

Российский физиологический журнал им. И.М. Сеченова, 2020, T. 106, № 1, стр. 3-16
Роль микроРНК, NO-синтаз, киназ, КАТФ-каналов в инфаркт-лимитирующем эффекте адаптации к гипоксии
Н. В. Нарыжная 1, Н. С. Воронков 1, 2, А. А. Скрябина 2, Ю. В. Бушов 2, Л. Н. Маслов 1, *
1 Научно-исследовательский институт кардиологии, Томский национальный
исследовательский медицинский центр РАН
Томск, Россия
2 Национальный исследовательский Томский государственный университет
Томск, Россия
* E-mail: Maslov@cardio-tomsk.ru
Поступила в редакцию 18.11.2019
После доработки 18.12.2019
Принята к публикации 18.12.2019
Аннотация
Установлено, что хроническая гипоксия (ХГ) существенно изменяет экспрессию микроРНК в миокарде. Показано, что miR-138 и miR-184 усиливают толерантность кардиомиоцитов к гипоксии, а microRNA‑199a‑5p и miR-23b снижают устойчивость этих клеток к гипоксии. ХГ усиливала экспрессию индуцибельной (iNOS) и эндотелиальной NO-синтазы (eNOS). Показано, что кардиопротекторный эффект ХГ связан с активацией iNOS. Установлено, что изоформы δ и ε протеинкиназы С участвуют в кардиопротекторном эффекте адаптации к гипоксии. Показано, что ХГ вызывает усиление экспрессии киназ CaMKII, p-ERK1/2, p-p38, p-Akt, гексокиназы-1, гексокиназы-2. Гипоксия усиливает транслокацию гексокиназы-2 в митохондрии. У животных, адаптированных к гипоксии, не удалось обнаружить увеличения экспрессии киназ: ПКA, p-GSK3β, AMPK и JNK. Представлены данные, указывающие на то, что киназы ERK1/2, MEK1/2 участвуют в кардиопротекторном эффекте адаптации к гипоксии. Гипертрофия миокарда, вызванная хронической гипоксией, связана с активацией Rho-киназы. Вопрос о роли PI3, Akt JNK, PKG, Rho-киназы, mTOR и p38 в защитном эффекте адаптации к гипоксии является спорным. Установлено, что инфаркт-лимитирующий эффект ХГ зависит от активации митоКАТФ-каналов. Цель обзора: анализ данных о роли микроРНК, NO-синтазы и киназ в кардиопротекторном эффекте ХГ.
Хорошо известно, что адаптация к хронической гипоксии (ХГ) усиливает толерантность сердца к аритмогенному влиянию ишемии/реперфузии (И/Р) [1–3]; ограничивает размер инфаркта, возникающего при коронароокклюзии и реперфузии [4, 5]; улучшает сократимость сердца в реперфузионном периоде [4, 6]. Установлено, что в регуляции толерантности сердца к действию И/Р важную роль играют NO-синтаза (NOS), киназы и АТФ-чувствительные К+-каналы (КАТФ-каналы) [7–9].
МикроРНК и ХГ. МикроРНК (miRNAs) составляют многочисленную группу небольших некодирующих РНК длиной в 21–22 нуклеотида, которые являются посттранскрипционными регуляторами экспрессии генов у животных, растений и простейших [10]. miRNAs происходят из областей транскриптов РНК, которые сворачиваются обратно в себя, образуя короткие шпильки [11]. miRNAs взаимодействуют с 3-нетранслируемой областью мРНК, ингибируя взаимодействие мРНК с рибосомами и ускоряя энзиматическую деградацию мРНК [10], что ведет к снижению синтеза молекулы белка, кодируемой мРНК. Полагают, что miRNAs участвуют в регуляции толерантности сердца к И/Р [12–15].
Установлено, что экспрессия microRNA-138 (miR-138) увеличена в правом желудочке пациентов с цианозным врожденным пороком сердца [16]. Было установлено, что miR-138 предупреждает апоптоз клеток H9C2 в условиях гипоксии за счет ингибирования киназы JNK [16]. Установлено, что хроническая периодическая гипоксия (ХПГ) вызывает снижение уровня miRNA-214 в миокарде крыс [17]. Однако неясно, имеет ли эта микроРНК отношение к толерантности сердца при И/Р. Установлено, что уровень miR-184 снижен в образцах правого желудочка пациентов с цианозом и дефектом сердечных клапанов (ДСК) [18]. Показано, что ингибитор miR-184 снижает выживаемость клеток H9C2 в условиях гипоксии и стимулирует апоптоз этих клеток за счет усиления экспрессии каспазы-3 и каспазы-9 [18]. Представленные данные косвенно указывают на то, что miR-184 обеспечивает толерантность кардиомиоцитов к гипоксии. Установлено, что уровень microRNA‑199a‑5p снижен в образцах правого желудочка пациентов с пороками сердца и в кардиомиоцитах человека, которые культивировали в условиях гипоксии [19]. Показано, что microRNA‑199a‑5p-mimic усиливает апоптоз кардиомиоцитов в условиях гипоксии, а microRNA‑199a‑5p-ингибитор подавляет апоптоз кардиомиоцитов в условиях гипоксии. Эти данные свидетельствуют, что microRNA‑199a‑5p играет негативную роль в регуляции толерантности кардиомиоцитов к гипоксии. Высокий уровень miR-23b был найден при биопсии правого желудочка у пациентов с цианозом и ДСК по сравнению с пациентами с ДСК без цианоза [20]. Продолжительная гипоксия вызывает увеличение экспрессии miR-23b в клетках H9C2. Было показано, что miR-23b вызывал индуцированный гипоксией апоптоз клеток H9C2. Авторы заключили, что избыточная экспрессия miR-23b может способствовать апоптозу кардиомиоцитов в условиях гипоксии [20]. ХПГ клеток H9C2 вызывает гипертрофию клеток, усиливает экспрессию miR-31 и протеинкиназы Сε [21]. Полагают, что miR-31 может индуцировать гипертрофию кардиомиоцитов. Было показано, что ХПГ вызывает увеличение уровня miR-21в ткани предсердий [22]. Однако остается неясным, имеет ли miR-21отношение к увеличению резистентности сердца к И/Р.
Таким образом, установлено, что ХГ существенно изменяет экспрессию м-икроРНК в миокарде. В условиях ХГ экспрессия miR-138, miR-23b miR-31 и miR-21в усиливается, а экспрессия miR-184, microRNA‑199a‑5p снижается. Показано, что miR-138 и miR-184 усиливают толерантность кардиомиоцитов к гипоксии, а microRNA‑199a‑5p и miR-23b усиливают апоптоз кардиомиоцитов в условиях гипоксии.
NO-синтаза. Экспрессия изоформ NOS. NO-синтаза катализирует синтез оксида азота (NO•), который играет роль внутриклеточного мессенджера и обеспечивает межклеточную сигнализацию [23, 24]. NOS участвует в реализации феномена ишемического пре- и посткондиционирования [7–9], поэтому были основания предполагать, что ХГ вызывает усиление экспрессии NOS. В ряде исследований было установлено, что ХГ стимулирует экспрессию индуцибельной (iNOS) [25–30] и эндотелиальной NOS (eNOS) в миокарде [31–39]. Есть сообщение о том, что ХГ увеличивает экспрессию митохондриальной NOS (mtNOS) в сердце [40]. Последняя является нейрональной NOS (nNOS), связанной с внутренней мембраной митохондрий [41]. P.H.La Padula и соавт. показали, что ХГ способствует усилению экспрессии nNOS [39]. Другие исследователи сообщили, что ХГ не влияет на экспрессию nNOS [33]. Есть данные о том, что ХГ не влияет на уровень eNOS в миокарде, но снижает экспрессию p-eNOS [4]. Приводятся данные о снижении экспрессии eNOS в миокарде после ХГ [30]. Установлено, что в качестве индуктора синтеза белка iNOS выступает транскрипционный фактор hypoxia inducing factor 1α (HIF-1α) [26].
