

Российский физиологический журнал им. И.М. Сеченова, 2020, T. 106, № 10, стр. 1251-1265
Оценка эффективности превентивной терапии с индуктором шаперонов U133 в модели доклинической стадии болезни Паркинсона у пожилых крыс
Д. В. Белан 1, *, С. Г. Полоник 2, И. В. Екимова 1
1 Институт эволюционной физиологии и биохимии им. И.М. Сеченова
Российской академии наук
Санкт-Петербург, Россия
2 Тихоокеанский институт биоорганической химии им. Г.Б. Елякова
Дальневосточного отделения Российской академии наук
Владивосток, Россия
* E-mail: daf205@yandex.ru
Поступила в редакцию 16.04.2020
После доработки 25.05.2020
Принята к публикации 12.07.2020
Аннотация
Болезнь Паркинсона (БП) – хроническое неуклонно прогрессирующее нейродегенеративное заболевание с преимущественным поражением дофамин (ДА-)ергических нейронов нигростриатной системы головного мозга. БП чаще всего поражает пожилых людей и относится к числу неизлечимых. Нарушение конформации белка α-синуклеина и его гиперфосфолирование, а также развитие хронического нейровоспаления являются основными звеньями патогенеза нейродегенерации при БП. Фармакотерапия, направленная на мобилизацию стресс-индуцируемого шаперона Hsp70, играющего решающую роль в контроле качества белковых молекул и обладающего иммуномодулирующей активностью, представляется перспективной для разработки превентивной терапии БП. В настоящем исследовании в модели доклинической стадии БП у крыс пожилого возраста, созданной путем интраназального введения ингибитора протеасом лактацистина, применена терапия, основанная на системном введении индуктора шаперона Hsp70 – низкомолекулярного хиноидного соединения U133 (ацетилированного 2,3,7-трис-О-глюкозида эхинохрома). Показано, что соединение U133 вызывает отставленное во времени повышение содержания Hsp70 в компактной части черной субстанции (кчЧС) у пожилых животных. Превентивная Hsp70-индуцирующая терапия с U133 в модели доклинической стадии БП у крыс пожилого возраста ослабляет процесс нейродегенерации в кчЧС и противодействует развитию нейровоспаления. При этом отмечается уменьшение количества агрегированного α-синуклеина и регрессия посттрансляционно модифицированного фосфорилированного по Ser129 α-синуклеина. Полученные данные свидетельствуют, что пролекарство, низкомолекулярный препарат U133, обладает значимым терапевтическим потенциалом при развитии Паркинсон-подобной патологии в пожилом возрасте. Результаты исследования имеют научно-практическую значимость для разработки инновационной технологии превентивной фармакотерапии БП на основе отечественного препарата U133.
Болезнь Паркинсона (БП) – это мультисистемное хроническое неизлечимое нейродегенеративное заболевание, которым страдают преимущественно пожилые люди. БП занимает второе место в мире по частоте встречаемости, уступая лишь болезни Альцгеймера [1]. Основным патоморфологическим признаком БП является прогрессирующая дегенерация дофамин (ДА)-ергических нейронов в компактной части черной субстанции, сопряженная с формированием телец Леви, содержащих в качестве одного из основных компонентов агрегированный α-синуклеин, и развитием нейровоспаления [2]. Помимо нейронов кчЧС среднего мозга, при БП поражаются вегетативные ядра ствола, голубое пятно, базальное ядро Мейнерта, обонятельные луковицы, кора больших полушарий. В нейронах вышеперечисленных структур также выявляются тельца Леви [3]. В настоящее время лечение БП носит симптоматический характер и не способно повлиять на патогенез нейродегенерации. Успех в лечении БП связывают с разработкой превентивной нейропротективной фармакотерапии, нацеленной на основные молекулярные мишени, определяющие гибель нейронов при БП. В настоящее время не существует средств, доказательно замедляющих прогрессирование БП.
В качестве одного из основных молекулярных звеньев патогенеза нейродегенерации при БП рассматривается нарушение конформации белка α-синуклеина, приводящее к его агрегированию [4, 5]. В результате агрегации формируются токсичные олигомеры или протофибриллы, которые затем превращаются в нерастворимые фибриллы α-синуклеина [6]. Наличие аберрантных форм α-синуклеина в нейронах кчЧС характерно как для наследственной, так и для спорадической формы БП [7]. Посттрансляционные модификации α-синуклеина (фосфорилирование, нитрирование и окисление) могут усиливать его способность к агрегации [5]. Большая часть (около 90%) α-синуклеина, входящего в состав телец Леви, фосфорилирована в положении Ser129, это указывает на то, что такая посттрансляционная модификация может быть связана с образованием и/или токсичностью агрегированных форм белка [8]. Олигомерные формы α-синуклеина способны покидать нейроны и распространяться между “здоровыми” нервными клетками по прион-подобному механизму; проникая в нейроны, аберрантный α-синуклеин инициирует переход внутриклеточного растворимого мономерного α-синуклеина в агрегированную патологическую форму [9]. Показано, что патологические формы α-синуклеина могут нарушать процесс синтеза ДА и его загрузки в везикулы, повышая тем самым уровень цитозольного ДА [7]. Помимо того, что цитозольный ДА увеличивает уровень внутриклеточного окислительного стресса, он способен связываться с токсичными α-синуклеиновыми протофибриллами и замедлять их превращение в нерастворимые, менее токсичные фибриллы [10]. Кроме этого, агрегаты α-синуклеина способны увеличивать частоту спонтанной спайковой активности ДА-ергических нейронов в кчЧС, истощая тем самым запасы АТФ в клетке и повышая уровень активных форм кислорода [11]. Также внеклеточные олигомеры α-синуклеина активируют систему микроглии через Toll-like рецепторы на поверхности микроглиоцитов, что приводит к повышению синтеза провоспалительных цитокинов и развитию хронического нейровоспаления, которое способствует прогрессированию нейродегенерации [12–14].
