 мин) и реперфузии (2 ч) [42]. Однако в случае использования антагониста СВ1-каннабиноидных рецепторов АМ251 (1 мг/кг) за 15 мин до окклюзии мезентериальных артерий кардиопротекторный эффект ДИПре сохранялся [42]. Таким образом, в реализации кардиопротекторного действия ДИПре, который проявляется ограничением размера инфаркта и снижением частоты возникновения реперфузионных желудочковых тахикардий и желудочковых фибрилляций, принимают участие эндогенные агонисты каннабиноидных СВ2-рецепторов, но не агонисты СВ1-рецепторов [42]. К сожалению авторы не проводили исследование содержания эндогенных каннабиноидов в сыворотке крови у крыс, подвергавшихся ДИПре.
Еще одной группой гуморальных факторов, опосредующих реализацию кардиопротекторного эффекта ДИПре, являются эндованилоиды – агонисты TRPV каналов (transient receptor potential cation channels for vanilloid, TRPV). TRPV-каналы являются механочувствительными ионными каналами, проводящими Na+ и Ca2+ [67], которые локализуются на сарколемме кардиомиоцитов [68, 69], на эндотелиальных клетках аорты, предсердий, желудочков и коронарных артерий [70–72], а также на окончаниях афферентных нервов в сердце, скелетной мускулатуре и других органах [73–75]. В работе P.K. Randhawa и соавт. [43] было продемонстрировано, что ДИПре с помощью четырех 5-минутных эпизодов инфляции-дефляции манжеты, расположенной на задней конечности крысы, способствует снижению степени постишемического некроза кардиомиоцитов, улучшает реперфузионное восстановление сократительной функции миокарда, а также уменьшает размер инфаркта в ответ на моделирование глобальной ишемии (30 мин) и реперфузии (120 мин) изолирован действия ДИПре использовался их блокатор рутений красный (4 или 8 мг/кг внутрибрюшинно за 30 мин до моделирования ДИПре). Исследование показало, что блокада TRPV-каналов устраняла антинекротическое, инотропное и инфаркт-лимитирующее действие ДИПре [43, 44]. Такой же эффект оказывал антагонист TRPV1-каналов капсазепин [76]. Активатор TRPV1-каналов капсаицин имитировал кардиопротекторный эффект ДИПре [77], поэтому можно утверждать, что активация TRPV1-каналов эндованилоидами играет важную роль в реализации кардиопротекторного эффекта дистантного ишемического прекондиционирования. Есть данные, что эти каналы участвуют в дистантном посткондиционировании [78].
Известно, что некоторые эндогенные каннабиноиды, например, анандамид и 2-арахидоноилглицерол, одновременно являются эндованилоидами [79]. Данные о фармакокинетике анандамида отсутствуют, но известно, что его гипотензивный эффект сохраняется в течение нескольких минут после внутривенного введение [79]. Установлено, что через 5 мин после добавления в плазму крови 2-арахидоноилглицерола его уровень в плазме составляет около 10% от исходного количества [80]. Представленные данные говорят о том, что у эндоканнабиноидов при внутривенном введении достаточно времени для того, чтобы оказать существенное влияние на сердце. Подобное утверждение справедливо, если дистантный орган секретирует эндоканнабиноиды 5–0 мин.
Таким образом, несмотря на немногочисленные данные о том, что формирование кардиопротекторного действия ДИПре зависит от активации каннабиноидных и ваниллоидных (TRPV1) рецепторов, все-таки можно говорить об участии эндогенных агонистов данных рецепторов в гуморальном механизме кардиопротекции при дистантном ишемическом прекондиционировании.
КАЛЬЦИТОНИН ГЕН-РОДСТВЕННЫЙ ПЕПТИД (CGRP)
Кальцитонин ген-родственный пептид (calcitonin gene related peptide, CGRP) является представителем семейства кальцитониновых пептидов, содержится в основном в капсаицин-чувствительных нервных окончаниях и существует в двух изоформах – α-CGRP и β-CGRP [81, 82]. Рецепторы к кальцитонин ген-связанному пептиду CGRP1 и CGRP2, которые относятся к семейству G-белок-сопряженных рецепторов и обнаружены в различных органах, преимущественно в нервной ткани головного и спинного мозга, а также на эндотелиальных клетках артерий, в том числе коронарных, и в кардиомиоцитах желудочков сердца и предсердий [83]. Высвобождение CGRP из афферентных нервных волокон стимулируется различными факторами, в том числе ишемией, снижением pH или лактат-ацидозом [84, 85]. Освобождение CGRP из афферентных нервных терминалей может происходить в ответ на стимуляцию TRPV1-каналов [86]. В частности, A.J. D’Alonzo и соавт. [87] показано, что перфузия изолированного сердца крысы раствором, содержащим 30 мкМ капсаицина, который способен стимулировать высвобождение CGRP из капсаицин-чувствительных нервных окончаний, оказывает антиаритмический эффект и способствует лучшему восстановлению сократительной функции миокарда после глобальной ишемии и реперфузии in vitro. В исследовании R. Lu и соавт. [88] продемонстрирована важная роль CGRP в реализации кардиопротекторного действия классического ИПре сердца. При этом блокада CGRP-рецепторов антагонистом CGRP8-37 устраняла проявления защитного эффекта классического ИПре сердца [88].
Участие CGRP как гуморального фактора реализации кардиопротекторного действия ДИПре было продемонстрировано S. Wolfrum и соавт. [45]. ДИПре моделировали у крыс с помощью окклюзии мезентериальной артерии (15 мин) и последующей реперфузии (15 мин). Кардиопротекторный эффект прекондиционирования проявлялся уменьшением ЗИ/ОР в 2.4 раза после коронароокклюзии (30 мин) и реперфузии (150 мин) in vivo и не наблюдался после селективной блокады CGRP-рецепторов антагонистом CGRP8-37 (3 нМ/кг = 9.38 мкг/кг внутривенно за 5 мин до ДИПре) [45]. Исследование концентрации CGRP в сыворотке крови у крыс после ДИПре показало ее увеличение на 40% по сравнению с исходными значениями (до моделирования ДИПре). Стоит отметить, что использование ганглиоблокатора гексаметония (20 мг/кг внутривенно после ДИПре) не влияло на концентрацию CGRP в сыворотке крови у крыс после ДИПре [45]. Этот факт подтверждает участие CGRP как гуморального фактора реализации ДИПре сердца.