Таким образом, большинство исследователей полагает, что ХГ увеличивает экспрессию eNOS и iNOS в миокарде (табл. 1).
Таблица 1.
Влияние хронической гипоксии на экспрессию NO-синтазы в миокарде Table 1. The effect of chronic hypoxia on the expression of NO-synthase in the myocardium
| Название изоформ NO-синтаза NO-synthase isoforms | Авторы Authors |
|---|---|
| nNOS↑, eNOS↑ | La Padula P.H. et al., 2018 [39] |
| eNOS↑ | Wang N. et al., 2017 [38] |
| eNOS↓, iNOS↑ | Yuan X. et al., 2015 [30] |
| p-eNOS/eNOS при ХНГ↓ | Milano G. et al., 2010 [4] |
| p-eNOS/eNOS нет эффекта при ХПГ | Milano G. et al., 2010 [4] |
| eNOS нет эффекта при ХПГ и ХНГ | Milano G. et al., 2010 [4] |
| iNOS ↑, nNOS нет эффекта | Thompson L. et al., 2009 [33] |
| mtNOS↑ | La Padula P. et al., 2008 [40] |
| eNOS↑ | Quing M et al., 2007 [37] |
| eNOS↑ | Thompson L.P., Dong Y., 2005 [32] |
| eNOS ↑ | Forkel J. et al., 2004 [36] |
| iNOS↑ | Grilli A. et al., 2003a [28] |
| iNOS↑ | Grilli A. et al., 2003b [29] |
| iNOS↑ | Ferreiro C.R. et al., 2001 [27] |
| eNOS↑ | Felaco M. et al., 2000 [35] |
| eNOS↑ | Shi Y. et al., 2000 [34] |
| eNOS↑ | Thompson L.P. et al, 2000 [31] |
| iNOS↑ | Jung F. et al., 2000 [26] |
| iNOS↑ | Rouet-Benzineb P. et al., 1999 [25] |
eNOS – эндотелиальная NO-синтаза; iNOS – индуцибельная NO-синтаза; nNOS – нейрональная NO-синтаза; mtNOS – митохондриальная NO-синтаза; ХПГ – хроническая периодическая гипоксия; ХНГ – хроническая непрерывная гипоксия. eNOS, endothelial NO-synthase; iNOS, inducible NO-synthase; nNOS, neuronal NO-synthase; mtNOS, mitochondrial NO-synthase; CIH, chronic intermittent hypoxia; CCH, chronic continuous hypoxia.
NO-синтаза и кардиопротекция при ХГ. Установлено, что ХГ вызывает увеличение активности NOS в миокарде [25, 42]. Увеличение активности NOS было найдено в миокарде детей с цианозными врожденными пороками сердца [27]. Активация продукции NO при ХГ имеет генерализованный характер, наблюдается не только в миокарде, но и в изолированных mesenteric arteries [43]. Мы обнаружили увеличение уровня метаболитов NO – нитритов и нитратов в сыворотке крови и миокарде крыс после хронической непрерывной гипоксии (ХНГ) [5]. Увеличение нитритов и нитратов в миокарде адаптированных к гипоксии крыс отмечали и другие исследователи [33].
Увеличение продукции NO обеспечивает повышение толерантности сердца к действию И/Р. Так, было показано, что ингибитор NOS L-NAME устраняет повышение толерантности изолированного сердца к действию И/Р у адаптированных к ХГ кроликов [44]. Мы установили, что L-NAME, неселективный ингибитор NOS, устраняет инфаркт-лимитирующий эффект ХНГ [5]. Также действует ингибитор iNOS S-methylisothiourea [5]. Ингибитор nNOS 7-nitroindazole не влиял на кардиопротекторный эффект ХНГ [5].
Таким образом, установлено, что ХГ вызывает увеличение продукции NO в миокарде. Инфаркт-лимитирующий эффект ХГ связан с активацией iNOS.
Протеинкиназа С (ПКС). Известно, что ПКС принимает участие в кардиопротекторном эффекте ишемического прекондиционирования и посткондиционирования [7, 8], поэтому исследователи обратили внимание на роль этого фермента в инфаркт-лимитирующем эффекте адаптации к гипоксии (рис. 1). У детей с пороком сердца и цианозом и у новорожденных кроликов, находившихся в условиях гипоксии, была обнаружена транслокация (активация) ПКСε в клеточные органеллы [45]. У крыс, находящихся длительное время в условиях гипоксии, наблюдается увеличение уровня ПКСδ, ПКСε, ПКСζ [46]. Установлено, что инфаркт-лимитирующий эффект хронической периодической гипоксии (ХПГ) не проявляется после блокады ПКС хелеритрином [47]. Селективной блокатор ПКСδ роттлерин уменьшал, но не устранял инфаркт-лимитирующий эффект ХПГ [47]. ХПГ индуцировала транслокацию ПКСδ в митохондрии и ядра клеток. Мы установили, что роттлерин устраняет адаптационное повышение толерантности кардиомиоцитов к действию аноксии/реоксигенации [48]. Эти факты указывают на важную роль ПКСδ в кардиопротекторном эффекте ХГ. Установлено, что ХГ способствует увеличению в миокарде уровня активатора ПКС диацилглицерола [49]. Возможно, что он индуцирует активацию ПКС. Вместе с тем, есть данные о том, что увеличение активности ПКСδ может быть результатом окислительного стресса, который наблюдается после воздействия ХПГ [50]. Так, было установлено, что ежедневное введение крысам антиоксиданта N-ацетилцистеина устраняет инфаркт-лимитирующий эффект ХПГ и устраняет транслокацию ПКСδ в клеточные органеллы [50]. Действительно, образование нитротирозина, который является маркером окислительного стресса, усиливается при ХПГ в миокарде, особенно в митохондриях [51]. Способность активных форм кислорода активировать ПКС является известным фактом [52], который хорошо согласуется с данными исследований, выполненных на адаптированных к ХПГ крысах [50]. Следует отметить, что ПКСδ принимает участие в кардиопротекторном эффекте хронической непрерывной гипоксии (ХНГ) [48]. Установлено, что уровень ПКСδ в мембранной фракции отрицательно коррелирует с размером инфаркта после адаптации к гипоксии [53]. Установлено, что ХПГ повышает устойчивость кардиомиоцитов к действию аноксии/реоксигенации и уменьшает Ca2+-перегрузку кардиомиоцитов после аноксии/реоксигенации [54]. Хелеритрин, ингибитор ПКС, устраняет эти защитные эффекты [54]. Представленные данные убедительно свидетельствуют об участии ПКСδ в кардиопротекторном эффекте ХГ (табл. 2).
Рис. 1.
Вовлечение киназ в кардиопротекторный эффект хронической гипоксии. Аббревиатуры: MEK1/2, mitogen-activated protein kinase kinase; ERK1/2, extracellular signal-regulated kinase; PKC, protein kinase C; JNK, c-Jun N-terminal kinase; p38, p38 kinase; PI3K, phosphatidylinositol-3-kinase; Akt, Akt kinase; CaMKII, Ca2+-calmodulin kinase II; mTOR, mammalian target of rapamycin; PKG, protein kinase G. Fig. 1. An involvement of kinases in cardioprotective effect of chronic hypoxia. Abbreviation: MEK1/2, mitogen-activated protein kinase kinase; ERK1/2, extracellular signal-regulated kinase; PKC, protein kinase C; JNK, c-Jun N-terminal kinase; p38, p38 kinase; PI3K, phosphatidylinositol-3-kinase; Akt, Akt kinase; CaMKII, Ca2+-calmodulin kinase II; mTOR, mammalian target of rapamycin; PKG, protein kinase G.
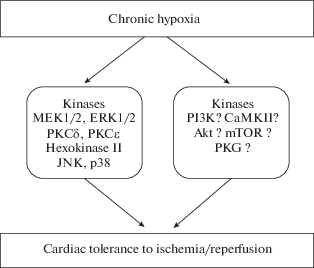
Таблица 2.
