

Российский физиологический журнал им. И.М. Сеченова, 2020, T. 106, № 10, стр. 1191-1208
Role of neprilysin in synaptic plasticity and memory
N.N. Nalivaeva 1, 2, *, D.S. Vasilev 1, N.M. Dubrovskaya 1, A.J. Turner 2, I.A. Zhuravin 1
1 Sechenov Institute of Evolutionary Physiology and Biochemistry, Russian Academy of Sciences
St. Petersburg, Russia
2 School of Biomedical Sciences, Faculty of Biological Sciences, University of Leeds
Leeds, United Kingdom
* E-mail: n.n.nalivaeva@leeds.ac.uk
Поступила в редакцию 28.07.2019
После доработки 24.08.2020
Принята к публикации 25.08.2020
Аннотация
Neprilysin (NEP, neutral endopeptidase, endopeptidase-24.11) is an important enzyme being most abundant in the kidney and liver and participating in the degradation of a variety of biologically active peptides. It is essential for the regulation of blood pressure and the normal function of practically all organs in the body, and its dysregulation contributes to the development of such diseases like heart failure, cancer, diabetes, and chronic inflammation. It is also a critical neuropeptidase that terminates the action of various neuropeptides such as enkephalins, substance P, somatostatin, which participate in signaling and regulatory events in the brain. NEP is a major amyloid-degrading enzyme, which makes it an important therapeutic target in Alzheimer’s disease (AD). Our studies have revealed that NEP expression declines with age in the brain structures participating in memory and which are mainly affected by AD pathogenesis. Moreover, NEP expression decreases under hypoxic conditions, and prenatal hypoxia leads to its deficit in some brain structures both during development and adulthood, which correlates with a reduced number of labile dendritic spines and cognitive deficit in rats. Using the model of prenatal hypoxia in rats, we have tested the effectiveness of several compounds (inhibitors of caspases, histone deacetylases, tyrosine kinases or nuclear retinoid X receptor agonists as well as an antioxidant, epigallocatechin gallate) which, via different mechanisms, were able to restore NEP expression and activity reduced after prenatal hypoxia and improve cognitive functions in rats. In this paper, we provide an overview of the literature and our data on the role of NEP in synaptic plasticity, neuronal activity, and cognitive functions and outline current approaches to regulate its expression and activity in the brain.
GENERAL CHARACTERISTICS OF NEP
A neutral endopeptidase, neprilysin (NEP, EC 3.4.24.11), is a type II integral membrane protein. It has a short N-terminal domain located in the cytoplasm, a single span membrane domain, and a large C-terminal ectodomain containing the active site, which is localized in the extracellular space where it cleaves various substrates. NEP is a zinc-dependent enzyme, and its zinc-binding domain is localized in the extracellular C-terminal part of the molecule [1]. There is also a soluble form of NEP detected in blood plasma [2], but its origin is still debated [3].
The highest content and activity of NEP is in the kidney, where it represents about 4% of all proteins of the renal brush border membrane from where it was initially isolated [4]. It is also enriched in liver and testis but can be found in many other organs (for review, see [5]). NEP is also expressed in the brain and was shown to cleave enkephalins, which gave it a trivial name enkephalinase. However, after the subsequent discovery of other substrates, NEP was given a more general name – endopeptidase-24.11 [6]. Yet, in the current literature, the enzyme is commonly named neutral endopeptidase or NEP. However, it can also be referred to as CD10 – a leukocyte cell surface antigen, or as CALLA – the common acute lymphoblastic leukemia antigen [7, 8].
The human NEP gene is located on chromosome 3 and spans more than 80 kb. It is composed of 24 exons and exists in a single copy [9]. It has two distinct promoters whose operation depends on cell type, although both promoters have similar characteristics and effectiveness for controlling NEP gene expression [10]. The NEP gene is highly conserved among mammalian species, and three distinct NEP mRNAs identified in humans and rats differ only in their 5'-noncoding regions [11], which makes rat NEP a useful model for studying its regulation and properties for therapeutic purposes. The NEP gene is widely distributed in diverse animal species from the nematode worm, Caenorhabditis elegans, and the fly, Drosophila melanogaster to humans [12]. In Aplysia californica, an enzyme similar, although not identical to the mammalian NEP (apNEP), was found both in the CNS and periphery suggesting that it could play a significant role in the control of synaptic transmission [13].