В исследовании A. Singh и соавт. [44] также изучалась роль CGRP в реализации кардиопротекции при ДИПре. Прекондиционирование осуществляли с помощью 4 циклов ишемии (5 мин) и реперфузии (5 мин) задней конечности крыс, после чего изолировали сердце животного и моделировали его глобальную ишемию (30 мин) и реперфузию (120 мин) [44]. ДИПре способствовало снижению соотношения ЗИ/ОР в 1.7 раз, улучшало реперфузионное восстановление сократимости миокарда, а также оказывало антинекротическое действие после ишемии/реперфузии изолированного сердца у крыс [44]. Все перечисленные проявления кардиопротекторного действия ДИПре не наблюдались в случае использования препарата суматриптан (8 мг/кг внутрибрюшинно за 30 мин до моделирования ДИПре), который применялся с целью блокирования высвобождения в кровь CGRP [44]. Антагонист CGRP рецепторов CGRP8-37 (1 мг/кг) также устранял кардиопротекторный эффект ДИПре [86], В совокупности эти данные подтверждают участие CGRP как гуморального фактора в реализации кардиопротекторного действия дистантного ишемического прекондиционирования.
ЛЕЙКОТРИЕНЫ
В работе B. Singh и соавт. [50] было проведено исследование участия лейкотриенов в реализации кардиопротекторного эффекта ДИПре. Рецепторы к лейкотриенам обнаружены в различных органах: сердце, бронхи, спинномозговые нервы, кишечник [89–91]. ДИПре моделировали на крысах с помощью раздувания/сдувания манжеты, расположенной вокруг задней конечности животного (четыре цикла 5 мин ишемии и 5 мин реперфузии) [50]. Кардиопротекторный эффект ДИПре оценивался на изолированном перфузируемом сердце крысы, которое подвергалось глобальной ишемии (30 мин) и реперфузии (120 мин). Прекондиционирование способствовало 4-х-кратному снижению соотношения ЗИ/ОР, уменьшало постишемический некроз кардиомиоцитов, ДИПре улучшало показатели реперфузионного восстановления сократительной функции миокарда (частота сердечных сокращений, давление, развиваемое левым желудочком, а также максимальные скорости сокращения и расслабления миокарда) [50]. Авторы использовали препарата монтелукаст – антагонист рецепторов цистеиновых лейкотриенов ЛТС4, ЛТD4, ЛТЕ4. Препарат вводили в дозе 10 или 20 мг/кг внутрибрюшинно за 30 мин до ДИПре. Монтелукаст устранял все проявления кардиопротекторного действие прекондиционирования [50]. Аналогичный эффект на сердце оказал препарат зилеутон, ингибитор 5-липооксигеназы, который вводили в дозе 2.5 или 5 мг/кг внутрибрюшинно за 30 мин до ДИПре. Он также устранял вышеперечисленные проявления кардиопротекции прекондиционирования [50]. Эти данные указывают на важную роль лейкотриенов в реализации кардиопротекторного эффекта ДИПре. Время полужизни лейкотриена С4 в крови составляет 79 с [92]. Следовательно, есть основания предполагать, что лейкотриены являются гуморальными факторами, участвующими в реализации защитного действия ДИПре.
НОРАДРЕНАЛИН
Гипотезу о роли норадреналина как гуморального фактора в 2000 г. выдвигали E.W. Dickson и соавт. [18] . Действительно, увеличение уровня норадреналина в крови крыс после ДИПре обнаружили D.W. McBride и соавт. [51]. Следует отметить, что стимуляция β-адренорецепторов в период, предшествующий ишемии, может способствовать повышению толерантности сердца к действию ишемии-реперфузии [93, 94], поэтому норадреналин может претендовать на роль нейрогенного и гуморального фактора, играющего важную роль в ДИПре.
АДРЕНОМЕДУЛЛИН
В 2018 г. W. Dong и соавт. [52] обнаружили, что через 24 ч после ДИПре в крови крыс увеличивается уровень адреномедуллина, сразу же после ДИПре изменений концентрации адреномедуллина в крови животных обнаружить не удалось. Известно, что адреномедулин уменьшает соотношение ЗИ/ОР [95]. Следовательно, есть основания предполагать, что адреномедулин может участвовать в отсроченном ДИПре.
ГЛИЦИН
Глицин является еще одним кандидатом на роль гуморального фактора, участвующего в ДИПре. У свиней воспроизводили ДИПре, которое приводило к увеличению в 10 раз уровня глицина в плазме крови [53]. Диализат плазмы крови использовали для перфузии изолированного сердца мыши, которое подвергали воздействию ишемии-реперфузии. Диализат вызывал уменьшение соотношения ЗИ/ОР на 18%, такой же эффект оказывал глицин. Антагонист рецепторов глицина стрихнин устранял протекторный эффект диализата [53]. Мы хотели обратить внимание читателя на слабый инфаркт-лимитирующий эффект диализата, что, на наш взгляд, ставит под сомнение роль глицина как гуморального фактора.
ЭКЗОСОМЫ И МикроРНК
Экзосомами принято называть микровезикулы чашеобразной формы диаметром 50–150 нм, которые секретируются большинством клеток млекопитающих [96, 97]. Экзосомы обеспечивают межклеточную сигнализацию и содержатся во всех жидкостях организма (плазма крови, спинномозговая жидкость, слюна, моча и др.) [97, 98]. Экзосомы переносят липиды, белки [98], мРНК [99], микроРНК [99–101].
В качестве еще одной группы гуморальных факторов, опосредующих реализацию кардиопротекторного эффекта ДИПре, рассматривают микроРНК. МикроРНК принято считать небольшие некодирующие последовательности РНК (20–23 нуклеотидов), которые подавляют экспрессию различных генов в эукариотических клетках [102]. Образование микроРНК происходит в результате транскрипции РНК ферментом РНК-полимеразой II [103], последовательного процессинга первичных транскриптов комплексами Drosha и DGCR8 (внутри ядра клетки) [104], миграции предшественников микроРНК (пре-микроРНК, 70 нуклеотидов) из ядра в цитоплазму с помощью белка exportin-5 [103]. В цитоплазме пре-микроРНК гидролизуются РНКазой III и затем связываются с комплексом RISC (microRNA-induced silencing complex), формируя зрелую молекулу микроРНК. По мнению некоторых авторов [105, 106], зрелая микроРНК вместе с комплексом белков RISC, связываясь с комплементарными сайтами на мРНК, блокирует трансляцию мРНК. Благодаря этой способности, микроРНК принимают участие в регуляции ряда клеточных функций и процессов, таких как рост, дифференцировка, пролиферация, метаболизм и апоптоз [107, 108].