Влияние хронической гипоксии на экспрессию протеинкиназ в миокарде Table 2. The effect of chronic hypoxia on the expression of protein kinases in the myocardium
| Название киназ The name of the kinases | Авторы Authors |
|---|---|
| ПКСδ↑, ПКСε↑, ПКСζ↑ | Morel O.E. et al., 2003 [46] |
| ПКСδ↑ | Hlaváčková M. et al., 2007 [53] |
| ПКСε↑ | Holzerova K. et al., 2015 [55] |
| ПКСα↑ | Micova P. et al., 2016 [56] |
| p-AMPK↓ | Xie S. et al., 2016 [58] |
| CaMKIIγ↑, CaMKIIδ↑ | Zhao P.J. et al., 2008 [60] |
| ERK2↑ | Strnisková M. et al., 2006 [63] |
| p-ERK1/2↑ | Milano G. et al., 2010 [64] |
| p-ERK1/2↑ | Micova P. et al., 2016 [56] |
| p-ERK1/2↑ | Zhang K. et al., 2018 [22] |
| p-Akt↑ | Strnisková M. et al., 2006 [63] |
| p-Akt↑ | Kolar D. et al., 2017 [67] |
| PI3K↓, p-Akt↓ | Zhang K. et al., 2018 [22] |
| ПKA нет эффекта | Larsen K.O. et al., 2008 [68] |
| p38↓, p-p38↓ | Morel S. et al., 2006 [69] |
| p-p38 в ПЖ↓, p-p38 в ЛЖ↑ | Strnisková M. et al., 2006 [63] |
| p-p38 в ЛЖ↑ | Micova P. et al., 2016 [56] |
| p-p38↑ | Quing M. et al., 2007 [37] |
| p-JNK в ЛЖ нет эффекта | Morel S. et al., 2006 [69] |
| JNK и p-JNK в ЛЖ нет эффекта | Strnisková M.et al., 2006 [63] |
| JNK и p-JNK в ПЖ нет эффекта | Strnisková M. et al., 2006 [63] |
| p-JNK↑ | He S. et al., 2016 [72] |
| p-JNK/JNK↑ | Zhao Y.S. et al., 2019 [73] |
| p-GSK3β нет эффекта | McCarthy J. et al., 2011 [76] |
| HK-1↑, HK-2↑ | Waskova-Arnostova P. et al., 2015 [77] |
| HK-1↑, HK-2↑ | Kolar D. et al., 2017 [67] |
| HK-1↑, HK-2↑ | Nedvedova I. et al., 2018 [78] |
ПКС – протеинкиназа С; AMPK – AMP-activated protein kinase; CaMKII – Ca2+-calmodulin kinase II; ERK – extracellular signal-regulated kinase; Akt – Akt-киназа; PI3K – phosphatidylinositol-3-kinase; ПKA – протеинкиназа А; p38 – p38-киназа; JNK – c-Jun N-terminal kinase; PKG – протеинкиназа G; GSK3β – glycogen synthase kinase 3 β; HK – hexokinase. PKC, protein kinase C; AMPK, AMP-activated protein kinase; CaMKII, Ca2+-calmodulin kinase II; ERK, extracellular signal-regulated kinase; Akt, Akt-kinase; PI3K, phosphatidylinositol-3-kinase; PKA, protein kinase A; p38, p38-kinase; JNK, c-Jun N-terminal kinase; PKG, protein kinase G; GSK3β, glycogen synthase kinase 3 β; HK, hexokinase.
Вместе с тем, есть данные о том, что ПКСε также участвует в повышении толерантности сердца к действию И/Р после адаптации к непрерывной гипоксии [55]. Адаптация крыс к гипоксии приводила к усилению толерантности кардиомиоцитов к 25-минутному метаболическому ингибированию, которое достигалось с помощью NaCN и 2-дезоксиглюкозы. Было обнаружено, что ХГ приводит к усилению экспрессии ПКСε. Селективный ингибитор ПКСε KP-1633 устранял цитопротекторный эффект ХГ [55]. Возможно, что в кардиопротекторном эффекте ХГ участвуют другие изоформы ПКС. Так, было обнаружено, что ХПГ приводит к усилению экспрессии в миокарде ПКСα и фосфорилированной (активной) ПКСα [56]. Хроническая непрерывная гипоксия (ХНГ) приводила к усилению транслокации PKCβII и PKCη в мембраны кардиомиоцитов [57] (табл. 2).
Таким образом, по меньшей мере, две изоформы ПКС участвуют в кардиопротекторном эффекте ХГ: ПКСε и ПКСδ.
AMPK. АМФ-активируемая протеинкиназа AMPK (AMP-activated protein kinase) играет важную роль в реализации феномена пре- и посткондиционирования [8] (рис.1). По всей видимости, AMPK не играет существенной роли в кардиопротекторном эффекте адаптации к гипоксии, поскольку у крыс, подвергнутых ХПГ, отмечается снижение уровня p-AMPK (активированная AMPK) в миокарде [58] (табл. 2).
CaMKII. Известно, что активация CaMKII (Ca2+-calmodulin kinase II) усугубляет И/Р повреждения сердца [59], поэтому были основания предполагать, что ее активность в миокарде снижается при адаптации к гипоксии (рис.1). Однако было обнаружено, что экспрессия мРНК, кодирующей кальмодулин, CaMKIIγ, CaMKIIδ, в миокарде крыс после воздействия ХГ усиливается [60] (табл. 2). Усиление экспрессии CaMKII после воздействия ХГ отмечали и другие исследователи [61]. Получены данные, что повышенная экспрессия CaMKII может препятствовать повреждению кардиомиоцитов при Ca2+-парадоксе [62], но может ли CaMKII обеспечивать инфаркт-лимитирующий эффект ХГ – неизвестно.
ERK и MEK. ERK (extracellular signal-regulated kinase) и MEK (mitogen-activated protein kinase ) играют важную роль в пре- и посткондиционировании сердца [8], поэтому были основания полагать, что они могут участвовать в инфаркт-лимитирующем эффекте ХГ (рис.1). Установлено, что ХПГ приводит к увеличению экспрессии ERK2 в миокарде крыс [63]. ХПГ вызывает увеличение в миокарде уровня фосфорилированной (активной) ERK1/2 (p-ERK1/2) [64]. Ингибиторы MEK1/2 U0126 и PD-98059 устраняли инфаркт-лимитирующий эффект ХПГ [64]. Увеличение количества p-ERK1/2 в миокарде крыс после ХПГ отмечают и другие исследователи [22, 56]. Представленные данные указывают на то, что ERK1/2 и MEK1/2 участвуют в кардиопротекторном эффекте ХПГ (табл. 2).
PI3K и Akt. PI3K (phosphatidylinositol-3-kinase) и Akt принимают участие в пре- и посткондиционировании сердца [8], поэтому можно было предположить, что они участвуют в кардиопротекторном эффекте ХГ. Показано, что ХПГ вызывает увеличение экспрессии p-Akt в левом желудочке крыс [63]. Установлено, что ингибитор PI3K LY294002 устраняет инфаркт-лимитирующий эффект ХПГ [65] (рис. 1). Хроническая умеренная гипоксия вызывает увеличение уровня p-Akt в кардиомиобластах H9C2 [66]. В исследовании, выполненном на изолированных перфузируемых сердцах крыс, подвергнутых И/Р, было показано, что инфаркт-лимитирующий эффект ХПГ связан с активацией PI3K [64]. Кроме того, показано, что ХНГ вызывает увеличение уровня p-Akt в миокарде крыс [67]. Вместе с тем, по данным некоторых авторов [22], ХПГ вызывает снижение в миокарде крыс уровня PI3K и p-Akt. Нам также не удалось подтвердить участие PI3K в инфаркт-лимитирующем эффекте ХНГ [5]. Нами было установлено, что блокада PI3K вортманнином не влияет на повышение толерантности изолированных кардиомиоцитов к действию аноксии/реоксигенации у адаптированных к ХНГ крыс [48]. Таким образом, вопрос о роли PI3K и Akt в кардиопротекторном эффекте хронической гипоксии является спорным (табл. 2).