Важнейшими молекулярными механизмами защиты нейронов от процесса агрегации белковых молекул являются механизмы, направленные на поддержание протеостаза, в которых участвуют около 2000 факторов, однако ключевыми являются убиквитин-протеасомная система и система белков теплового шока HSP70. Эти системы составляют основные функциональные модули протеостазной сети, которые уравновешивают биосинтез белков, фолдинг и рефолдинг, транслокацию, сборку, разборку и утилизацию белков; неисправности в этих системах могут привести к развитию ряда нейродегенеративных заболеваний. Одним из прямых свидетельств, подтверждающих вовлечение молекулярных шаперонов в патогенез БП, явились результаты исследований тканей головного мозга (postmortem) у пациентов с БП, которые указывают не только на присутствие шаперонов семейства HSP70 в тельцах Леви [15], но и на их колокализацию с агрегатами α-синуклеина [16]. Кроме того, показано низкое содержание некоторых шаперонов семейства HSP70 в секционном материале кчЧС при БП, что отражает ослабление механизмов конформационного контроля белков и клеточной защиты при развитии заболевания [17]. Ряд данных, имеющихся в литературе, указывает, что увеличение содержания стресс-индуцируемого белка Hsp70 в ДА-ергических нейронах кчЧС приводит к повышению их выживаемости в моделях БП у животных [18, 19]. Поэтому в последние 20 лет идет поиск препаратов, способных мобилизовать шаперонный механизм Hsp70 в нейронах головного мозга. Особое внимание уделяется так называемым химическим шаперонам, т.е. малым молекулам, хорошо проникающим через гематоэнцефалический барьер и не обладающим побочными токсическими эффектами. В этом отношении одним из перспективных индукторов Hsp70 является активатор транскрипционного фактора белков теплового шока HSF1, нетоксичное низкомолекулярное соединение трисацетилглюкозид эхинохрома U133 [20, 21]. Ранее нами показано, что препарат U133, вызывающий повышение уровней Hsp70 и HSP40 в кчЧС, значительно ослабляет процесс нейродегенерации в нигростриатной системе в модели клинической стадии БП у крыс среднего возраста [22]. При этом возрастание уровня Hsp70 сопряжено с уменьшением количества агрегатов α‑синуклеина и нейровоспаления в кчЧС. Однако спорадическая форма БП обычно развивается в позднем возрасте [23], для которого характерна потеря функции шаперонной и убиквитин-протеасомной систем [24]. У пожилых лиц отмечено снижение уровня и активности транскрипционного фактора HSF1, скорости транскрипции генов белка Hsp70 и синтеза шаперонов [24–26]. Оставалось неясным, будет ли проявляться Hsp70-индуцирующий эффект соединения U133 в пожилом возрасте и каковы перспективы применения препарата U133 для превентивной нейропротективной терапии БП при старении. Задача настоящего исследования – оценить нейропротективный эффект препарата U133 и его способность противодействовать развитию α-синуклеиновой патологии и хронического нейровоспаления у крыс в пожилом возрасте в лактацистиновой модели доклинической стадии БП.
МЕТОДЫ ИССЛЕДОВАНИЯ
Эксперименты проводились на крысах-самцах линии Вистар пожилого возраста (20–22 мес.), которых содержали в виварии при постоянной температуре окружающей среды 23 ± 1°C и свободном доступе к воде и пище. Манипуляции с животными осуществлялись в соответствии с принципами Базельской декларации и протоколом, утвержденным этическим комитетом Института эволюционной физиологии и биохимии им. И.М. Сеченова РАН, находящимся в соответствии с национальными и международными требованиями.
Для моделирования нейродегенеративной патологии нигростриатной системы, характерной для БП, использовали специфический ингибитор ферментативной активности протеасом лактацистин (ЛЦ, Enzo, США), который растворяли в апирогенном фосфатном буфере (pH 7.4) и вводили в дозе 100 мкг/8 мкл в каждую ноздрю крысы (n = 16) дважды с недельным интервалом; общая доза на одну крысу составила 400 мкг. Методология интраназального введения ЛЦ описана нами ранее [27]. Препарат U133 (5 мг/кг) растворяли в DMSO и вводили внутрибрюшинно животным (n = 8) через 4 ч после каждого введения ЛЦ и через 7 дней после последнего введения ЛЦ. Контрольной группе животных (n = 10) вводили эквивалентный объем растворителя ЛЦ и U133.
Отечественный препарат U133 получен в лаборатории органического синтеза природных соединений Тихоокеанского института биоорганической химии им. Г.Б. Елякова ДВО РАН путем глюкозилирования эхинохрома (2,3,5,7,8-пентагидрокси-1,4-нафтохинона) – природного пигмента морских ежей – и является ацетилированным 2,3,7-трис-О-глюкозидом эхинохрома [28, 29]. Соединение U133 относится к классу малых молекул (M = 1257 Да) и хорошо проникает через гематоэнцефалический барьер. В микромолярных концентрациях U133 способен вызывать активацию транскрипционного фактора теплового шока HSF1, что приводит к индукции синтеза белков теплового шока Hsp70 и HSP40 (Hsp40/Hdj1) [21, 22]. U133 не обладает токсическим свойством и проявляет разнообразную биологическую активность, в частности, антиоксидантную и антиканцерогенную [22, 30, 31]. Тестирование нейропротективного эффекта препарата U133 и его способности противодействовать развитию α-синуклеиновой патологии и хронического нейровоспаления у крыс пожилого возраста в модели доклинической стадии БП ранее никем не проводилось.
На 21-й день после первой микроинъекции ЛЦ крыс декапитировали, головной мозг извлекали из черепной коробки и фиксировали в 4%-ном растворе параформальдегида при 4°С в течение 5 дней, промывали в PBS и переносили в 20%-ный раствор сахарозы при 4°С для криопротекции. Спустя 3–5 дней мозг замораживали в изопентане при температуре –45°С и хранили при –80°С. При помощи криостата Leica (Германия) при –20°С приготавливали чередующиеся серии фронтальных срезов головного мозга толщиной 10 мкм, содержащие кчЧС. Для проведения анализа Вестерн-блот после декапитации из головного мозга выделяли кчЧС, замораживали и хранили при –80°С.