Принято считать, что свое защитное действие микроРНК (миРНК) реализуют путем миграции с током крови к клеткам-мишеням внутри экзосом (микровезикул) [109]. Микровезикулы, в свою очередь, могут высвобождаться из клеток, ткань которых подвергалась кратковременной транзиторной ишемии/реперфузии [109]. В сердце источниками экзосом могут быть кардиомиоциты [110], фибробласты [111], а также эндотелиальные клетки сосудов [112 ] . Впервые Y. Zheng и соавт. [113 ] сообщили, что совместная инкубация кардиомиоцитов с эндотелиальными клетками линии HUVEC (Human umbilical vein endothelial cells) уменьшает последующую гибель кардиомиоцитов после моделирования аноксии/реоксигенации in vitro с 80 ± 11% до 51 ± 4% [113 ] . Предварительная инкубация кардиомиоцитов с экзосомами, полученными в результате центрифугирования инкубационной среды клеток HUVEC, также уменьшала гибель кардиомиоцитов после воздействия аноксии/реоксигенации с 88 ± 4% до 55 ± 3% [113 ] . Интересно, что прекондиционирование эндотелиоцитов HUVEC путем предварительного воздействия аноксии/реоксигенации (30 мин) способствовало двукратному увеличению продукции экзосом этой линией клеток [113 ] . При этом инкубация кардиомиоцитов с экзосомами, полученными от “прекондиционированных” клеток HUVEC, уменьшала процент гибели клеток миокарда до 45 ± 10% [113 ] . Таким образом, это первые данные, указывающие на то, что эндотелиальные экзосомы способны повышать устойчивость кардиомиоцитов к аноксии/реоксигенации [113 ] . Спустя год, T. Baranyai и соавт. сообщили [114 ] , что перфузионный раствор, оттекающий от изолированных сердец крыс-доноров, подвергавшихся ИПре [3 цикла глобальной ишемии (5 мин) и реперфузии (5 мин)], способен повышать устойчивость изолированных сердец интактных крыс-реципиентов к действию глобальной ишемии (30 мин) и реперфузии (120 мин). В случае перфузии изолированных сердец крыс-реципиентов перфузатом, лишенным экзосом (они удалялись из раствора предварительным центрифугированием), кардиопротекторное действие ИПре не наблюдалось [114 ] . Кроме того, иммуноблоттинг-анализ, в котором определялась экспрессия белка теплового шока 60 (HSP60, связан с мембраной исследуемых экзосом и является их маркером) показал значительное увеличение содержания экзосом в перфузате после моделирования ИПре изолированного сердца у крыс [114 ] . Таким образом, в данных исследованиях впервые продемонстрировано участие экзосом в реализации кардиопротекторного действия ДИПре.
В исследовании X. Duan и соавт. [55] ДИПре у крыс [3 цикла окклюзии (5 мин) и реперфузии (5 мин) бедренных артерий] ограничивало размер инфаркта миокарда (после 30 мин глобальной ишемии и 60 мин реперфузии изолированного сердца крысы), а также в 2 раза уменьшало количество TUNEL-позитивных кардиомиоцитов, что характеризует снижение количества клеток, подвергающихся апоптозу. Такое защитное действие ДИПре характеризовалось снижением экспрессии миРНК-1 в миокарде у прекондиционированных крыс [55]. Экспрессия белка теплового шока 70 (HSP70), активность гена которого регулируется миРНК-1, снижалась в миокарде левого желудочка у крыс после моделирования ДИПре [55]. Исследование экспрессии миРНК-21 у крыс после ДИПре не показало достоверных различий с крысами из контрольной группы (без ДИПре) [55]. Однако при этом экспрессия проапоптотического белка PDCD4 (programmed cell death protein 4, его экспрессию подавляет микроРНК-21) в миокарде левого желудочка снижалась в 1.5 раза у прекондиционированных особей [55]. В опубликованной двумя годами позже работе T. Brandenburger и соавт. [54] сообщается, что моделирование ДИПре у крыс с помощью 4 циклов ишемии (5 мин) и реперфузии (5 мин) задней конечности, во-первых, оказывает инфаркт-лимитирующий эффект при коронароокклюзии (35 мин) и реперфузии (2 ч или 6 ч) in vivo; во-вторых, характеризуется, спустя 2 ч реперфузии миокарда, уменьшением экспрессии миРНК-1 в миокарде, а спустя 6 ч реперфузии – увеличением экспрессии миРНК-1 в миокарде. К сожалению, в исследовании in vivo авторами работы не было обнаружено корреляции между экспрессией миРНК-1 и содержанием в миокарде белка BDNF (brain-derived neurotrophic factor protein), обладающего кардиопротекторным действием, и экспрессия которого регулируется миРНК-1 [54].
В исследовании, выполненном на крысах с ДИПре, было установлено, что сыворотка, обогащенная экзосомами, повышает толерантность кардиомиоцитов HL-1 к гипоксии и реоксигенации [115 ] . В исследовании J. Li и соавт. [57] сообщается, что моделирование ДИПре с помощью 4 циклов ишемии (5 мин) и реперфузии (5 мин) путем перевязки нижней конечности у мышей или раздувания манжеты, расположенной на нижней конечности у людей, сопровождалось значительным увеличением содержания миРНК-144 в плазме крови, в то время как моделирование глобальной ишемии (30 мин) и последующей реперфузии (60 мин) изолированного сердца мыши приводило к значительному снижению экспрессии миРНК-144 в миокарде. Моделирование ДИПре у мышей, а также внутривенное введение миРНК-144 (8 мг/кг) оказывали инфаркт-лимитирующий эффект [57]. В то же время внутривенное введение антисенс-олигонуклеотида миРНК-144 (8 мг/кг) устраняло кардиопротекторное действие как использования самой миРНК-144, так и ДИПре [57]. Стоит отметить, что наряду с увеличением уровня миРНК-144 в плазме крови после моделирования ДИПре, исследователям не удалось обнаружить изменения в содержании микровезикул в плазме крови, а также увеличения концентрации миРНК-144 внутри самих экзосом [57]. Однако в гранулах экзосом было обнаружено почти четырехкратное увеличение содержания предшественника миРНК-144, а также белка-переносчика миРНК argonaute-2 [57]. Следовательно, увеличение экспрессии миРНК-144 играет важную роль в механизме кардиопротекции при ДИПре. Установлено, что ДИПре вызывает у детей повышение уровня миРНК-21 в крови и моче [56]. Авторы полагают, что протекторный эффект миРНК-21 связан с изменением экспрессии киназы p38 [56]. Показано, что ДИПре вызывает увеличение уровня миРНК-24 в экзосомах, выделенных из плазмы крови адаптированных крыс [58]. В исследовании, выполненном in vitro на кардиомиобластах H9c2, было показано, что миРНК-24 ингибирует H2O2-индуцированный апоптоз и некроз H9c2-клеток. Крыс подвергали коронароокклюзии (45 мин) и реперфузии (24 ч). Интрамиокардиальная инъекция ДИПре-экзосом способствовала уменьшению соотношения ЗИ/ОР и улучшала насосную функцию сердца [56, 58]. Кроме того, ДИПре-экзосомы препятствовали апоптозу кардиомиоцитов. Экзосомы, выделенные из крови неадаптированных крыс, не оказывали подобных эффектов. Защитный эффект миРНК-24 связывают с даунрегуляцией проапоптотического белка Bim [58]. Остается неясным механизм антинекротического действия экзосом.