ПКA. Протеинкиназа A (ПKA) является цАМФ-зависимой киназой, которая, по мнению некоторых авторов [8], принимает участие в кардиопротекторном эффекте пре- и посткондиционирования (рис.1). У мышей, подвергнутых воздействию ХНГ, не удалось обнаружить изменение количества изоформ ПKA [68] (табл. 2). Однако это пока единственная работа, в которой оценивали влияние длительной гипоксии на уровень ПKA, поэтому делать вывод роли ПKA в кардиопротекторном эффекте адаптации к гипоксии пока рано.
p38 также участвует в пре- и посткондиционировании [8]. Установлено, что ХПГ вызывает снижение уровня киназы p38 и p-p38 в миокарде [69]. ХНГ не оказывала подобного эффекта [69] (рис. 1). В то же время, согласно данным M. Strnisková и соавт. [63], при ХПГ уровень p-p38 в правом желудочке снижается, а в левом – повышается, уровень общей p38 не изменяется. Повышение уровня p-p38 в миокарде левого желудочка после ХПГ было отмечено в более поздней работе того же коллектива исследователей [56]. Показано, что уровень p-p38 увеличен в миокарде у детей с пороками сердца и цианозом, но не у больных с пороками сердца без цианоза [37] (табл. 2). Установлено, что блокирование p38 ингибитором SB203580 устраняет кардиопротекторный эффект ХНГ [45]. Представленные данные говорят о повышения уровня p-p38 в миокарде левого желудочка при ХПГ, что способствует повышению толерантности сердца к действию И/Р. Вместе с тем, следует отметить, что позитивная роль p38 в кардиопротекторном эффекте хронической гипоксии показана только в одной статье [45].
JNK. Принято считать, что JNK (c-Jun N-terminal kinase) играет негативную роль в регуляции толерантности сердца к действию И/Р [70] (рис. 1). Вместе с тем, есть данные о том, что этот фермент участвует в кардиопротекторном эффекте дистантного прекондиционирования [71]. Установлено, что куркумин, ингибитор JNK, устраняет кардиопротекторный эффект ХНГ у кроликов [45]. При ХПГ не удалось обнаружить увеличение уровня p-JNK в левом желудочке крыс [69]. В то же время другим исследователям не удалось обнаружить изменение уровня общей JNK и p-JNK в правом и левом желудочке адаптированных к ХПГ крыс [63]. Пребывание кардиомиобластов H9C2 в среде, содержащей 1% O2, в течение 72 ч способствовало увеличению уровня p-JNK [72]. Установлено, что ХПГ приводит к увеличению соотношения p-JNK/JNK в миокарде [73] (табл. 2). Представленные данные свидетельствуют о том, что ХГ может приводить к увеличению уровня активной p-JNK в миокарде. Вместе с тем, данные об участии этой киназы в кардиопротекторном эффекте адаптации ограничены пока одной статьей [45], поэтому вопрос о роли JNK в защитном эффекте ХПГ остается открытым.
mTOR. Есть данные о том, что рапамицин, ингибитор mTOR (mammalian target of rapamycin), устраняет кардиопротекторный эффект ишемического прекондиционирования [74] (рис.1). Следовательно, были основания предполагать, что mTOR может принимать участие в кардиопротекторном эффекте хронической гипоксии. Установлено, что рапамицин устраняет гипертрофию сердца, вызванную ХПГ [58]. Эти данные указывают на то, что mTOR при ХПГ активируется. Вместе с тем, они не дают ответа на вопрос о роли mTOR в инфаркт-лимитирующем эффекте ХГ.
PKG. Известно, что PKG (cGMP-dependent protein kinase G) участвует в кардиопротекторном эффекте пре- и посткондиционирования [8], поэтому были основания предполагать, что эта киназа участвует в инфаркт-лимитирующем эффекте ХГ (рис. 1). Было установлено, что хроническая гипобарическая гипоксия вызывает увеличение в миокарде уровня цГМФ, активатора PKG [61]. Пока остается неизвестным, ведет ли увеличение содержания цГМФ в миокарде к повышению активности PKG. Неизвестно, какова роль этой киназы в инфаркт-лимитирующем эффекте хронической гипоксии.
GSK3β. Известно, что фосфорилирование GSK3β (glycogen synthase kinase 3 β) способствует инактивации этой киназы и повышению устойчивости сердца к действию И/Р [75] (рис.1). Установлено, что ХГ не влияет на уровень p-GSK3β в миокарде мышей [76] (табл. 2). Следовательно, GSK3β не играет существенной роли в инфаркт-лимитирующем эффекте ХГ. Вместе с тем, это пока единственное исследование, в котором оценивали роль GSK3β в кардиопротекторном эффекте адаптации к гипоксии.
Гексокиназа 2 (HK-2, hexokinase 2). Полагают, что связывание HK-2 с митохондриями предупреждает апоптоз кардиомиоцитов [8] (рис.1). Установлено, что ХПГ вызывает транслокацию гексокиназы в митохондрии [77]. Кроме того, было установлено, что ХПГ вызывает увеличение экспрессии HK-1 и HK-2 в миокарде [77]. ХНГ также повышала экспрессию HK-1 и HK-2 в миокарде, усиливала ассоциацию HK-2 с митохондриями [67]. Аналогичные эффекты ХНГ оказывала у крыс линии SHR [78] (табл. 2).
Rho-киназа (ROCK, Rho-associated protein kinase). Известно, Rho-киназа участвует в апоптозе кардиомиоцитов [79], а ее ингибирование способствует уменьшению размера инфаркта при И/Р сердца [80] (рис.1). Можно было ожидать, что экспрессия Rho-киназы снизится в миокарде крыс при адаптации. Однако оказалось, что гипертрофия правого желудочка при хронической гипоксии связана с активацией Rho-киназы [81]. Показано, что при ХПГ гипертрофия левого желудочка также связана активацией Rho-киназы [82]. Имеет ли указанная киназа отношение к кардиопротекторному эффекту хронической гипоксии, пока неясно.
Роль КАТФ-каналов в кардиопротекторном эффекте ХГ. Известно, что в кардиомиоцитах существует два основных пула АТФ-чувствительных К+-каналов (КАТФ-каналов): сарколеммальные КАТФ-каналы (саркКАТФ-каналы) и митохондриальные КАТФ-каналы (митоКАТФ-каналы) [83–85]. Оба субтипа КАТФ-каналов участвуют в кардиопротекторном эффекте ишемического пре- и посткондиционирования [9, 84, 85]. Активаторы КАТФ-каналов повышают устойчивость сердца к И/Р [83]. Следовательно, были основания предполагать, что КАТФ-каналы могут принимать участие в кардиопротекторном эффекте ХГ.