NEP is a glycoprotein and has up to six N-linked glycosylation sites depending on its source [14] and can also be sialylated [15]. The NEP molecule can undergo various modifications within the cytoplasmic domain, which regulate its transport and functions. NEP myristoylation at the N-terminal site (Gly2) affects the membrane targeting of the enzyme [16]. NEP phosphorylation by casein kinase II (CKII) blocks focal adhesion kinase signaling [17], while phosphorylation at Ser 6 inhibits its interaction with a tumour suppressor – phosphatase and tensin homolog (PTEN) – which acts as a regulator of the cell cycle [18]. As such, NEP serves dual functions in extracellular and intracellular signal transduction.
The list of NEP substrates and its functions is continuing to expand. NEP cleaves a wide range of peptides containing up to 40–50 amino acids (for review, see [19]). Among the most efficiently hydrolyzed substrates, in addition to enkephalins, are tachykinins (substance P), calcitonin gene-related peptide (CGRP), somatostatin, adrenomedullin, glucagon, thymopentin, endothelins, the atrial natriuretic peptide family, bradykinin, members of the vasoactive intestinal peptide family, etc. Moreover, NEP is the major amyloid-degrading enzyme cleaving β amyloid (Aβ)-peptide, which makes it an important player in the pathogenesis and therapeutics of Alzheimer’s disease (AD) (for review, see [20, 21]).
The pH optimum for NEP activity is around 6.0 [22]. Since it is exclusively a zinc-dependent enzyme with the zinc-binding domain located in the extracellular C-terminal part of the molecule [1], it is inhibited by zinc-chelating reagents. There is a range of synthetic inhibitors widely used for studying NEP properties and for clinical application. Among them, the most widely used NEP inhibitors are thiorphan [23] and phosphoramidon [24], although the latter is less specific and inhibits other metallopeptidases. Because of the role of NEP in the regulation of blood pressure and heart failure, various NEP inhibitors have been designed. However, the observations that loss of NEP activity can promote the accumulation of Aβ in the brain or enhance the activity of mitogenic peptides urged caution for the chronic application of NEP inhibitors in humans [25]. Apart from synthetic inhibitors, there are natural endogenous NEP inhibitors, such as sialorphin – the stress-response peptide [26] and “opiorphins” with strong analgesic properties [27] which also have a medical application.
LOCALIZATION OF NEP IN THE BRAIN
In the brain, NEP expression was confirmed in the majority of the structures [6, 28] as well as in the choroid plexus [29]. It is also present in the cerebrospinal fluid, although NEP activity is much lower than in the brain [2]. The highest protein levels of NEP are found in the striatum [30]. NEP is primarily located on neuronal cells, especially in the striatonigral pathway [28], and in Schwann cells in the peripheral nervous system [31]. Immunofluorescence analyses confirmed that NEP is localized along axons and at the synapses, which suggests that after synthesis in the soma NEP is transported to the synapses via the anterograde axonal transport system [32]. Even in Aplysia CNS ganglia, expression of NEP-like peptidase (apNEP) mRNA was found both in the cell bodies and in neurites of the neuropil and peripheral nerves [13]. Application of NEP inhibitors resulted in a prolonged physiological delay in response of slugs to stimulation, which suggests that the role of NEP-like endopeptidases in the regulation of behavior was established early in evolution. Axonal and dendritic localization of NEP was also shown in the Drosophila nervous system [33].