ДИПре может вызывать не только увеличение уровня микроРНК, но и снижать уровень микроРНК [116 ] . ДИПре воспроизводили с помощью ишемии-реперфузии задних конечностей. Оказалось, что ДИПре вызывает снижение уровня миРНК-29а/b/c в скелетных мышцах. Указаная миРНК ингибирует экспрессию индуцибельной NO-синтазы (iNOS) [116 ] . Авторы полагают, что снижение уровня миРНК-29а/b/c в плазме крови способствует усилению экспрессии iNOS в дистантных органах и обеспечивает геапатопротекторный эффект ДИПре.
Таким образом, микровезикулы (экзосомы), содержимое которых представлено различными белками, а также мРНК, миРНК могут служить важным гуморальным компонентом реализации кардиопротекторного эффекта ДИПре. В отношении молекулярных мишеней миРНК пока полной ясности нет, в связи с чем необходимы дополнительные исследования.
ЗАКЛЮЧЕНИЕ
Реализация кардиопротекторного действия дистантного ишемического прекондиционирования возможна за счет высвобождения в кровь циркулирующих гуморальных факторов, передающих сигнал от органа, который подвергался кратковременной транзиторной ишемии, к органу-мишени (сердцу). Первые доказательства подобного “переноса защитного сигнала” появились в экспериментах с перекрестной перфузией сердца кровью от организма-донора (с моделированным ишемическим прекондиционированием) к организму-реципиенту [17] . Гуморальный путь реализации кардиопротекции при ДИПре продемонстрирован на различных моделях прекондиционирования. В качестве основных гуморальных компонентов реализации ДИПре выступают: гидрофобные пептиды, эндогенные опиоиды, эндоканнабиноиды, эндованилоиды, простаноиды, норадреналин, адреномедуллин, лейкотриены, а также кальцитонин ген-связанный пептид и микроРНК как компонент содержимого экзосом. Несмотря на очевидные доказательства участия активации аденозиновых рецепторов в механизме кардиопротекции при ДИПре, о самом аденозине скорее уместно говорить, как о посреднике между гуморальным фактором и кардиомиоцитами. Дальнейшие исследования гуморальных факторов, участвующих в реализации кардиопротекторного действия ДИПре, позволят разработать лекарственные средства, повышающие устойчивость сердца к действию ишемии-реперфузии.
Список литературы
Gedik N., Maciel L., Schulte C., Skyschally A., Heusch G., Kleinbongard P. Cardiomyocyte mitochondria as targets of humoral factors released by remote ischemic preconditioning. Arch. Med. Sci. 13(2): 448–458. 2017.
Heusch G., Rassaf T. Time to give up on cardioprotection? A critical appraisal of clinical studies on ischemic pre-, post-, and remote conditioning. Circ. Res. 119(5): 676–695. 2016.
Heusch G. Remote ischemic conditioning in cardiovascular surgery. J. Cardiovasc. Pharmacol. Ther. 22(4): 297–301. 2017.
Kleinbongard P., Skyschally A., Gent S., Pesch M., Heusch G. STAT3 as a common signal of ischemic conditioning: A lesson on “rigor and reproducibility” in preclinical studies on cardioprotection. Basic Res. Cardiol. 113(1): 3. 2017.
Singh L., Kulshrestha R., Singh N., Jaggi A. S. Mechanisms involved in adenosine pharmacological preconditioning-induced cardioprotection. Korean J. Physiol. Pharmacol. 22(3): 225–234. 2018.
Huda R., Chung D.H., Mathru M. Ischemic preconditioning at a distance: Altered gene expression in mouse heart and other organs following brief occlusion of the mesenteric artery. Heart Lung Circ. 14(1): 36–43. 2005.
Wagner R., Piler P., Bedanova H., Adamek P., Grodecka L., Freiberger T. Myocardial injury is decreased by late remote ischaemic preconditioning and aggravated by tramadol in patients undergoing cardiac surgery: A randomised controlled trial. Interact. Cardiovasc. Thorac. Surg. 11(6): 758–762. 2010.
Xiao L., Lu R., Hu C.P., Deng H.W., Li Y.J. Delayed cardioprotection by intestinal preconditioning is mediated by calcitonin gene-related peptide. Eur. J. Pharmacol. 427(2): 131–135. 2001.
Abdul-Ghani S., Fleishman A.N., Khaliulin I., Meloni M., Angelini G.D., Suleiman M.S. Remote ischemic preconditioning triggers changes in autonomic nervous system activity: Implications for cardioprotection. Physiol. Rep. 5(3): pii: e13085. 2017.
Aulakh A.S., Randhawa P.K., Singh N., Jaggi A.S. Neurogenic pathways in remote ischemic preconditioning induced cardioprotection: Evidences and possible mechanisms. Korean J. Physiol. Pharmacol. 21(2): 145–152. 2017.
Gill R., Kuriakose R., Gertz Z.M., Salloum F.N., Xi L., Kukreja R.C. Remote ischemic preconditioning for myocardial protection: Update on mechanisms and clinical relevance. Mol. Cell Biochem. 402(1–2): 41–49. 2015.
Liem D.A., Verdouw P.D., Ploeg H., Kazim S., Duncker D.J. Sites of action of adenosine in interorgan preconditioning of the heart. Am. J. Physiol. Heart Circ. Physiol. 283(1): H29–H37. 2002.
Gho B.C., Schoemaker R.G., van den Doel M.A., Duncker D.J., Verdouw P.D. Myocardial protection by brief ischemia in noncardiac tissue. Circulation. 94(9): 2193–2200. 1996.
Liem D.A., Verdouw P.D., Duncker D.J. Transient limb ischemia induces remote ischemic preconditioning in vivo. Circulation. 107(24): e218–e219. 2003.
Weinbrenner C., Nelles M., Herzog N., Sarvary L., Strasser R.H. Remote preconditioning by infrarenal occlusion of the aorta protects the heart from infarction: A newly identified non-neuronal but PKC-dependent pathway. Cardiovasc. Res. 55(3): 590–601. 2002.
Oxman T., Arad M., Klein R., Avazov N., Rabinowitz B. Limb ischemia preconditions the heart against reperfusion tachyarrhythmia. Am. J. Physiol. 273(4 Pt 2): H1707–H1712. 1997.
Dickson E.W., Reinhardt C.P., Renzi F.P., Becker R.C., Porcaro W.A., Heard S.O. Ischemic preconditioning may be transferable via whole blood transfusion: Preliminary evidence. J. Thromb. Thrombolysis. 8(2): 123–129. 1999.
Dickson E.W., Porcaro W.A., Fenton R.A., Heard S.O., Reinhardt C.P., Renzi F.P., Przyklenk K. “Preconditioning at a distance” in the isolated rabbit heart. Acad. Emerg. Heart. 7(4): 311–317. 2000.