В 1997 г. было установлено, что ХГ вызывает укорочение потенциала действия в волокнах Пуркинье сердца кролика в результате активации КАТФ-каналов [86]. Факт активации саркКАТФ-каналов в ответ на ХГ был подтвержден в исследовании, выполненном на изолированных кардиомиоцитах мышей с dominant negative suppression of Kir6.2/SUR2A [87]. Установлено, что ХПГ повышает устойчивость изолированного сердца крыс к повреждению, вызванному Ca2+-парадоксом за счет активации митоКАТФ-каналов [62]. Длительная умеренная гипоксия кардиомиобластов H9C2 приводила к повышению устойчивости этих клеток к гипоксии/реоксигенации [88]. Селективный ингибитор саркКАТФ-каналов HMR 1098 устранял цитопротекторный эффект умеренной гипоксии [88]. Было показано, что умеренная гипоксия приводила к усилению экспрессии SUR2A, регуляторной субъединицы КАТФ-канала, но не Kir6.2, субъединицы, формирующей пору КАТФ-канала. Избыточная экспрессия HIF-1α не влияла на уровень SUR2A [88]. Этот факт указывает на то, что HIF-1α не участвует в транскрипции мРНК, кодирующей SUR2A. Ингибитор mTOR рапамицин не влиял на экспрессию SUR2A в условиях умеренной гипоксии. LY 294002, ингибитор PI3K и PD 184352, ингибитор MEK устраняли подъем уровня SUR2A, вызванный гипоксией. Эти данные указывают на то, что экспрессия SUR2A в условиях гипоксии активируется при участии PI3K и MEK. Показано, что неселективный ингибитор КАТФ-каналов глибенкламид устраняет повышенную устойчивость изолированного сердца адаптированных кроликов к И/Р [44]. MCC-134, блокатор митоКАТФ-каналов и “открыватель” саркКАТФ-каналов, устранял инфаркт-лимитирующий и антиаритмический эффект ХПГ [2]. Эти данные указывают на важную роль митоКАТФ-каналов в кардиопротекторном эффекте ХГ. Важная роль КАТФ-каналов в инфаркт-лимитирующем и антиаритмическом эффекте ХГ была подтверждена в нашем более позднем исследовании [89, 90]. Нами было установлено, что глибенкламид и селективный ингибитор митоКАТФ-каналов 5-гидроксидеканоат устраняют инфаркт-лимитирующий эффект ХНГ [5, 91].
Следует отметить, что не все исследователи смогли подтвердить важную роль КАТФ-каналов в адаптационном повышении толерантности сердца к И/Р. Так, согласно данным J. Forkel и соавт. [36], глибенкламид не устраняет повышение толерантности правого желудочка к И/Р у крыс, адаптированных к ХГ.
Таким образом, большинство исследований указывает, что инфаркт-лимитирующий эффект ХГ связан с активацией митоКАТФ-каналов. Вместе с тем, мы не можем исключить возможность того, что другие кардиопротекторные эффект могут быть связаны с саркКАТФ-каналами, которые также активируются при ХГ.
ЗАКЛЮЧЕНИЕ
Установлено, что ХГ существенно изменяет экспрессию микроРНК в миокарде. В условиях ХГ экспрессия miR-138, miR-23b, miR-31 и miR-21в усиливается, а экспрессия miR-184, microRNA‑199a‑5p снижается. Показано, что miR-138 и miR-184 усиливают толерантность кардиомиоцитов к гипоксии, а microRNA‑199a‑5p и miR-23b усиливают апоптоз кардиомиоцитов в условиях гипоксии. Показано, что ХГ вызывает увеличение продукции NO в миокарде. Инфаркт-лимитирующий эффект ХГ связан с активацией iNOS. Представленные данные свидетельствуют, что при ХГ активируются саркКАТФ-каналы и митоКАТФ-каналы. ХГ приводит к усилению экспрессии в миокарде следующих киназ: ПКСδ, ПКСε, ПКСζ, ПКСα, CaMKII, p-ERK1/2, p-p38, p-Akt, HK-1, HK-2. Адаптация к гипоксии усиливает ассоциацию HK-2 с митохондриями и вызывает транслокацию ПКСδ, PKCβII и PKCη в мембраны кардиомиоцитов. В исследовании, выполненном на адаптированных к гипоксии животных, не удалось обнаружить усиления экспрессии киназ: ПKA, p-GSK3β, AMPK и JNK. Доказано, что ПКСδ, ПКСε, ERK1/2, MEK1/2 участвуют в кардиопротекторном эффекте ХГ. Роль киназ JNK, PKG, Rho-киназы, mTOR и p38 в кардиопротекторном эффекте адаптации к гипоксии требует дальнейшего изучения. Инфаркт-лимитирующий эффект ХГ связан с активацией митоКАТФ-каналов.
Список литературы
Meerson F.Z., Ustinova E.E., Manukhina E.B. Prevention of cardiac arrhythmias by adaptation to hypoxia: regulatory mechanisms and cardiotropic effect. Biomed. Biochim. Acta. 48(2–3): S83–S88. 1989.
Kolar F., Neckár J., Ostádal B. MCC-134, a blocker of mitochondrial and opener of sarcolemmal ATP-sensitive K+ channels, abrogates cardioprotective effects of chronic hypoxia. Physiol. Res. 54(4): 467–471. 2005.
Naryzhnaya N.V., Mukhamedzyanov A.V., Lasukova T.V., Maslov L.N. Involvement of autonomic nervous system in antiarrhythmic effect of intermittent hypobaric hypoxia. Bull. Exp. Biol. Med. 163(3): 299–301. 2017.
Milano G., Corno A.F., Samaja M., Morel S., Vassalli G., von Segesser L.K. Daily reoxygenation decreases myocardial injury and improves post-ischaemic recovery after chronic hypoxia. Eur. J. Cardiothorac. Surg. 37(4): 942–949. 2010.
Tsibulnikov S.Y., Maslov L.N., Naryzhnaya N.V., Ma H., Lishmanov Y.B., Oeltgen P.R., Garlid K. Role of protein kinase C, PI3 kinase, tyrosine kinases, NO-synthase, KATP channels and MPT pore in the signaling pathway of the cardioprotective effect of chronic continuous hypoxia. Gen. Physiol. Biophys. 37(5): 537–547. 2018.
Maslov L.N., Naryzhnaya N.V., Prokudina E.S., Kolar F., Gorbunov A.S., Zhang Y., Wang H., Tsibulnikov S.Yu., Portnichenko A.G., Lasukova T.V., Lishmanov Yu.B. Preserved cardiac mitochondrial function and reduced ischemia/reperfusion injury afforded chronic continuous hypoxia: role of opioid receptors. Clin. Exp. Pharmacol. Physiol. 42(5): 496–501. 2015.
Yellon D.M., Downey J.M. Preconditioning the myocardium: from cellular physiology to clinical cardiology. Physiol. Rev. 83(4): 1113–1151. 2003.
Heusch G. Molecular basis of cardioprotection: signal transduction in ischemic pre-, post-, and remote conditioning. Circ. Res. 116(4): 674–699. 2015.
Cohen M.V., Downey J.M. Signalling pathways and mechanisms of protection in pre- and postconditioning: historical perspective and lessons for the future. Br. J. Pharmacol. 172(8): 1913–3132. 2015.
Fabian M.R., Sonenberg N., Filipowicz W. Regulation of mRNA translation and stability by microRNAs. Annu. Rev. Biochem. 79: 351–379. 2010.
McCreight J.C., Schneider S.E., Wilburn D.B., Swanson W.J. Evolution of microRNA in primates. PLoS One. 12(6): e0176596. 2017.
Han M., Toli J., Abdellatif M. MicroRNAs in the cardiovascular system. Curr. Opin. Cardiol. 26(3): 181–189. 2011.
Liu F., Li Y., Liu G. MicroRNA-200c exacerbates the ischemia/reperfusion injury of heart through targeting the glutaminase (GLS)-mediated glutamine metabolism. Eur. Rev. Med. Pharmacol. Sci. 21(14): 3282–3289. 2017.
Chen Y., Ye X., Yan F. MicroRNA 3113-5p is a novel marker for early cardiac ischemia/reperfusion injury. Diagn. Pathol. 14(1): 121. 2019.
Tsibulnikov S.Y., Maslov L.N., Gorbunov A.S., Voronkov N.S., Boshchenko A.A., Popov S.V., Prokudina E.S., Singh N., Downey J.M. A review of humoral factors in remote preconditioning of the heart. J. Cardiovasc. Pharmacol. Ther. 24(5): 403–421. 2019.
He S., Liu P., Jian Z., Li J., Zhu Y., Feng Z., Xiao Y. miR-138 protects cardiomyocytes from hypoxia-induced apoptosis via MLK3/JNK/c-jun pathway. Biochem. Biophys. Res. Commun. 441(4): 763–769. 2013.
Chen Q., Huang J., Wang H., Zhao J., Chen G., Qi J., Lin X., Chen L., Lin Q. Effects of chronic intermittent hypoxia on regulation of miRNA-214 in myocardial apoptosis in rats. Zhonghua Yi Xue Za Zhi. 95(16): 1214–1247. 2015.