In the mouse hippocampus, NEP is mostly present in the CA1-3 fields and the molecular layer of the dentate gyrus and is subcellularly localized along axons and at synapses [34]. NEP expression was observed in GABAergic neurons but not in catecholaminergic or cholinergic neurons [34]. Recently it was confirmed that, in mice, GABAergic neurons are especially enriched in NEP and that NEP mRNA is detectable in the parvalbumin-expressing interneurons. At the same time, NEP protein is localized in the perisomatic synapses [35]. Intriguingly, cholinergic neurons, which are the most vulnerable in AD brains, lack NEP expression [34, 35].
Although detectable NEP levels have not been reported in astrocytes and microglia in the normal brain under some pathological conditions, NEP expression can be up-regulated in these types of cells in vitro. For example, under hypoxic conditions, astrocytes were shown to increase NEP protein levels [36] while inflammatory conditions (IL-4 administration) activated NEP expression in type 2 microglia [37]. Some authors suggest that NEP expressed by astrocytes and microglia plays an important role in AD pathogenesis (for review see [38]).
NEP localization in the brain is, to a great extent, dictated by the distribution of its major substrates. Thus, there is a strong correlation between the distribution of NEP and µ and δ opioid receptors in different rat brain regions [39], and NEP was shown to selectively control opioid biosynthesis in the hypothalamus and spinal cord [40]. Immunochemical analysis has also confirmed the colocalization of NEP and substance P in the substantia nigra [41]. In the striatum and hippocampus, NEP was found to be the major somatostatin-degrading enzyme [31], and co-expression of multiple somatostatin receptor subtypes and NEP was shown in the meningeal cells which possess a high capacity to inactivate somatostatin by proteolytic degradation [42]. Somatostatin receptors are widely expressed in the olfactory bulbs and all other olfactory structures, where somatostatin regulates olfactory discrimination responses [43].
NEP IN BRAIN DEVELOPMENT
In the developing brain, the only structure distinctly labeled for NEP before birth, as shown by in vitro autoradiography in rats, is the external layer of the olfactory bulbs [44]. According to our data, in other brain structures, such as the striatum and cortex, NEP protein level is steadily increasing during the first month after birth and then either stays at a very high level (in the striatum) or declines with age (in the cortex) [30]. In mice, high NEP expression in the embryonic brain was reported in the nigrostriatal axis, nucleus accumbens, and globus pallidus at E9 with a steady decrease until birth and then remaining at that level during postnatal ontogenesis [45]. This suggests that in different species, distinct cerebral localization of NEP during ontogenesis could reflect certain specificity in the development of the peptidergic system.
In the peripheral nervous system, NEP expression was detected in rat sciatic nerve at E16. However, the levels of immunoreactive protein and enzyme activity declined with age, suggesting that in the Schwann cells, NEP regulation is also age-dependent [46]. It was also observed that NEP expression increases in the Schwann cells after axonal damage, which suggests that it plays an essential role in nerve development and regeneration [47]. Interestingly, in patients with Charcot–Marie–Tooth disease, a loss-of-function NEP mutation leads to the development of axonal neuropathy in the peripheral nerves correlating with muscle weakness and sensory disturbance, but these patients do not demonstrate symptoms of dementia [48].
NEP is expressed in stem cells and has been used as a marker of stem cells in many tissues (for review, see [49]. It is also expressed in some types of neuroblastoma cells, which makes them convenient models for studying NEP gene regulation [10].
NEP gene knockout in mice is not lethal, and animals develop normally. Still, they are highly sensitive to endotoxic shock, which confirms that NEP plays an essential role in the metabolism of proinflammatory peptides [50]. NEP gene deletion also increases the thermo-nociceptive threshold in mice suggesting that NEP controls opioid metabolism in the hypothalamus and spinal cord [51]. NEP knockouts also have enhanced aggressive behavior in the resident-intruder paradigm and altered locomotor activity [52]. They are also prone to increased consumption of alcohol compared to the wild type mice [53]. NEP deficit in the knockout mice also enhances pain symptoms and neurogenic inflammation after nerve injury [54], supporting the role of NEP in pain signaling.