Bartekova M., Styk J., Pancza D., Kukan M., Sebokova J., Breier A. Proteins released from liver after ischaemia induced an elevation of heart resistance against ischaemia-reperfusion injury: 1. Beneficial effect of protein fraction isolated from perfusate after ischaemia and reperfusion of liver. Gen. Physiol. Biophys. 22(4): 567–577. 2003.
Bartekova M., Sulova Z., Pancza D., Ravingerova T., Stankovicova T., Styk J., Breier A. Proteins released from liver after ischaemia induced an elevation of heart resistance against ischaemia-reperfusion injury: 2. Beneficial effect of liver ischaemia. Gen. Physiol. Biophys. 23(4): 489–497. 2004.
Serejo F.C., Rodrigues L.F., da Silva Tavares K.C., de Carvalho A.C., Nascimento J.H. Cardioprotective properties of humoral factors released from rat hearts subject to ischemic preconditioning. J. Cardiovasc. Pharmacol. 49(4): 214–220. 2007.
Wang L., Oka N., Tropak M., Callahan J., Lee J., Wilson G., Redington A., Caldarone C.A. Remote ischemic preconditioning elaborates a transferable blood-borne effector that protects mitochondrial structure and function and preserves myocardial performance after neonatal cardioplegic arrest. J. Thorac. Cardiovasc. Surg. 136(2): 335–342. 2008.
Leung C.H., Wang L., Nielsen J.M., Tropak M.B., Fu Y.Y., Kato H., Callahan J., Redington A.N., Caldarone C.A. Remote cardioprotection by transfer of coronary effluent from ischemic preconditioned rabbit heart preserves mitochondrial integrity and function via adenosine receptor activation. Cardiovasc. Drugs. Ther. 28(1): 7–17. 2014.
Konstantinov I.E., Li J., Cheung M.M., Shimizu M., Stokoe J., Kharbanda R.K., Redington A.N. Remote ischemic preconditioning of the recipient reduces myocardial ischemia-reperfusion injury of the denervated donor heart via a Katp channel-dependent mechanism. Transplantation. 79(12): 1691–1695. 2005.
Shimizu M., Tropak M., Diaz R. J., Suto F., Surendra H., Kuzmin E., Li J., Gross G., Wilson G.J., Callahan J., Redington A.N. Transient limb ischaemia remotely preconditions through a humoral mechanism acting directly on the myocardium: Evidence suggesting cross-species protection. Clin. Sci. (Lond.) 117(5): 191–200. 2009.
Maciel L., de Oliveira D.F., Verissimo da Costa G.C., Bisch P.M., Nascimento J.H. M. Cardioprotection by the transfer of coronary effluent from ischaemic preconditioned rat hearts: Identification of cardioprotective humoral factors. Basic Res. Cardiol. 112(5): 52. 2017.
Zhao Y., Zheng Z.N., Cheung C.W., Zuo Z.Y., Jin S.Q. Transfusion of plasma collected at late phase after preconditioning reduces myocardial infarct size induced by ischemia-reperfusion in rats in vivo. Chin. Med. J. 130(3): 303–308. 2017.
Zhao Y., Zheng Z. N., Pi Y.N., Liang X., Jin S.Q. Cardioprotective effects of transfusion of late-phase preconditioned plasma may be induced by activating the reperfusion injury salvage kinase pathway but not the survivor activating factor enhancement pathway in rats. Oxid. Med. Cell. Longev. 2017: 8526561. 2017.
Hausenloy D.J., Yellon D.M. Reperfusion injury salvage kinase signalling: Taking a RISK for cardioprotection. Heart Fail. Rev. 12(3-4): 217–234. 2007.
Hausenloy D.J., Yellon D.M. Ischaemic conditioning and reperfusion injury. Nat. Rev. Cardiol. 13(4): 193–209. 2016.
Zhao Y., Zheng Z.N., Liu X., Dai G., Jin S.Q. Effects of preconditioned plasma collected during the late phase of remote ischaemic preconditioning on ventricular arrhythmias caused by myocardial ischaemia reperfusion in rats. J. Int. Med. Res. 46(4): 1370–1379. 2018.
Gedik N., Kottenberg E., Thielmann M., Frey U.H., Jakob H., Peters J., Heusch G., Kleinbongard P. Potential humoral mediators of remote ischemic preconditioning in patients undergoing surgical coronary revascularization. Sci. Rep. 7(1): 12660. 2017.
Heinen A., Behmenburg F., Aytulun A., Dierkes M., Zerbin L., Kaisers W., Schaefer M., Meyer-Treschan T., Feit S., Bauer I., Hollmann M. W., Huhn R. The release of cardioprotective humoral factors after remote ischemic preconditioning in humans is age- and sex-dependent. J. Transl. Med. 16(1): 112. 2018.
Ney J., Hoffmann K., Meybohm P., Goetzenich A., Kraemer S., Benstöm C., Weber N.C., Bickenbach J., Rossaint R., Marx G., Zacharowski K., Bernhagen J., Stoppe C. Remote ischemic preconditioning does not affect the release of humoral factors in propofol-anesthetized cardiac surgery patients: A secondary analysis of the RIPHeart study. Int. J. Mol. Sci. 19(4): pii: E1094. 2018.
Dickson E.W., Blehar D.J., Carraway R.E., Heard S.O., Steinberg G., Przyklenk K. Naloxone blocks transferred preconditioning in isolated rabbit hearts. J. Mol. Cell. Cardiol. 33(9): 1751–1756. 2001.
Dickson E.W., Ludwig P.S., Ackermann L.W., Buresh C.T., Denning G. M. Met5-enkephalin-Arg6-Phe7 (MEAP): A cardioprotective hormonal opioid. Acad. Emerg. Med. 13(8): 813–819. 2006.
Dickson E.W., Tubbs R.J., Porcaro W.A., Lee W.J., Blehar D.J., Carraway R.E., Darling C.E., Przyklenk K. Myocardial preconditioning factors evoke mesenteric ischemic tolerance via opioid receptors and KATP channels. Am. J. Physiol. Heart Circ. Physiol. 283(1): H22–H28. 2002.
Surendra H., Diaz R.J., Harvey K., Tropak M., Callahan J., Hinek A., Hossain T., Redington A., Wilson G.J. Interaction of δ and κ opioid receptors with adenosine A1 receptors mediates cardioprotection by remote ischemic preconditioning. J. Mol. Cell Cardiol. 60: 142–150. 2013.
Weinbrenner C., Schulze F., Sarvary L., Strasser R.H. Remote preconditioning by infrarenal aortic occlusion is operative via δ1-opioid receptors and free radicals in vivo in the rat heart. Cardiovasc. Res. 61(3): 591–599. 2004.