Для сравнительного анализа содержания в кчЧС белка Hsp70 и различных форм α-синуклеина (общей и фосфорилированной в положении Ser129) применяли метод Вестерн-блот. Предварительно образцы тканей головного мозга лизировали с использованием High RIPA буфера и гомогенизировали металлической палочкой. Белки разделяли с помощью электрофореза в полиакриламидном геле, затем переносили на PVDF-мембрану Immobilon-P (Millipore Corporation, США). Для предотвращения неспецифического связывания антител мембрану инкубировали в PBS с добавлением 0.1% Tween-20 и 3% бычьего сывороточного альбумина в течение 1 ч при комнатной температуре. Иммуномечение проводили с использованием первичных моноклональных антител мыши к индуцибельному Hsp70 (клон 3C5 [32], любезно предоставлен г.н.с. лаборатории защитных механизмов клетки Института цитологии РАН Маргулисом Б.А.), поликлональных антител кролика к общей водорастворимой форме α-синуклеина (1 : 500; Abcam, Великобритания) или к α-синуклеину, фосфорилированному в положении Ser129 (1 : 700; Abcam, Великобритания). В качестве лоадинг-контроля использовали GAPDH (моноклональные антитела мыши, 1 : 1000; Abcam, Великобритания). Денситометрический анализ количества белка осуществляли с помощью программы ImageJ. Уровень серого бэндов, иммунореактивных к Hsp70, общему α-синуклеину и α-синуклеину, фосфорилированному в положении Ser129, скорректирован по фоновому сигналу и нормирован на сигнал GAPDH, выявляемый для определения общего количества белка в пробах.
Биотин-стрептавидиновый метод использовали для иммуномечения ДА-ергических нейронов и микроглиоцитов в кчЧС. Срезы промывали в PBS, затем в течение 10 мин выполняли термическую демаскировку антигенов в цитратном буфере (96–98°С; pH 6.0). Для гашения активности эндогенной пероксидазы срезы 20 мин выдерживали в 0.3%-ном растворе пероксида водорода на метаноле. После трехкратной промывки в PBS срезы на 10 мин помещали в PBS с добавлением Triton Х-100 (Sigma, США) для повышения проницаемости плазматических мембран и затем преинкубировали в блокирующем растворе (3%-ная сыворотка козы (Sigma, США) с 0.01% Triton X-100 на ФБ) в течение 1 ч при комнатной температуре, после чего инкубировали в течение 24 ч при температуре 4°С с первичными поликлональными антителами кролика против тирозингидроксилазы (ТГ; 1 : 900; Abcam, Великобритания) или Iba1 (маркер микроглии; 1 : 500, Sigma, США), затем последовательно в течение 1 ч во вторичных антителах против IgG кролика, конъюгированных с биотином (1 : 450; VectorLab, США), и в комплексе стрептавидин-пероксидаза (1 : 400; VectorLab, США). После синхронной визуализации реакции с помощью 0.05%-ного раствора диаминобензидина срезы заключали под покровное стекло с помощью среды BioMount (Италия) и хранили при комнатной температуре. Специфичность иммуногистохимической реакции проверяли негативным контролем без первичных или вторичных антител. Изображения кчЧС получали с помощью микроскопа KarlZeiss (Германия) с встроенной видеокамерой и программного обеспечения для захвата изображения. Подсчет количества иммунопозитивных к ТГ нейронов в кчЧС и Iba1-иммунопозитивных микроглиоцитов проводили с помощью программы PhotoM (Черниговский, http://t-lambda.chart.ru). По каждому животному анализировали 10–12 снимков кчЧС.
Методом двойного иммуномечения с применением антител против общей или фосфорилированной в положении Ser129 форм α-синуклеина и ТГ изучена локализация различных форм α-синуклеина и наличие их агрегатов в ДА-ергических нейронах кчЧС. После стандартной предварительной обработки, описанной выше, срезы инкубировали с поликлональными антителами кролика к общему α-синуклеину (1 : 500; Abcam, Великобритания) или α-синуклеину, фосфорилированному в положении Ser129 (1 : 700; Abcam, Великобритания) в течение 24 ч при температуре 4°С, затем в течение 1 ч при комнатной температуре во вторичных антителах козы против IgG кролика, конъюгированных с флуоресцентной меткой CFTM488 (1 : 600; Sigma, США), после чего срезы промывали в PBS и инкубировали с первичными моноклональными антителами мыши против ТГ (1 : 1000; Sigma, США) в течение ночи при комнатной температуре, затем в течение 1 ч во вторичных антителах козы против IgG мыши, конъюгированных с флуоресцентной меткой CFTM568 (1 : 600; Sigma, США). Cрезы заключали под покровное стекло с помощью среды Mowiol (Sigma, США) и хранили при температуре 4°С. Специфичность всех иммуногистохимических реакций проверяли негативным контролем без первичных или вторичных антител.
Флуоресцентную микроскопию проводили с помощью конфокального микроскопа DMI6000 и лазерной установки SP5 II (Leiсa Microsystems, Германия). Использовали иммерсионный объектив ×63 и лазеры с длиной волны возбуждения 488 и 568 нм. Изображения анализировали с помощью пакета программ Leica LAS AF. Подготовку к демонстрации проводили c помощью программы Photoshop CS5.
Анализ всех полученных данных осуществлялся с помощью программы Statistica 7.0. Статистический анализ данных, полученных посредством Вестерн-блота или иммуногистохимических исследований, выполняли с помощью параметрического дисперсионного анализа ANOVA для несвязанных выборок. Полученные результаты считались статистически значимыми при p ≤ 0.05.
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ И ИХ ОБСУЖДЕНИЕ
Для решения задачи работы использована ранее разработанная в лаборатории сравнительной термофизиологии Института эволюционной физиологии и биохимии РАН модель доклинической стадии БП у крыс, созданная на основе угнетения активности протеасом в головном мозге [27]. Данная модель характеризуется: а) допороговым (30–35%) уровнем гибели ДА-ергических нейронов в кчЧС по сравнению с уровнем клинической стадии (60–70%); б) развитием нейродегенеративного процесса в экстранигральных структурах (обонятельные луковицы, вентральная тегментальная область, голубое пятно); в) наличием α-синуклеиновой патологии и хронического нейровоспаления в кчЧС и экстранигральных структурах; г) нарушением сна, эмоционального поведения и когнитивных функций [33–37].