Huang J., Li X., Li H., Su Z., Wang J., Zhang H. Down-regulation of microRNA-184 contributes to the development of cyanotic congenital heart diseases. Int. J. Clin. Exp. Pathol. 8(11): 14221–14227. 2015.
Zhou Y., Jia W.K., Jian Z., Zhao L., Liu C.C., Wang Y., Xiao Y.B. Downregulation of mic-roRNA‑199a‑5p protects cardiomyocytes in cyanotic congenital heart disease by attenuating endoplasmic reticulum stress. Mol. Med. Rep. 16(3): 2992–3000. 2017.
He W., Che H., Jin C., Ge S. Effects of miR-23b on hypoxia-induced cardiomyocytes apoptosis. Biomed. Pharmacother. 96: 812–817. 2017.
Ren J., Liu W., Li G.C., Jin M., You Z.X., Liu H.G., Hu Y. Atorvastatin attenuates myocardial hypertrophy induced by chronic intermittent hypoxia in vitro partly through miR-31/PKCε pathway. Curr. Med. Sci. 38(3): 405–412. 2018.
Zhang K., Ma Z., Wang W., Liu R., Zhang Y., Yuan M., Li G. Beneficial effects of tolvaptan on atrial remodeling induced by chronic intermittent hypoxia in rats. Cardiovasc. Ther. 36(6): e12466. 2018.
Меньщикова Е.Б., Ланкин В.З., Зенков Н.К., Бондарь И.А., Круговых Н.Ф., Труфакин В.А. Окислительный стресс.Прооксиданты и антиоксиданты. Москва. 2006. [Menshchikova E.B., Lankin V.Z., Zenkov N.K., Bondar I.A., Krugovikh N.F., Trufakin V.A. Okislitel`ny`j stress. Prooksidanty` i antioksidanty. [Oxidative Stress. Prooxidants and Antioxidants]. Moscow. 2006].
Santolini J. What does “NO-Synthase” stand for? Front. Biosci. (Landmark Ed). 24: 133–171. 2019.
Rouet-Benzineb P., Eddahibi S., Raffestin B., Laplace M., Depond S., Adnot S., Crozatier B. Induction of cardiac nitric oxide synthase 2 in rats exposed to chronic hypoxia. J. Mol. Cell Cardiol. 31(9): 1697–1708. 1999.
Jung F., Palmer L.A., Zhou N., Johns R.A. Hypoxic regulation of inducible nitric oxide synthase via hypoxia inducible factor-1 in cardiac myocytes. Circ. Res. 86(3): 319–325. 2000.
Ferreiro C.R., Chagas A.C., Carvalho M.H., Dantas A.P., Jatene M.B., Bento De Souza L.C., Lemos Da Luz P. Influence of hypoxia on nitric oxide synthase activity and gene expression in children with congenital heart disease: a novel pathophysiological adaptive mechanism. Circulation. 103(18): 2272–2276. 2001.
Grilli A., De Lutiis M.A., Patruno A., Speranza L., Gizzi F., Taccardi A.A., Di Napoli P., De Caterina R., Conti P., Felaco M. Inducible nitric oxide synthase and heme oxygenase-1 in rat heart: direct effect of chronic exposure to hypoxia. Ann. Clin. Lab. Sci. 33(2): 208–215. 2003.
Grilli A., De Lutiis M.A., Patruno A., Speranza L., Cataldi A., Centurione L., Taccardi A.A., Di Napoli P., De Caterina R., Barbacane R., Conti P., Felaco M. Effect of chronic hypoxia on inducible nitric oxide synthase expression in rat myocardial tissue. Exp. Biol. Med. (Maywood). 228(8): 935–942. 2003.
Yuan X., Zhu D., Guo X.L., Deng Y., Shang J., Liu K., Liu H.G. Telmisartan attenuates myocardial apoptosis induced by chronic intermittent hypoxia in rats: modulation of nitric oxide metabolism and inflammatory mediators. Sleep Breath. 19(2): 703–709. 2015.
Thompson L.P., Aguan K., Pinkas G., Weiner C.P. Chronic hypoxia increases the NO contribution of acetylcholine vasodilation of the fetal guinea pig heart. Am. J. Physiol. Regul. Integr. Comp. Physiol. 279(5): R1813–R1820. 2000.
Thompson L.P., Dong Y. Chronic hypoxia decreases endothelial nitric oxide synthase protein expression in fetal guinea pig hearts. J. Soc. Gynecol. Investig. 12(6): 388–395. 2005.
Thompson L., Dong Y., Evans L. Chronic hypoxia increases inducible NOS-derived nitric oxide in fetal guinea pig hearts. Pediatr. Res. 65(2): 188–192. 2009.
Shi Y., Pritchard K.A., Holman P., Rafiee P., Griffith O.W., Kalyanaraman B., Baker J.E. Chronic myocardial hypoxia increases nitric oxide synthase and decreases caveolin-3. Free Radic. Biol. Med. 29(8): 695–703. 2000.
Felaco M., Grilli A., Gorbunov N., Di Napoli P., De Lutiis M.A., Di Giulio C., Taccardi A.A., Barsotti A., Barbacane R.C., Reale M., Conti P. Endothelial NOS expression and ischemia-reperfusion in isolated working rat heart from hypoxic and hyperoxic conditions. Biochim. Biophys. Acta. 1524(2–3): 203–211. 2000.
Forkel J., Chen X., Wandinger S., Keser F., Duschin A., Schwanke U., Frede S., Massoudy P., Schulz R., Jakob H., Heusch G. Responses of chronically hypoxic rat hearts to ischemia: KATP channel blockade does not abolish increased RV tolerance to ischemia. Am J Physiol. Heart Circ. Physiol. 286(2): H545–H551. 2004.
Quing M., Görlach A., Schumacher K., Wöltje M., Vazquez-Jimenez J.F., Hess J., Seghaye M.C. The hypoxia-inducible factor HIF-1 promotes intramyocardial expression of VEGF in infants with congenital cardiac defects. Basic Res. Cardiol. 102(3): 224–232. 2007.
Wang N., Chang Y., Chen L., Guo Y.J., Zhao Y.S., Guo Q.H., Ji E.S. Tanshinone IIA protects against chronic intermittent hypoxia-induced myocardial injury via activating the endothelin 1 pathway. Biomed. Pharmacother. 95: 1013–1020. 2017.
La Padula P.H., Etchegoyen M., Czerniczyniec A., Piotrkowski B., Arnaiz S.L., Milei J., Costa L.E. Cardioprotection after acute exposure to simulated high altitude in rats. Role of nitric oxide. Nitric Oxide. 73: 52–59. 2018.
La Padula P., Bustamante J., Czerniczyniec A., Costa L.E. Time course of regression of the protection conferred by simulated high altitude to rat myocardium: correlation with mtNOS. J. Appl. Physiol. (1985). 105(3): 951–957. 2008.
Ghafourifar P., Cadenas E. Mitochondrial nitric oxide synthase. Trends Pharmacol. Sci. 26(4): 190–195. 2005.
Baker J.E., Holman P., Kalyanaraman B., Griffith O.W., Pritchard K.A. Adaptation to chronic hypoxia confers tolerance to subsequent myocardial ischemia by increased nitric oxide production. Ann. N. Y. Acad. Sci. 874: 236–253. 1999.
Earley S., Walker B.R. Increased nitric oxide production following chronic hypoxia contributes to attenuated systemic vasoconstriction. Am. J. Physiol. Heart Circ. Physiol. 284(5): H1655–H1661. 2003.
Fitzpatrick C.M., Shi Y., Hutchins W.C., Su J., Gross G.J., Ostadal B., Tweddell J.S., Baker J.E. Cardioprotection in chronically hypoxic rabbits persists on exposure to normoxia: role of NOS and KATP channels. Am. J. Physiol. Heart Circ. Physiol. 288(1): H62–H68. 2005.