Our multidisciplinary studies on the role of prenatal hypoxia in development of motor and cognitive functions in rats (for review, see [55]) have demonstrated a significant reduction of NEP mRNA, protein levels and enzyme activity in the cortex and hippocampus of the offspring which correlated with morphological changes in these brain structures and cognitive impairment during postnatal development [56–58]. These animals are also characterized by a decrease in the number of labile synaptopodin-positive dendritic spines in the cortex and hippocampus [58, 59]. The reduction of the number of dendritic spines in the molecular level of the parietal cortex might be related to the prenatal hypoxia-induced death of the pyramidal neurons in the lower layers of the cortex whose apical dendrites form the spines. One of the reasons for reduced spine density in the stratum pyramidale of the hippocampus might be impaired CA1 innervation coming from the neurons of the entorhinal cortex [60], which was observed in rats subjected to prenatal hypoxia [61]. Because of the primary localization of NEP in the synapses, the decrease in the number of the dendritic spines, and as such of the number of synaptic contacts, correlates well with reduced NEP protein levels and activity in these brain areas. In agreement with our studies are reports that prenatal hypoxia in AD mice (APPSwe/PS1A246E) results not only in a significant decrease in synapses and impaired learning and memory of the offspring but also in lower levels of NEP and enhanced accumulation of the amyloid Aβ peptide in their brain [62].
ROLE OF NEP IN MEMORY AND SYNAPTIC PLASTICITY
Because of the important role of NEP in peptidergic neurotransmission and its synaptic localization, it is not surprising that this enzyme is involved in various processes underlying synaptic plasticity and memory (Fig. 1). High levels of NEP expression in the first month of rodent development, as shown in our studies, coincides with the period of active formation of the neuronal network, which shapes the whole repertoire of animal behavior in later life. Any disruptive conditions which result in decreased levels of NEP expression and activity in the brain will lead to memory deficits and cognitive impairment, as shown in our studies using the model of prenatal hypoxia [30, 55, 57, 58].
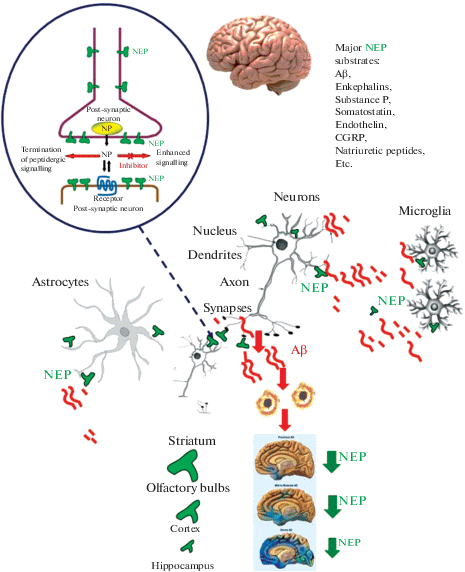
Fig. 1.
Schematic representation of NEP brain distribution, signaling, and localization in relation to the sites of Aβ cleavage and AD pathology. The inserts at the top of the figure give the names of major NEP substrates and a scheme of NEP action at the synapses (NP – neuropeptides). The inserts in the bottom part show the spread of amyloid load in the AD brain, which inversely correlates with the levels of NEP content and activity in brain structures. The highest levels of NEP expression and activity are observed in the striatum and olfactory bulbs with much lower levels in the cortex and hippocampus, where NEP expression also significantly decreases with aging. The size of the letters and arrows in the bottom panel reflects the levels of NEP expression and activity.
The role of NEP in neuronal signaling and memory has been established in invertebrates using Drosophila melanogaster, which has four NEP genes. It was shown that protein products of all four genes are involved in the middle- and long-term memory in flies [63] and are required for cleaving neuropeptides in the mushroom bodies and closely connected pairs of neurons. Most recently, these authors have shown that neprilysin 1 (Nep1) is terminating the signaling function of a peptide Amnesiac required for specific memory phases in the olfactory memory center in fly mushroom bodies [64].