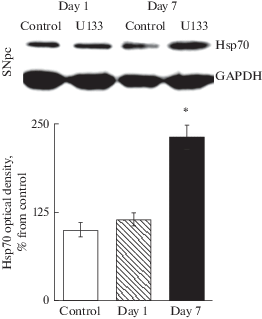
В предыдущих исследованиях мы показали, что введение препарата U133 крысам среднего возраста приводит к значительному возрастанию уровня стресс-индуцируемого Hsp70 в кчЧС через 1 сутки с момента инъекции [22]. В данной работе методом Вестерн-блота установлено, что после введения U133 пожилым крысам прирост Hsp70 в кчЧС составил 130% по сравнению с контролем. При этом повышение уровня Hsp70 отмечалось только через 7 сут с момента введения U133; в 1-е сутки изменений в содержании Hsp70 не обнаружено (рис. 1). Отставленное во времени повышение синтеза Hsp70 может быть связано с возрастным снижением уровня и активности транскрипционного фактора белков теплового шока HSF1 и, как следствие, уменьшением скорости экспрессии генов и синтеза белка Hsp70 в головном мозге [38–40]. Показано, что базальный уровень HSF1, а также скорость транскрипции генов белка Hsp70 при действии теплового шока значительно ниже в старых Т-лимфоцитах человека по сравнению с молодыми [26].
Рис. 1.
Содержание Hsp70 в компактной части черной субстанции у пожилых крыс в контроле (Control), через 1 (Day 1) и 7 дней (Day 7) после однократного внутрибрюшинного введения U133 в дозе 5 мг/кг. В верхней части рисунка представлены репрезентативные иммуноблоты компактной части черной субстанции (SNpc). На графике по вертикали – оптическая плотность Hsp70, % от контроля. Достоверность различий в опыте относительно контроля * – p < 0.05. Fig. 1. Hsp70 content in the substantia nigra pars compacta in elderly rats in the control condition, day 1 and day 7 after a single intraperitoneal administration of U133 at a dose of 5 mg/kg. The upper part of the figure shows representative immunoblots of the substantia nigra pars compacta (SNpc). The significance of differences relative to control * – p < 0.05.
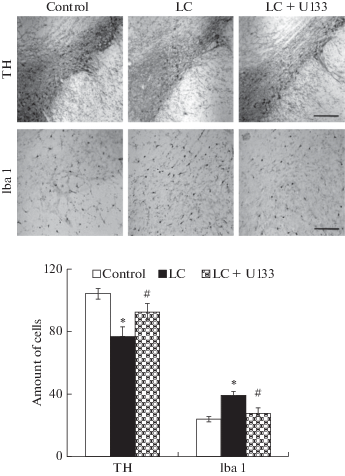
Методом иммуногистохимии установлено, что интраназальное введение ЛЦ вызывает у пожилых крыс уменьшение количества тел ТГ-позитивных нейронов в кчЧС на 27% (p < 0.05) по сравнению с контролем, что сопряжено с развитием хронического нейровоспаления, о чем свидетельствует возрастание количества активированных микроглиоцитов в кчЧС (рис. 2). Проведенная терапия с помощью U133 привела к ослаблению процесса нейродегенерации в кчЧС, на что указывает увеличение на 16% (p < 0.05) количества выживших ДА-ергических нейронов (уменьшение в 2.4 раза уровня нейродегенерации), и препятствовала возрастанию количества активированных микроглиоцитов в кчЧС (рис. 2). Следовательно, трисацетилглюкозид эхинохрома U133 уменьшает воспаление в кчЧС, ведущее к гибели нейронов и прогрессии БП. В предыдущих исследованиях мы экспериментально доказали, что повышение уровня стресс-индуцируемого шаперона Hsp70 лежит в основе защитного действия соединения U133: нокдаун Hsp70 c помощью специфической shRNA в С6 клетках глиомы устранял нейропротективный эффект U133 от токсичности, вызванной ЛЦ [22]. С другой стороны, интраназальная доставка в мозг рекомбинантного Hsp70 препятствовала прогрессии нейродегенерации в нигростриатной системе в модели БП у крыс среднего возраста [18]. Противовоспалительный эффект шаперона Hsp70 может быть связан с его негативным влиянием на экспрессию транскрипционного фактора STAT3, активирующего транскрипцию провоспалительных цитокинов, и ингибированием JNK- и NF-κB-опосредованного сигналлинга [41, 42]. Помимо этого, Hsp70 способен модулировать активность макрофагов, связываясь с Тoll-like рецепторами на их поверхности, и влиять на синтез и секрецию провоспалительных цитокинов IL-1β, IL-6 и TNF-α [43, 44]. Противовоспалительный потенциал Hsp70 продемонстрирован и другими авторами при системном введении Hsp70 в 6-гидроксидофаминовой модели БП у мышей [45].
Рис. 2.
Изменение количества дофаминергических нейронов и микроглиоцитов в компактной части черной субстанции у пожилых крыс в модели болезни Паркинсона при терапии с U133. Световая микроскопия: ТГ-иммунореактивные нейроны в компактной части черной субстанции (верхний ряд, TH; масштаб 50 мкм); Iba1-иммунореактивные микроглиоциты в компактной части черной субстанции (нижний ряд, Iba1; масштаб 20 мкм). На графике по вертикали – количество клеток. Control – введение контрольного раствора, LC – модель болезни Паркинсона, LC + U133 – терапия с U133 в модели болезни Паркинсона. Достоверность различий в опыте относительно: контроля * – p < 0.05; LC + U133 по сравнению с действием LC # – p < 0.05. Fig. 2. The change in the number of dopaminergic neurons and microgliocytes in the substantia nigra pars compacta in elderly rats in the Parkinson’s disease model after the therapy with U133. Microphotographs: tyrosine hydroxylase-immunoreactive neurons in the substantia nigra pars compacta (upper panel, TH; scale 50 μm); Iba1-immunoreactive microgliocytes in substantia nigra pars compacta (bottom panel, Iba1; scale 20 μm). Control – the administration of a control solution, LC – the model of Parkinson’s disease, LC + U133 – therapy with U133 in the model of Parkinson’s disease. The significance of differences relative to: control * – p < 0.05; LC + + U133 compared with the LC # – p < 0.05.