Zhang S.Z., Wang N.F., Xu J., Gao Q., Lin G.H., Bruce I.C., Xia Q. Kappa-opioid receptors mediate cardioprotection by remote preconditioning. Anesthesiology. 105(3): 550–556. 2006.
Sharma R., Randhawa P.K., Singh N., Jaggi A. S. Possible role of thromboxane A2 in remote hind limb preconditioning-induced cardioprotection. Naunyn-Schmiedeberg’s Arch. Pharmacol. 389(1): 1–9. 2016.
Hajrasouliha A.R., Tavakoli S., Ghasemi M., Jabehdar-Maralani P., Sadeghipour H., Ebrahimi F., Dehpour A.R. Endogenous cannabinoids contribute to remote ischemic preconditioning via cannabinoid CB2 receptors in the rat heart. Eur. J. Pharmacol. 579(1–3): 246–252. 2008.
Randhawa P.K., Jaggi A.S. Gadolinium and ruthenium red attenuate remote hind limb preconditioning-induced cardioprotection: Possible role of TRP and especially TRPV channels. Naunyn-Schmiedeberg’s. Arch. Pharmacol. 389(8): 887–896. 2016.
Singh A., Randhawa P.K., Bali A., Singh N., Jaggi A.S. Exploring the role of TRPV and CGRP in adenosine preconditioning and remote hind limb preconditioning-induced cardioprotection in rats. Cardiovasc. Drugs Ther. 31(2): 133–143. 2017.
Wolfrum S., Nienstedt J., Heidbreder M., Schneider K., Dominiak P., Dendorfer A. Calcitonin gene related peptide mediates cardioprotection by remote preconditioning. Regul. Pept. 127(1–3): 217–224. 2005.
Dong J.H., Liu Y.X., Ji E.S., He R.R. Limb ischemic preconditioning reduces infarct size following myocardial ischemia-reperfusion in rats. Sheng Li Xue Bao. 56(1): 41–46. 2004. [In Chinese].
Pell T.J., Baxter G.F., Yellon D.M., Drew G.M. Renal ischemia preconditions myocardium: Role of adenosine receptors and ATP-sensitive potassium channels. Am. J. Physiol. 275(5 Pt 2): H1542–H1547. 1998.
Schulte G., Sommerschild H., Yang J., Tokuno S., Goiny M., Lovdahl C., Johansson B., Fredholm B.B., Valen G. Adenosine A1 receptors are necessary for protection of the murine heart by remote, delayed adaptation to ischaemia. Acta. Physiol. Scand. 182(2): 133–143. 2004.
Takaoka A., Nakae I., Mitsunami K., Yabe T., Morikawa S., Inubushi T., Kinoshita M. Renal ischemia/reperfusion remotely improves myocardial energy metabolism during myocardial ischemia via adenosine receptors in rabbits: Effects of “remote preconditioning”. J. Am. Coll. Cardiol. 33(2): 556–564. 1999.
Singh B., Randhawa P.K., Singh N., Jaggi A. S. Investigations on the role of leukotrienes in remote hind limb preconditioning-induced cardioprotection in rats. Life Sci. 152: 238–243. 2016.
McBride D.W., Reis C., Zhang J.H., Applegate R., Tang J. Remote limb ischemic preconditioning attenuates cerebrovascular depression during sinusoidal galvanic vestibular stimulation via α1-adrenoceptor-protein kinase Cε-endothelial NO synthase pathway in rats. J. Am. Heart Assoc. 7(7): pii: e007105. 2018.
Dong W., Yu P., Zhang T., Zhu C., Qi J., Liang J. Adrenomedullin serves a role in the humoral pathway of delayed remote ischemic preconditioning via a hypoxia-inducible factor-1α-associated mechanism. Mol. Med. Rep. 17(3): 4547–4553. 2018.
Jose Alburquerque-Béjar J., Barba I., Valls-Lacalle L., Ruiz-Meana M., Pecoraro M., Rodríguez-Sinovas A., García-Dorado D. Remote ischemic conditioning provides humoural cross-species cardioprotection through glycine receptor activation. Cardiovasc. Res. 113(1): 52–60. 2017.
Brandenburger T., Grievink H., Heinen N., Barthel F., Huhn R., Stachuletz F., Kohns M., Pannen B., Bauer I. Effects of remote ischemic preconditioning and myocardial ischemia on microRNA-1 expression in the rat heart in vivo. Shock. 42(3): 234–238. 2014.
Duan X., Ji B., Wang X., Liu J., Zheng Z., Long C., Tang Y., Hu S. Expression of microRNA-1 and microRNA-21 in different protocols of ischemic conditioning in an isolated rat heart model. Cardiology. 122(1): 36–43. 2012.
Kang Z., Li Z., Huang P., Luo J., Liu P., Wang Y., Xia T., Zhou Y. Remote ischemic preconditioning upregulates microRNA-21 to protect the kidney in children with congenital heart disease undergoing cardiopulmonary bypass. Pediatr. Nephrol. 33(5): 911–919. 2018.
Li J., Rohailla S., Gelber N., Rutka J., Sabah N., Gladstone R.A., Wei C., Hu P., Kharbanda R.K., Redington A.N. MicroRNA-144 is a circulating effector of remote ischemic preconditioning. Basic Res. Cardiol. 109(5): 423. 2014.
Minghua W., Zhijian G., Chahua H., Qiang L., Minxuan X., Luqiao W., Weifang Z., Peng L., Biming Z., Lingling Y., Zhenzhen W., Jianqing X., Huihui B., Xiaozhong W., Xiaoshu C. Plasma exosomes induced by remote ischaemic preconditioning attenuate myocardial ischaemia/reperfusion injury by transferring miR-24. Cell Death Dis. 9(3): 320. 2018.
Patel H.H., Moore J., Hsu A.K., Gross G.J. Cardioprotection at a distance: Mesenteric artery occlusion protects the myocardium via an opioid sensitive mechanism. J. Mol. Cell Cardiol. 34(10): 1317–1323. 2002.
Xie R.Q., Cui W., Hao Y.M., Liu F., Li B.H., Wu J.F., Du G.Y., Zhang T. Effects of remote preconditioning induced by skeletal muscle ischemia on myocardial cells apoptosis and roles of opioid receptors in pigs. Zhongguo Ying Yong Sheng Li Xue Za Zhi. 22(4): 474–478. 2006. [In Chinese].
Maslov L.N., Khaliulin I., Oeltgen P.R., Naryzhnaya N.V., Pei J.-M., Brown S.A., Lishmanov Y.B., Downey J.M. Prospects of creation of cardioprotective and antiarrhythmic drugs based on opioid receptor agonists. Med. Res. Rev. 36(5): 871–923. 2016.