Rafiee P., Shi Y., Kong X., Pritchard K.A., Tweddell J.S., Litwin S.B., Mussatto K., Jaquiss R.D., Su J., Baker J.E. Activation of protein kinases in chronically hypoxic infant human and rabbit hearts: role in cardioprotection. Circulation. 106(2): 239–245. 2002.
Morel O.E., Buvry A., Le Corvoisier P., Tual L., Favret F., León-Velarde F., Crozatier B., Richalet J.P. Effects of nifedipine-induced pulmonary vasodilatation on cardiac receptors and protein kinase C isoforms in the chronically hypoxic rat. Pflugers Arch. 446(3): 356–364. 2003.
Neckar J., Marková I., Novák F., Nováková O., Szárszoi O., Ost'ádal B., Kolár F. Increased expression and altered subcellular distribution of PKC-δ in chronically hypoxic rat myocardium: involvement in cardioprotection. Am. J. Physiol. Heart Circ. Physiol. 288(4): H1566–H1572. 2005.
Нарыжная Н.В., Маслов Л.Н., Халиулин И.Г., Пей Ж.-М., Жанг Н., Цепокина А.В., Хуторная М.В., Кутихин А.Г., Лишманов Ю.Б. Адаптация с помощью хронической нормобарической гипоксии увеличивает толерантность кардиомиоцитов крыс к аноксии-реоксигенации: роль протеинкиназ. Рос. физиол. журн. им. И.М. Сеченова. 102(12): 1462–1471. 2016. [Naryzhnaia N.V., Maslov L.N., Khaliulin I.G., Pei J.M., Zhang Y., Tsepokina A.V., Khutornaya M.V., Kutikhin A.G., Lishmanov Y.B. Chronic continuous normobaric hypoxia augments cell tolerance to anoxia/reoxygenation: the role of protein kinases. Russ. J. Physiol. 102(12): 1462–1471. 2016. (In Russ)].
El Alwani M., Usta J., Nemer G., El Sabban M., Nasser M., Bitar H., Souki R., Dbaibo G.S., Bitar F.F. Regulation of the sphingolipid signaling pathways in the growing and hypoxic rat heart. Prostaglandins Other Lipid Mediat. 78(1–4): 249–263. 2005.
Kolar F., Jezková J., Balková P., Breh J., Neckár J., Novák F., Nováková O., Tomásová H., Srbová M., Ost'ádal B., Wilhelm J., Herget J. Role of oxidative stress in PKC-δ upregulation and cardioprotection induced by chronic intermittent hypoxia. Am. J. Physiol. Heart Circ. Physiol. 292(1): H224–H230. 2007.
Hlaváčková M., Kožichová K., Neckář J., Kolář F., Musters R.J., Novák F., Nováková O. Up-regulation and redistribution of protein kinase C-δ in chronically hypoxic heart. Mol. Cell. Biochem. 345(1–2): 271–282. 2010.
Krylatov A.V., Maslov L.N., Voronkov N.S., Boshchenko A.A., Popov S.V., Gomez L., Wang H., Jaggi A.S., Downey J.M. Reactive oxygen species as intracellular signaling molecules in the cardiovascular system. Curr. Cardiol. Rev. 14(4): 290–300. 2018.
Hlaváčková M., Neckár J., Jezková J., Balková P., Stanková B., Nováková O., Kolár F., Novák F. Dietary polyunsaturated fatty acids alter myocardial protein kinase C expression and affect cardioprotection induced by chronic hypoxia. Exp. Biol. Med. (Maywood). 232(6): 823–832. 2007.
Ma H.J., Li Q., Ma H.J., Guan Y., Shi M., Yang J., Li D.P., Zhang Y. Chronic intermittent hypobaric hypoxia ameliorates ischemia/reperfusion-induced calcium overload in heart via Na+/Ca2+ exchanger in developing rats. Cell Physiol. Biochem. 34(2): 313–324. 2014.
Holzerova K., Hlaváčková M., Žurmanová J., Borchert G., Neckář J., Kolář F., Novák F., Nováková O. Involvement of PKCε in cardioprotection induced by adaptation to chronic continuous hypoxia. Physiol. Res. 64(2): 191–201. 2015.
Micova P., Hahnova K., Hlavackova M., Elsnicova B., Chytilova A., Holzerova K., Zurmanova J., Neckar J., Kolar F., Novakova O., Novotny J. Chronic intermittent hypoxia affects the cytosolic phospholipase A2α/cyclooxygenase 2 pathway via β2-adrenoceptor-mediated ERK/p38 stimulation. Mol. Cell Biochem. 423(1-2): 151–163. 2016.
Zeng C., Liang B., Jiang R., Shi Y., Du Y. Protein kinase C isozyme expression in right ventricular hypertrophy induced by pulmonary hypertension in chronically hypoxic rats. Mol. Med. Rep. 16(4): 3833–3840. 2017.
Xie S., Deng Y., Pan Y.Y., Ren J., Jin M., Wang Y., Wang Z.H., Zhu D., Guo X.L., Yuan X., Shang J., Liu H.G. Chronic intermittent hypoxia induces cardiac hypertrophy by impairing autophagy through the adenosine 5'-monophosphate-activated protein kinase pathway. Arch. Biochem. Biophys. 606: 41–52. 2016.
Ling H., Gray C.B., Zambon A.C., Grimm M., Gu Y., Dalton N., Purcell N.H., Peterson K., Brown J.H. CaMKIIδ mediates myocardial ischemia/reperfusion injury through nuclear factor-κB. Circ. Res. 112(6): 935–944. 2013.
Zhao P.J., Pan J., Li F., Sun K. Effects of chronic hypoxia on the expression of calmodulin and calcicum/calmodulin-dependent protein kinase II and the calcium activity in myocardial cells in young rats. Zhongguo Dang Dai Er Ke Za Zhi. 10(3): 381–385. 2008.
Nehra S., Bhardwaj V., Kar S., Saraswat D. Chronic hypobaric hypoxia induces right ventricular hypertrophy and apoptosis in rats: therapeutic potential of nanocurcumin in improving adaptation. High Alt. Med. Biol. 17(4): 342–352. 2016.
Xie Y., Zhu W.Z., Zhu Y., Chen L., Zhou Z.N., Yang H.T. Intermittent high altitude hypoxia protects the heart against lethal Ca2+ overload injury. Life Sci. 76(5): 559–772. 2004.
Strnisková M., Ravingerová T., Neckár J., Kolár F., Pastoreková S., Barancík M. Changes in the expression and/or activation of regulatory proteins in rat hearts adapted to chronic hypoxia. Gen. Physiol. Biophys. 25(1): 25–41. 2006.
Milano G., von Segesser L.K., Morel S., Joncic A., Bianciardi P., Vassalli G., Samaja M. Phosphorylation of phosphatidylinositol-3-kinase-protein kinase B and extracellular signal-regulated kinases 1/2 mediate reoxygenation-induced cardioprotection during hypoxia. Exp. Biol. Med. (Maywood). 235(3): 401–410. 2010.
Ravingerová T., Matejíková J., Neckár J., Andelová E., Kolár F. Differential role of PI3K/Akt pathway in the infarct size limitation and antiarrhythmic protection in the rat heart. Mol. Cell. Biochem. 297(1–2): 111–120. 2007.
Jia W., Jian Z., Li J., Luo L., Zhao L., Zhou Y., Tang F., Xiao Y. Upregulated ATF6 contributes to chronic intermittent hypoxia-afforded protection against myocardial ischemia/reperfusion injury. Int. J. Mol. Med. 37(5): 1199–1208. 2016.
Kolar D., Gresikova M., Waskova-Arnostova P., Elsnicova B., Kohutova J., Hornikova D., Vebr P., Neckar J., Blahova T., Kasparova D., Novotny J., Kolar F., Novakova O., Zurmanova J.M. Adaptation to chronic continuous hypoxia potentiates Akt/HK2 anti-apoptotic pathway during brief myocardial ischemia/reperfusion insult. Mol. Cell Biochem. 432(1–2): 99–108. 2017.