Поскольку в основе патогенеза нейродегенерации при БП лежит накопление в ДА-ергических нейронах кчЧС агрегированных и модифицированных форм α-синуклеина, мы провели морфологическое и биохимическое исследование, применив антитела против общего водорастворимого и фосфорилированного по Ser129 α-синуклеина, чтобы выяснить эффективность Hsp70-индуцирующей терапии на α-синуклеиновую патологию. С помощью конфокальной микроскопии установлено, что в контрольных условиях α-синуклеин распределен диффузно в цитоплазме нейронов кчЧС и не обнаруживается в ядре (рис. 3A). В модели доклинической стадии БП у пожилых крыс на результирующем изображении кчЧС прослеживается накопление α-синуклеина в виде крупных агрегатов белка в цитоплазме и в ядрах ДА-ергических нейронов (рис. 3B). При этом, агрегаты α-синуклеина у пожилых животных были более крупного размера по сравнению с агрегатами у крыс среднего возраста в модели БП [46]. Эти факты могут свидетельствовать об усилении процесса агрегации белка α-синуклеина по мере старения вследствие ослабления работы протеасом в кчЧС. У пожилых пациентов с БП postmortem в нейронах кчЧС также обнаружены крупные агрегаты α-синуклеина подобной морфологии, локализованные как в цитоплазме, так и в ядре ДА-ергических нейронов [47, 48]. Применение Hsp70-индуцирующей терапии с U133 привело к значительному сокращению количества крупных агрегатов α-синуклеина в ДА-ергических нейронах кчЧС, судя по анализу изображения на конфокальных микрофотографиях (рис. 3C). Это может указывать на дезагрегационную активность Hsp70. Сходные результаты при морфологическом анализе кчЧС были получены нами ранее при лечении крыс среднего возраста с помощью U133 в модели клинической стадии БП [22]. Также эти данные были подтверждены методом ловушки на фильтре (Filter Trap Assay), с помощью которого показано, что терапия с U133 сопровождалась двукратным снижением оптической плотности иммунореактивного вещества к α-синуклеину в нерастворимом осадке проб кчЧС крыс в модели БП [22]. Следует отметить, что снижение агрегации α-синуклеина и связанной с ним токсичности продемонстрировал и другой перспективный индуктор шаперонов, препарат FLZ, в модели α-синуклеиновой патологии у мышей [49].
Рис. 3.
Влияние терапии с индуктором белков теплового шока U133 на развитие α-синуклеиновой патологии в компактной части черной субстанции (кчЧС) в модели болезни Паркинсона у крыс пожилого возраста. Немодифицированная общая форма растворимого α-синуклеина (α-syn); пострансляционно модифицированная форма α-синуклеина, фосфорилированного по Ser129 (α-syn (Ser129)). Control – введение контрольного раствора, LC – модель болезни Паркинсона, LC + U133 – терапия с U133 в модели болезни Паркинсона. Конфокальные микрофотографии препаратов кчЧС: (A)–(C). Двойное иммуномечение с применением антител против тирозингидроксилазы (TH) и α-syn; масштаб 10 мкм. Стрелками показаны агрегаты α-синуклеина. (D)–(F). Двойное иммуномечение с применением антител против тирозингидроксилазы (TH) и α-syn (Ser129); масштаб 10 мкм. Стрелками показаны агрегаты α-синуклеина, фосфорилированного в положении Ser129. Fig. 3. The effect of therapy with heat shock proteins inducer U133 on the development of α-synuclein pathology in the substantia nigra pars compacta (SNpc) in the Parkinson’s disease model in elderly rats. Unmodified form of soluble α-synuclein (α-syn); post-translationally modified form of α-synuclein phosphorylated at Ser129 (αрsyn (Ser129)). Control – the administration of a control solution, LC – the model of Parkinson’s disease, LC + U133 – therapy with U133 in the model of Parkinson’s disease. Confocal microphotographs of SNpc: (A)–(C). Double immunolabeling using antibodies against tyrosine hydroxylase (TH) and α-syn; scale 10 μm. Arrows indicate α-synuclein aggregates. (D)–(F). Double immunolabeling with antibodies against TH and α-syn (Ser129); scale 10 μm. Arrows indicate aggregates of α-synuclein phosphorylated at Ser129.

Одной из основных патологических модификаций α-синуклеина является его фосфорилирование по Ser129, которое может влиять на растворимость этого белка и способствовать его патогенной агрегации [5, 50]. Поэтому важным представлялось исследовать влияние U133 на содержание фосфорилированного α-синуклеина (Ser129) в кчЧС при моделировании БП у пожилых крыс. Проведенный морфологический анализ показал, что в контрольных условиях α-синуклеин (Ser129) представлен скудно в ДА-ергических нейронах кчЧС, в то время как в модели доклинической стадии БП отмечается его накопление и агрегирование в телах ДА-ергических нейронов кчЧС (рис. 3D, E). Судя по результирующему изображению, терапия с U133 препятствовала избыточному образованию модифицированной формы α-синуклеина и ее агрегированию в модели БП у пожилых животных (рис. 3F).