Маслов Л.Н., Хедрик Дж.П., Мешоулам Р., Крылатов А.В., Лишманов А.Ю., Барзах Е.И., Нарыжная Н.В., Жанг И. Роль трансактивации рецепторов в кардиопротекторных эффектах прекондиционирования и посткондиционирования. Рос. физиол. журн. 98(3): 305–317. 2012. [Maslov L.N., Headrick J.P., Mechoulam R., Krylatov A.V., Lishmanov A.Yu., Barzakh E.I., Naryzhnaya N.V., Zhang Y. The role of transactivation of receptors in the cardioprotective effects of preconditioning and postconditioning. Russ. J. Physiol. 98(3): 305–317. 2012. (In Russ.)].
Klabunde R.E. Dipyridamole inhibition of adenosine metabolism in human blood. Eur. J. Pharmacol. 93(1–2): 21–26. 1983.
Mahaffey K.W., Puma J.A., Barbagelata N.A., DiCarli M.F., Leesar M.A., Browne K.F., Eisenberg P.R., Bolli R., Casas A.C., Molina-Viamonte V., Orlandi C., Blevins R., Gibbons R.J., Califf R.M.,Granger C.B. Adenosine as an adjunct to thrombolytic therapy for acute myocardial infarction: Results of a multicenter, randomized, placebo-controlled trial: the Acute Myocardial Infarction STudy of ADenosine (AMISTAD) trial. J. Am. Coll. Cardiol. 34(6): 17111–720. 1999.
Mullane K.M., Dusting G.J., Salmon J.A., Moncada S., Vane J.R. Biotransformation and cardiovascular effects of arachidonic acid in the dog. Eur. J. Pharmacol. 54(3): 217–228. 1979.
Arad M., Oxman T., Leor R., Rabinowitz B. Prostaglandins and the antiarrhythmic effect of preconditioning in the isolated rat heart. Mol. Cell. Biochem. 160–161: 249–255. 1996.
Zheng J. Molecular mechanism of TRP channels. Compar. Physiol. 3(1): 221–242. 2013.
Friedrich O., Wagner S., Battle A.R., Schurmann S., Martinac B. Mechamo-regulation of the beating heart at the cellular level – mechanosensitive channels in normal and diseased heart. Prog. Biophys. Mol. Biol. 110(2–3): 226–238. 2012.
Kim D. A mechanosensitive K+ channel in heart cells. Activation by arachidonic acid. J. Gen. Pysiol. 100(6): 1021–1040. 1992.
Craelius W., Chen V., el-Sherif N. Stretch activated ion channels in ventricular myocytes. Biosci. Rep. 8(5): 407–414. 1988.
Lehoux S., Tedgui A. Cellular mechanis and gene expression in blood vessels. J. Biomech. 36(5): 631–643. 2003.
Nilius B., Viana F., Droogmans G. Ion channels in vascular endothelium. Annu. Rev. Physiol. 59: 145–170. 1997.
Fischer M.J., Reeh P.W., Sauer S.K. Proton-induced calcitonin gene-related peptide release from rat sciatic nerve axons, in vitro, involving TRPV1. Eur. J. Neurosci. 18(4): 803–810. 2003.
Shenton F.C., Pyner S. Expression of transient receptor potential channels TRPC1 and TRPV4 in venoatrial endocardium of the rat heart. Neuroscience. 267: 195–204. 2014.
Zahner M.R., Li D.P., Chen S.R., Pan H.L. Cardiac vanilloid receptor 1-expressing afferent nerves and their role in the cardiogenic sympathetic reflex in rats. J. Physiol. 551(Pt 2): 515–523. 2003.
Randhawa P.K., Jaggi A.S. Investigating the involvement of TRPV1 ion channels in remote hind limb preconditioning-induced cardioprotection in rats. Naunyn-Schmiedeberg’s Arch. Pharmacol. 390(2): 117–126. 2017.
Randhawa P.K., Jaggi A.S. Investigating the involvement of glycogen synthase kinase-3β and gap junction signaling in TRPV1 and remote hind preconditioning-induced cardioprotection. Eur. J. Pharmacol. 814: 9–17. 2017.
Gao Y., Song J., Chen H., Cao C., Lee C. TRPV1 activation is involved in the cardioprotection of remote limb ischemic postconditioning in ischemia-reperfusion injury rats. Biochem. Biophys. Res. Commun. 463(4): 1034–1039. 2015.
Malinowska B., Kwolek G., Gothert M. Anandamide and methanandamide induce both vanilloid VR1- and cannabinoid CB1 receptor-mediated changes in heart rate and blood pressure in anaesthetized rats. Naunyn-Schmiedeberg’s Arch. Pharmacol. 364(6): 562–569. 2001.
Kozak K.R., Crews B.C., Ray J.L., Tai H.H., Morrow J.D., Marnett L.J. Metabolism of prostaglandin glycerol esters and prostaglandin ethanolamides in vitro and in vivo. J. Biol. Chem. 276(40): 36993–36998. 2001.
Amara S.G., Jonas V., Rosenfeld M.G., Ong E.S., Evans R.M. Alternative RNA processing in calcitonin gene expression generates mRNAs encoding different polypeptide products. Nature. 298(5871): 240–244. 1982.
Muff R., Born W., Lutz T.A., Fischer J.A. Biological importance of the peptides of the calcitonin family as revealed by disruption and transfer of corresponding genes. Peptides. 25(11): 2027–2038. 2004.
Zaidi M., Breimer L.H., Maclntyre I. Biology of peptides from the calcitonin genes. Q. J. Exp. Physiol. 72(4): 371–408. 1987.
Franco-Cereceda A. Calcitonin gene-related peptide and tachykinins in relation to local sensory control of cardiac contractility and coronary vascular tone. Acta. Physiol. Scand. Suppl. 569: 1–63. 1988.
Kallner G. Release and effects of calcitonin gene-related peptide in myocardial ischaemia. Scand. Cardiovasc. J. Suppl. 49: 1–35. 1998.
Randhawa P.K., Jaggi A.S. Exploring the putative role of TRPV1-dependent CGRP release in remote hind preconditioning-induced cardioprotection. Cardiovasc. Ther. 35(5): 2017.
D’Alonzo A.J., Grover G.J., Darbenzio R.B., Hess T.A., Sleph P.G., Dzwonczyk S., Zhu J.L., Sewter J.C. In vitro effects of capsaicin: Antiarrhythmic and antiischemic activity. Eur. J. Pharmacol. 272(2–3): 269–278. 1995.
Lu R., Li Y.J., Deng H.W. Evidence for calcitonin gene-related peptide-mediated ischemic preconditioning in the ray heart. Regul. Pept. 82(1–3): 53–57. 1999.
Barajas-Espinosa A., Ochoa-Cortes F., Moos M.P., Ramirez F.D., Vanner S.J., Funk C.D. Characterizatuion of the cysteinyl leukotriene 2 receptor in novel expression sites of the gastrointestinal tract. Am. J. Pathol. 178(6): 2682–2689. 2011.