Larsen K.O., Lygren B., Sjaastad I., Krobert K.A., Arnkvaern K., Florholmen G., Larsen A.K., Levy F.O., Taskén K., Skjønsberg O.H., Christensen G. Diastolic dysfunction in alveolar hypoxia: a role for interleukin-18-mediated increase in protein phosphatase 2A. Cardiovasc. Res. 80(1): 47–54. 2008.
Morel S., Milano G., Ludunge K.M., Corno A.F., Samaja M., Fleury S., Bonny C., Kappenberger L., von Segesser L.K., Vassalli G. Brief reoxygenation episodes during chronic hypoxia enhance posthypoxic recovery of LV function: role of mitogen-activated protein kinase signaling pathways. Basic Res. Cardiol. 101(4): 336–345. 2006.
Milano G., Morel S., Bonny C., Samaja M., von Segesser L.K., Nicod P., Vassalli G. A peptide inhibitor of c-Jun NH2-terminal kinase reduces myocardial ischemia-reperfusion injury and infarct size in vivo. Am. J. Physiol. Heart Circ. Physiol. 292(4): H1828–H1835. 2007.
Heidbreder M., Naumann A., Tempel K., Dominiak P., Dendorfer A. Remote vs. ischaemic preconditioning: the differential role of mitogen-activated protein kinase pathways. Cardiovasc. Res. 78(1): 108–115. 2008.
He S., Liu S., Wu X., Xin M., Ding S., Xin D., Ouyang H., Zhang J. Protective role of downregulated MLK3 in myocardial adaptation to chronic hypoxia. J. Physiol. Biochem. 73(3): 371–380. 2016.
Zhao Y.S., An J.R., Yang S., Guan P., Yu F.Y., Li W., Li J.R., Guo Y., Sun Z.M., Ji E.S. Hydrogen and oxygen mixture to improve cardiac dysfunction and myocardial pathological changes induced by intermittent hypoxia in rats. Oxid. Med. Cell Longev. 2019: 7415212. 2019.
Wagner C., Tillack D., Simonis G., Strasser R.H., Weinbrenner C. Ischemic post-conditioning reduces infarct size of the in vivo rat heart: role of PI3-K, mTOR, GSK-3beta, and apoptosis. Mol. Cell Biochem. 339(1–2): 135–147. 2010.
Miura T., Miki T. GSK-3β, a therapeutic target for cardiomyocyte protection. Circ. J. 73(7): 1184–1192. 2009.
McCarthy J., Lochner A., Opie L.H., Sack M.N., Essop M.F. PKCε promotes cardiac mitochondrial and metabolic adaptation to chronic hypobaric hypoxia by GSK3β inhibition. J. Cell Physiol. 226(9): 2457–2468. 2011.
Waskova-Arnostova P., Elsnicova B., Kasparova D., Hornikova D., Kolar F., Novotny J., Zurmanova J. Cardioprotective adaptation of rats to intermittent hypobaric hypoxia is accompanied by the increased association of hexokinase with mitochondria. J. Appl. Physiol. (1985). 119(12): 1487–1493. 2015.
Nedvedova I., Kolar D., Elsnicova B., Hornikova D., Novotny J., Kalous M., Pravenec M., Neckar J., Kolar F., Zurmanova J.M. Mitochondrial genome modulates myocardial Akt/Glut/HK salvage pathway in spontaneously hypertensive rats adapted to chronic hypoxia. Physiol. Genomics. 50(7): 532–541. 2018.
Zhang J., Li X.X., Bian H.J., Liu X.B., Ji X.P., Zhang Y. Inhibition of the activity of Rho-kinase reduces cardiomyocyte apoptosis in heart ischemia/reperfusion via suppressing JNK-mediated AIF translocation. Clin. Chim. Acta. 401(1–2): 76–80. 2009.
Bao W., Hu E., Tao L., Boyce R., Mirabile R., Thudium D.T., Ma X.L., Willette R.N., Yue T.L. Inhibition of Rho-kinase protects the heart against ischemia/reperfusion injury. Cardiovasc. Res. 61(3): 548–558. 2004.
Gosal K., Dunlop K., Dhaliwal R., Ivanovska J., Kantores C., Desjardins J.F., Connelly K.A., McNamara P.J., Jain A., Jankov R.P. Rho kinase mediates right ventricular systolic dysfunction in rats with chronic neonatal pulmonary hypertension. Am. J. Respir. Cell Mol Biol. 52(6): 717–727. 2015.
Wang Z.H., Zhu D., Xie S., Deng Y., Pan Y., Ren J., Liu H.G. Inhibition of Rho-kinase attenuates left ventricular remodeling caused by chronic intermittent hypoxia in rats via suppressing myocardial inflammation and apoptosis. J. Cardiovasc. Pharmacol. 70(2): 102–109. 2017.
Grover G.J., Garlid K.D. ATP-Sensitive potassium channels: a review of their cardioprotective pharmacology. J. Mol. Cell Cardiol. 32(4): 677–695. 2000.
Peart J.N., Gross G.J. Sarcolemmal and mitochondrial KATP channels and myocardial ischemic preconditioning. J. Cell Mol. Med. 6(4): 453–464. 2002.
Gross G.J., Peart J.N. KATP channels and myocardial preconditioning: an update. Am. J. Physiol. Heart Circ. Physiol. 285(3): H921–H930. 2003.
Baker J.E., Contney S.J., Gross G.J., Bosnjak Z.J. KATP channel activation in a rabbit model of chronic myocardial hypoxia. J. Mol. Cell Cardiol. 29(2): 845–848. 1997.
Zhu Z., Burnett C.M., Maksymov G., Stepniak E., Sierra A., Subbotina E., Anderson M.E., Coetzee W.A., Hodgson-Zingman D.M., Zingman L.V. Reduction in number of sarcolemmal KATP channels slows cardiac action potential duration shortening under hypoxia. Biochem. Biophys. Res. Commun. 415(4): 637–641. 2011.
Crawford R.M., Jovanović S., Budas G.R., Davies A.M., Lad H., Wenger R.H., Robertson K.A., Roy D.J., Ranki H.J., Jovanović A. Chronic mild hypoxia protects heart-derived H9c2 cells against acute hypoxia/reoxygenation by regulating expression of the SUR2A subunit of the ATP-sensitive K+ channel. J. Biol. Chem. 278(33): 31444-31455. 2003.
Kolar F., Nekar J., Ostadal B., Maslov L.N., Stakheev D.L., Tayurskaya A.S., Lishmanov Y.B. Role of ATP-sensitive K+-channels in antiarrhythmic and cardioprotective action of adaptation to intermittent hypobaric hypoxia. Bull. Exp. Biol. Med. 145(4): 418–421. 2008.
Нарыжная Н.В., Некар Я., Маслов Л.Н., Лишманов Ю.Б., Колар Ф., Ласукова Т.В. Роль сарколеммальных и митохондриальных KATф-каналов в реализации кардиопротективного и антиаритмического эффектов разных режимов гипобарической адаптации. Рос физиол журн им. И.М. Сеченова. 95(8): 837–849. 2009. [Naryzhnaia N.V., Neckar J., Maslov L.N., Lishmanov Iu.B., Kolar F., Lasukova T.V. The role of sarcolemmal and mitochondrial KATP-channels in realization of the cardioprotection and antiarrhythmic effect of different regimens of hypobaric adaptation. Russ. J. Physiol. 95(8): 837–849. 2009. (In Russ].
Lishmanov Y.B., Naryzhnaya N.V., Tsibul’nikov S.Y., Wang H., Maslov L.N. Role of ATP-Sensitive K+ channels in myocardial infarct size-limiting effect of chronic continuous normobaric hypoxia. Bull. Exp. Biol. Med. 163(1): 22–24. 2017.
Дополнительные материалы отсутствуют.
Инструменты
Российский физиологический журнал им. И.М. Сеченова