Результаты морфологического анализа были подтверждены Вестерн-блоттингом. Показано, что у крыс пожилого возраста в модели БП содержание общей водорастворимой формы α-синуклеина возрастает в 1.4 раза (p < 0.01) в кчЧС по сравнению с контролем, что является следствием ослабления работы протеасом. Накопление α-синуклеина в телах ДА-ергических нейронов кчЧС сопряжено с возрастанием в 2.9 раза (p < 0.01) уровня фосфорилированного α-синуклеина (Ser129) по сравнению с контролем (рис. 4). Курсовое введение индуктора шаперонов U133 модельным животным приводило к приросту уровня водорастворимого α-синуклеина в кчЧС, что может указывать на уменьшение агрегированных водонерастворимых форм α-синуклеина. При этом содержание фосфорилированного α-синуклеина (Ser129) снижалось до контрольных значений в кчЧС (рис. 4). Можно полагать, что повышение выживаемости ДА-ергических нейронов в кчЧС при применении Hsp70-индуцирующей терапии связано со способностью шаперона стабилизировать основное состояние молекулы α-синуклеина, что препятствует его химической модификации, которая усиливает агрегационную способность α-синуклеина и его токсичность. Экспериментально доказано, что накопление фосфорилированного α-синуклеина (Ser129) токсично для клетки и сопряжено с усилением нейродегенеративного процесса в нигростриатной системе при моделировании у крыс наследственной формы БП [51]. Уменьшению содержания растворимых форм α-синуклеина может способствовать глубокий медленный сон, во время которого создаются условия для клиренса амилоидогенных белков посредством глимфатической системы [52]. Установлено, что при проведении терапии с U133 в модели доклинической стадии БП глубокий медленный сон значимо возрастает у пожилых крыс [37]. Однако вследствие того, что созданная нами модель БП основана на ингибировании работы протеасом, повышенный уровень водорастворимой неагрегированной формы α-синуклеина в кчЧС сохраняется. Это указывает на то, что для полноценной терапии α-синуклеиновой патологии при БП нужен комплексный подход, нацеленный как на молекулярные механизмы, противодействующие образованию конформационно дефектных белков (активация шаперонной системы белков теплового шока), так и на механизмы их утилизации (использование активаторов протеасом и шаперон-опосредованной аутофагии). В совокупности данные морфологического и биохимического анализа указывают на то, что Hsp70-индуцирующая терапия с помощью низкомолекулярного препарата U133 в пожилом возрасте способствует уменьшению количества агрегатов α-синуклеина и препятствует посттрансляционной модификации α-синуклеина в фосфорилированную форму в нейронах кчЧС в модели доклинической стадии БП.
Рис. 4.
Содержание общего α-синуклеина (α-syn) и его фосфорилированной по Ser129 фракции (α-syn (Ser129)) в компактной части черной субстанции у крыс в модели болезни Паркинсона при терапии с U133. В верхней части рисунка представлены репрезентативные иммуноблоты компактной части черной субстанции. На графике по вертикали – оптическая плотность, у.е. Control – введение контрольного раствора, LC – модель болезни Паркинсона, LC + U133 – терапия с U133 в модели болезни Паркинсона. Достоверность различий в опыте относительно: контроля * – p < 0.05, ** – p < 0.01; LC + U133 по сравнению с действием LC # – p < 0.05. Fig. 4. The content of total α-synuclein (α-syn) and its Ser129-phosphorylated fraction (α-syn (Ser129)) in the substantia nigra pars compacta of rats in the Parkinson’s disease model after the therapy with U133. The upper panel of the figure shows representative immunoblots of the substantia nigra pars compacta. Control – the administration of a control solution, LC – the model of Parkinson’s disease, LC + U133 - therapy with U133 in the model of Parkinson’s disease. The significance of differences relative to: control * – p < 0.05, ** – p < 0.01; LC + U133 compared with the LC # – p < 0.05.

ЗАКЛЮЧЕНИЕ
Таким образом, проведенное исследование демонстрирует, что фармакологическая активация транскрипционного фактора HSF1 в стареющем мозге с помощью трисацетилглюкозида эхинохрома U133 способна вызвать Hsp70-индуцирующий ответ в кчЧС, являющейся наиболее значимой структурой головного мозга при БП. Проведение Hsp70-индуцирующей терапии в модели доклинической стадии БП у пожилых крыс приводит к уменьшению прогрессирования нейродегенерации в нигростриатной системе, сопряженной с ослаблением признаков нейровоспаления и α-синуклеиновой патологии. В совокупности полученные факты свидетельствуют, что пролекарство – низкомолекулярный препарат U133 – обладает значительным терапевтическим потенциалом при Паркинсон-подобной патологии. Результаты исследования имеют научно-практическую значимость для разработки инновационной технологии превентивной фармакотерапии БП на основе отечественного препарата U133 у людей как среднего, так и пожилого возраста.
Список литературы
Erkkinen M.G., Kim M.O., Geschwind M.D. Clinical neurology and epidemiology of the major neurodegenerative diseases. Cold Spring Harb. Perspect. Biol. 10(4): a033118. 2018.
Fujita K.A., Ostaszewski M., Matsuoka Y., Ghosh S., Kitano H., Balling R. Integrating pathways of parkinson’s disease in a molecular interaction map. Mol. Neurobiol. 49(1): 88–102. 2014.
Braak H., Ghebremedhin E., Rьb U., Bratzke H., Del Tredici K. Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res. 318(1): 121–134. 2004.
Verma M., Basu S., Singh M., Rachana R., Kaur S., Goel N. Molecular Interactions of a-Synuclein, Mitochondria, and Cellular Degradation Pathways in Parkinson’s Disease. Qual. Control Cell. Protein Neurodegener. Disord. 212–234. 2020.
Rocha E.M., De Miranda B., Sanders L.H. Alpha-synuclein: Pathology, mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurobiol. Dis. 109: 249–257. 2018.
Giasson B.I., Duda J.E., Murray I.V.J., Chen Q., Souza J.M., Hurtig H.I., Ischiropoulos H., Trojanowski J.Q., Lee V.M.-Y. Oxidative Damage Linked to Neurodegeneration by Selective α-Synuclein Nitration in Synucleinopathy Lesions. Science. 290(5493): 985–989. 2000.
Lotharius J., Brundin P. Pathogenesis of parkinson’s disease: Dopamine, vesicles and α-synuclein. Nat. Rev. Neurosci. 3(12): 932–942. 2002.
Hara S., Arawaka S., Sato H., MacHiya Y., Cui C., Sasaki A., Koyama S., Kato T. Serine 129 phosphorylation of membrane-associated α-synuclein modulates dopamine transporter function in a G protein-coupled receptor kinase-dependent manner. Mol. Biol. Cell. 24(11): 1649–1660. 2013.
Marques O., Outeiro T.F. Alpha-synuclein: From secretion to dysfunction and death. Cell Death Dis. 3(7): e350. 2012.
Rochet J.C., Outeiro T.F., Conway K.A., Ding T.T., Volles M.J., Lashuel H.A., Bieganski R.M., Lindquist S.L., Lansbury P.J. Interactions among α-synuclein, dopamine, and biomembranes: Some clues for understanding neurodegeneration in Parkinson’s disease. J. Mol. Neurosci. 23(1–2): 23–33. 2004.