Ni N.C., Ballantyne L.L., Mewburn J.D., Funk C.D. Multiple-site activation of the cysteinyl leukotriene receptor 2 is required for exacerbation of ischemia/reperfusion injury. Arterioscler. Thromb. Vasc. Biol. 34(2): 321–330. 2014.
Noguchi K., Okubo M. Leukotrienes in nociceptive pathway and neuropathic/ inflammatory pain. Biol. Pharm. Bull. 34(8): 1163–1169. 2011.
Huber M., Guhlmann A., Jansen P.L., Keppler D. Hereditary defect of hepatobiliary cysteinyl leukotriene elimination in mutant rats with defective hepatic anion excretion. Hepatology. 7(2): 224–228. 1987.
Khaliulin I., Parker J.E., Halestrap A.P. Consecutive pharmacological activation of PKA and PKC mimics the potent cardioprotection of temperature preconditioning. Cardiovasc. Res. 88(2): 324–333. 2010.
Lewis M., Szobi A., Balaska D., Khaliulin I., Adameova A., Griffiths E., Orchard C.H., Suleiman M.S. Consecutive isoproterenol and adenosine treatment confers marked protection against reperfusion injury in adult but not in immature heart: A role for glycogen. Int. J. Mol. Sci. 19(2): pii: E494. 2018.
Nishida H., Sato T., Miyazaki M., Nakaya H. Infarct size limitation by adrenomedullin: Protein kinase A but not PI3-kinase is linked to mitochondrial KCa channels. Cardiovasc. Res. 77(2): 398–405. 2008.
Sluijter J.P.G., Davidson S.M., Boulanger C.M., Buzás E.I., de Kleijn D.P.V., Engel F.B., Giricz Z., Hausenloy D.J., Kishore R., Lecour S., Leor J., Madonna R., Perrino C., Prunier F., Sahoo S., Schiffelers R.M., Schulz R., Van Laake L.W., Ytrehus K., Ferdinandy P. Extracellular vesicles in diagnostics and therapy of the ischaemic heart: Position Paper from the Working Group on Cellular Biology of the Heart of the European Society of Cardiology. Cardiovasc Res. 114(1): 19–34. 2018.
van der Pol E., Böing A.N., Harrison P., Sturk A., Nieuwland R. Classification, functions, and clinical relevance of extracellular vesicles. Pharmacol. Rev. 64(3): 676–705. 2012.
Colombo M., Raposo G., Théry C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell. Dev. Biol. 30: 255–289. 2014.
Valadi H., Ekström K., Bossios A., Sjöstrand M., Lee J.J., Lötvall J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell. Biol. 9(6): 654–659. 2007.
Hunter M.P., Ismail N., Zhang X., Aguda B.D., Lee E.J., Yu L., Xiao T., Schafer J., Lee M.L., Schmittgen T. D., Nana-Sinkam S. P., Jarjoura D., Marsh C. B. Detection of microRNA expression in human peripheral blood microvesicles. PLoS One. 3(11): e3694. 2008.
Skog J., Würdinger T., van Rijn S., Meijer D.H., Gainche L., Sena-Esteves M., Curry W.T., Carter B.S., Krichevsky A.M., Breakefield X.O. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell. Biol. 10(12): 1470–1476. 2008.
Salloum F.N., Yin C., Kukreja R.C. Role of microRNA in cardiac preconditioning. J. Cardiovasc. Pharmacol. 56(6): 581–588. 2010.
Lee Y., Kim M., Han J., Yeom K.H., Lee S., Baek S.H., Kim V.N. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 23(20): 4051–4060. 2004.
Lee Y., Ahn C., Han J., Choi H., Kim J., Yim J., Lee J., Provost P., Radmark O., Kim S., Kim V. N. The nuclear RNAse Drosha initiates microRNA processing. Nature. 415(6956): 415–419. 2003.
Bartel D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell. 116(2): 281–297. 2004.
Eulalio A., Huntzinger E., Izaurralde E. Getting to the root of miRNA-mediated gene silencing. Cell. 132(1): 9–14. 2008.
Bushati N., Cohen S.M. MicroRNA functions. Annu. Rev. Cell. Dev. Biol. 23: 175–205. 2007.
Chang T.C., Mendell J.T. MicroRNAs in vertebrate physiology and human disease. Annu. Rev. Genomics Hum. Genet. 8: 215–239. 2007.
Barile L., Moccetti T., Marban E., Vassalli G. Roles of exosomes in cardioprotection. Eur. Heart J. 38(13): pii: ehw304. 2017.
Gupta S., Knowlton A.A. HSP60 traffiking in adult cardiac myocytes: Role of the exosomal pathway. Am. J. Physiol. Heart Circ. Physiol. 292(6): H3052–H3056. 2007.
Bang C., Batkai S., Dangwal S., Gupta S. K., Foinquinos A., Holzmann A., Just A., Remke J., Zimmer K., Zeug A., Ponimaskin E., Schmiedl A., Yin X., Mayr M., Halder R., Fischer A., Engelhardt S., Wei Y., Schober A., Fiedler J., Thum T. Cardiac fibroblast-derived microRNA passenger strand-enriched exosomes mediate cardiomyocyte hypertrophy. J. Clin. Invest. 124(5): 2136–2146. 2014.
van Balkom B.W., de Jong O.G., Smits M., Brummelman J., den Ouden K., de Bree P.M., van Eijndhoven M.A., Pegtel D.M., Stoorvogel W., Wurdinger T., Verhaar M.C. Endotelial cells require miR-214 to secrete exosomes that suppress senescence and induce angiogenesis in human and mouse endothelial cells. Blood. 121(19): 3997–4006. 2013.
Zheng Y., Vicencio J.M., Yellon D.M., Davidson S.M. Exosomes released from endothelial cells are cardioprotective. Heart. 100 Suppl 1: A10. 2014.
Baranyai T., Giricz Z., Varga Z., Sipos P., Paloczi K., Kittel A., Buzas E., Ferdinandy P. Extracellular vesicles mediate cardioprotection exerted by remote ischemic preconditioning in rats. Cardiovasc. Res. 103. Suppl 1: 435. 2014.
Wider J., Undyala V.V.R., Whittaker P., Woods J., Chen X., Przyklenk K. Remote ischemic preconditioning fails to reduce infarct size in the Zucker fatty rat model of type-2 diabetes: role of defective humoral communication. Basic Res. Cardiol. 113(3): 16. 2018.
Duan Y.F., Sun D.L., Chen J., Zhu F., An Y. MicroRNA-29a/b/c targets iNOS and is involved in protective remote ischemic preconditioning in an ischemia-reperfusion rat model of non-alcoholic fatty liver disease. Oncol. Lett. 13(3): 1775–1782. 2017.
Дополнительные материалы отсутствуют.
Инструменты
Российский физиологический журнал им. И.М. Сеченова