Subramaniam M., Althof D., Gispert S., Schwenk J., Auburger G., Kulik A., Fakler B., Roeper J. Mutant α-synuclein enhances firing frequencies in dopamine substantia nigra neurons by oxidative impairment of A-type potassium channels. J. Neurosci. 34(41): 13586–13599. 2014.
Ferrari C.C., Pott Godoy M.C., Tarelli R., Chertoff M., Depino A.M., Pitossi F.J. Progressive neurodegeneration and motor disabilities induced by chronic expression of IL-1β in the substantia nigra. Neurobiol. Dis. 24(1): 183–193. 2006.
Perez-Alvarez S., Solesio M.E., Manzanares J., Jordбn J., Galindo M.F. Lactacystin requires reactive oxygen species and Bax redistribution to induce mitochondria-mediated cell death. Br. J. Pharmacol. 158(4): 1121–1130. 2009.
Karpenko M.N., Muruzheva Z.M., Pestereva N.S., Ekimova I. V. An Infection Hypothesis of Parkinson’s Disease. Neurosci. Behav. Physiol. 49(5): 555–561. 2019.
Auluck P.K., Chan H.Y.E., Trojanowski J.Q., Lee V.M.Y., Bonini N.M. Chaperone suppression of α-synuclein toxicity in a Drosophila model for Parkinson’s disease. Science. 295(5556): 865–868. 2002.
Leverenz J.B., Umar I., Wang Q., Montine T.J., McMillan P.J., Tsuang D.W., Jin J., Pan C., Shin J., Zhu D., Zhang J. Proteomic Identification of Novel Proteins in Cortical Lewy Bodies. Brain Pathol. 17(2): 139–145. 2007.
Chu Y., Dodiya H., Aebischer P., Olanow C.W., Kordower J.H. Alterations in lysosomal and proteasomal markers in Parkinson’s disease: Relationship to alpha-synuclein inclusions. Neurobiol. Dis. 35(3): 385–398. 2009.
Pastukhov Y.F., Plaksina D.V., Lapshina K.V., Guzhova I.V., Ekimova I.V. Exogenous protein HSP70 blocks neurodegeneration in the rat model of the clinical stage of Parkinson’s disease. Dokl. Biol. Sci. 457(1). 2014.
Белан Д.В., Екимова И.В. Белки теплового шока при конформационных болезнях мозга. Рос. физиол. журн. им. И.М. Сеченова. 105(12): 1465–1485. 2019. [Belan D.V., Ekimova I.V. Heat shock proteins in conformational diseases of the brain. Russ. J. Physiol. 105(12): 1465–1485. 2019. (In Russ)].
Eremenko E.M., Antimonova O.I., Shekalova O.G., Polonik S.G., Margulis B.A., Guzhova I.V. Novel compounds that increase expression of Hsp70 and their biological activity. Cell Tissue Biol. 4(3): 251–257. 2010.
Lazarev V.F., Onokhin K.V., Antimonova O.I., Polonik S.G., Guzhova I.V., Margulis B.A. Kinetics of chaperone activity of proteins Hsp70 and Hdj1 in human leukemia u-937 cells after preconditioning with thermal shock or compound u-133. Biochemistry. 76(5): 590. 2011.
Ekimova I.V., Plaksina D.V., Pastukhov Y.F., Lapshina K.V., Lazarev V.F., Mikhaylova E.R., Polonik S.G., Pani B., Margulis B.A., Guzhova I.V., Nudler E. New HSF1 inducer as a therapeutic agent in a rodent model of Parkinson’s disease. Exp. Neurol. 306: 199–208. 2018.
de Lau L.M., Breteler M.M. Epidemiology of Parkinson’s disease. Lancet Neurol. 5(6): 525–535. 2006.
Labbadia J., Morimoto R.I. The Biology of Proteostasis in Aging and Disease. Annu. Rev. Biochem. 84(1): 435–464. 2015.
Soti C., Csermely P. Chaperones and aging: Role in neurodegeneration and in other civilizational diseases. Neurochem. Int. 41(6): 383–389. 2002.
Jurivich D.A., Choo M., Welk J., Qiu L., Han K., Zhou X. Human aging alters the first phase of the molecular response to stress in T-cells. Exp. Gerontol. 40(12): 948–958. 2005.
Ekimova I.V., Simonova V.V., Guzeev M.A., Lapshina K.V., Chernyshev M.V., Pastukhov Y.F. Changes in sleep characteristics of rat preclinical model of Parkinson’s disease based on attenuation of the ubiquitin–proteasome system activity in the brain. J. Evol. Biochem. Physiol. 52(6): 463–474. 2016.
Полоник С., Толкач А., Уварова Н. Гликозилирование эхинохрома и родственных гидроксинафтазаринов ортоэфирным методом. Журн. орган. химии. 30(2): 248–253. 1994. [Polonik S., Tolkach A., Uvarova N. Glycosylation of echinochrome and related hydroxynaphthazarines by the orthoester method. Zh. Organ. Himii. 30(2): 248–253. 1994. (In Russ)].
Mishchenko N.P., Fedoreyev S.A., Bagirova V.L. Histochrome: a New Original Domestic Drug. Pharm. Chem. J. 37(1): 48–52. 2003.
Lazarev V.F., Nikotina A.D., Mikhaylova E.R., Nudler E., Polonik S.G., Guzhova I.V., Margulis B.A. Hsp70 chaperone rescues C6 rat glioblastoma cells from oxidative stress by sequestration of aggregating GAPDH. Biochem. Biophys. Res. Commun. 470(3): 766–771. 2016.
Yurchenko E.A., Menchinskaya E.S., Polonik S.G., Agafonova I.G., Guzhova I. V., Margulis B.A., Aminin D.L. Hsp70 Induction and Anticancer Activity of U-133, the Acetylated Trisglucosydic Derivative of Echinochrome. Med. Chem. 5(6): 263–271. 2015.
Lasunskaia E.B., Fridlianskaia I., Arnoldt A.V., Kanashiro M., Guzhova I., Margulis B.A. Sub-lethal heat shock induces plasma membrane translocation of 70-kDa heat shock protein in viable, but not in apoptotic, U-937 leukaemia cells. APMIS. 118(3): 179–187. 2010.
Plaksina D.V., Chernyshev M.V., Karpenko M.N., Gazizova A.R., Pazi M.B., Ekimova I.V. Experimental modeling of a preclinical Parkinson’s disease stage in rats by intranasal lactacystin administration. Neurodegen. Dis. (Suppl). 17(1): 1655. 2017.
Ekimova I.V., Gazizova A.R., Karpenko M.N., Plaksina D.V. Signs of anhedonia and destructive changes in the ventral tegmental area of the midbrain in the model of the preclinical Parkinson’s disease stage in experiment. Zh. Nevrol. Psikhiatrii Im. S.S. Korsakova. 118(9): 61–67. 2018.
Abdurasulova I.N., Ekimova I.V., Matsulevich A.V., Gazizova A.R., Klimenko V.M., Pastukhov Y.F. Impairment of non-associative learning in a rat experimental model of preclinical stage of Parkinson’s disease. Dokl. Biol. Sci. 476(1): 188–190. 2017.
Pastukhov Y.F., Simonova V.V., Chernyshev M.V., Guzeev M.A., Shemyakova T.S., Ekimova I.V. Signs of sleep and behavior disorders indicating the initial stage of neurodegeneration in a rat model of Parkinson’s disease. J. Evol. Biochem. Physiol. 53(5): 431–434. 2017.
Pastukhov Y.F., Simonova V.V., Shemyakova T.S., Guzeev M.A., Polonik S.G., Ekimova I.V. U-133, a heat shock proteins inducer, precludes sleep disturbances in a model of the preclinical stage of Parkinson’s disease in aged rats. Adv. Gerontol. 32(6): 935. 2019.
Calderwood S.K., Murshid A., Prince T. The Shock of Aging: Molecular Chaperones and the Heat Shock Response in Longevity and Aging. Gerontology. 55(5): 550–558. 2009.
Shemesh N., Ben-Zvi A. HSF1 Regulation in Aging and Its Role in Longevity. Heat Shock Factor. 93–113. 2016.
Chesnokova A.Y., Ekimova I.V., Pastukhov Y.F. Parkinson’s Disease and Aging. Adv. Gerontol. 9(2): 164–173. 2019.
Yu W.W., Cao S.N., Zang C.X., Wang L., Yang H.Y., Bao X.Q., Zhang D. Heat shock protein 70 suppresses neuroinflammation induced by α-synuclein in astrocytes. Mol. Cell. Neurosci. 86: 58–64. 2018.
Guzhova I.V., Darieva Z.A., Melo A.R., Margulis B.A. Major stress protein Hsp70 interacts with NF-kB regulatory complex in human T-lymphoma cells. Cell Stress Chaperones. 2(2): 132. 1997.
Asea A., Kraeft S.K., Kurt-Jones E.A., Stevenson M.A., Chen L.B., Finberg R.W., Koo G.C., Calderwood S.K. HSP70 stimulates cytokine production through a CD 14-dependant pathway, demonstrating its dual role as a chaperone and cytokine. Nat. Med. 6(4): 435–442. 2000.
Calderwood S.K., Mambula S.S., Gray P.J., Theriault J.R. Extracellular heat shock proteins in cell signaling. FEBS Lett. 581(19): 3689–3694. 2007.
Tunesi M., Raimondi I., Russo T., Colombo L., Micotti E., Brandi E., Cappelletti P., Cigada A., Negro A., Ambrosio L., Forloni G., Pollegioni L., Gloria A., Giordano C., Albani D. Hydrogel-based delivery of Tat-fused protein Hsp70 protects dopaminergic cells in vitro and in a mouse model of Parkinson’s disease. NPG Asia Mater. 11(1): 1–15. 2019.
Plaksina D.V., Ekimova I.V. Age-Related Features of α-Synuclein Pathology in the Brain on Modeling the Preclinical Stage of Parkinson’s Disease in Rats. Neurosci. Behav. Physiol. 50(1): 109–114. 2020.
Ma K.L., Song L.K., Yuan Y.H., Zhang Y., Han N., Gao K., Chen N.H. The nuclear accumulation of alpha-synuclein is mediated by importin alpha and promotes neurotoxicity by accelerating the cell cycle. Neuropharmacology. 82: 132–142. 2014.
Raiss C.C., Braun T.S., Konings I.B.M., Grabmayr H., Hassink G.C., Sidhu A., Le Feber J., Bausch A.R., Jansen C., Subramaniam V., Claessens M.M.A.E. Functionally different α-synuclein inclusions yield insight into Parkinson’s disease pathology. Sci. Rep. 6(1): 1–13. 2016.
Bao X.Q., Wang X.L., Zhang D. FLZ Attenuates α-Synuclein-Induced Neurotoxicity by Activating Heat Shock Protein 70. Mol. Neurobiol. 54(1): 349–361. 2017.
Anderson J.P., Walker D.E., Goldstein J.M., De Laat R., Banducci K., Caccavello R.J., Barbour R., Huang J., Kling K., Lee M., Diep L., Keim P.S., Shen X., Chataway T., Schlossmacher M.G., Seubert P., Schenk D., Sinha S., Gai W.P., Chilcote T.J. Phosphorylation of Ser-129 is the dominant pathological modification of α-synuclein in familial and sporadic lewy body disease. J. Biol. Chem. 281(40): 29739–29752. 2006.
Sato H., Arawaka S., Hara S., Fukushima S., Koga K., Koyama S., Kato T. Authentically phosphorylated α-synuclein at Ser129 accelerates neurodegeneration in a rat model of familial Parkinson’s disease. J. Neurosci. 31(46): 16884–16894. 2011.
Xie L., Kang H., Xu Q., Chen M.J., Liao Y., Thiyagarajan M., O’Donnell J., Christensen D.J., Nicholson C., Iliff J.J., Takano T., Deane R., Nedergaard M. Sleep drives metabolite clearance from the adult brain. Science. 342(6156): 373–377. 2013.
Дополнительные материалы отсутствуют.
Инструменты
Российский физиологический журнал им. И.М. Сеченова