The role of NEP in memory has been studied in various research paradigms. Our studies on the effects of i.c. administration of the NEP inhibitors thiorphan or phosphoramidon to the sensorimotor cortex of rats have demonstrated significant impairment of short-term memory tested in a two-level radial maze even after a single injection of either inhibitor [65]. These results were in good agreement with the effect of chronic inhibition of NEP by continuous administration of thiorphan via an osmo-pump during four weeks, which led to significant cognitive dysfunction manifested by reduced ability to discriminate objects in the recognition test and spatial memory in the water maze [66]. Intracortical injection of phosphoramidon to rats also resulted in a reduced number of dendritic spines in the molecular layer of the brain cortex, which correlated with impaired short-term working memory tested in the two-level radial maze [67]. Continuous administration of thiorphan into rat hippocampus also led to cognitive impairment, which correlated with the decreased nicotine-stimulated release of acetylcholine, indicating that NEP inhibition reduces the cholinergic activity of hippocampal neurons [68].
NEP AND ALZHEIMER’S DISEASE
Among the broad spectrum of NEP substrates, probably the most studied is Aβ peptide, which is causative of AD pathology [69, 70]. The first demonstration that NEP can cleave Aβ in vitro [71] was also confirmed in vivo in the rat brain parenchyma [72] and NEP knockout mice [73]. Chronic administration of the NEP inhibitor thiorphan into the rat hippocampus was also shown to result in the accumulation of Aβ40 and Aβ42 in the hippocampal areas [68].
NEP inhibition or gene knockout in animal models recapitulate the pathological hallmarks of AD resulting in the accumulation of Aβ in the areas affected in human brains [73, 74]. Interestingly, NEP gene knockout in APPSw mice modeling AD led to the changes in expression of more than 600 proteins, including some which were not previously reported to be linked to AD but associated with increased Aβ levels [75]. These mice also exhibited enhanced amyloid pathology compared with only APP transgenics and had more severe learning and memory deficit [76].
In the human brain, reduced NEP levels were reported in the vasculature [77] as well as in the hippocampus and temporal gyrus, which are mostly affected in AD [78]. A meta-analysis of NEP levels in AD suggests that NEP expression and activity are decreased in the cortex of elderly AD patients [79]. Using the NEP inhibitor thiorphan and Aβ administration, an AD-like pathology model has been established in nonhuman primates [80].
Transfection of the NEP gene or recombinant NEP to the brain of AD mice was shown to clear amyloid load and improve animal memory and cognition [81, 82]. This confirmed a significant role of NEP in the clearance of Aβ from the brain as well as making it an important therapeutic target in AD (for review, see [20, 21]).
Further manipulations with NEP expression in AD transgenic mice led to a deeper understanding of the role which this enzyme plays in neuronal signaling and cognitive functions. As shown recently, knockout of the NEP gene in APPSW mice results in dramatic changes in the brain proteome affecting not only the major proteins participating in amyloid metabolism but also in neurotransmission, in particular of the excitatory amino acid transporter 2 (EAA2) which was significantly increased in the NEP KO AD mice [75]. On the other hand, the AD transgenic mice (5XFAD) per se have reduced NEP expression, and further reduction of its expression by knocking out the NEP gene results in an even more severe AD phenotype [83]. Cross-breeding of APP23 mice with NEP deficient mice has shown that NEP activity deficit in the brain elevated oligomeric forms of Aβ at the synapses and impaired hippocampal synaptic plasticity and cognitive function even before the appearance of amyloid plaques [84]. This testifies that reduced NEP activity might be causative and at least partly responsible for the memory deficit symptoms in early AD, which also supports the strategy to enhance NEP for alleviating these symptoms. On the other hand, engrafting stem cells expressing NEP in the brain of 3xTg-AD and Thy1-APP AD mice not only reduced the Aβ pathology but increased the synaptic density in both strains of animals. Importantly, the Aβ loads were reduced not only in the hippocampus and subiculum, which were adjacent to the engrafts but also within the amygdala and medial septum, which receive afferent projections from the engrafted region [85].
One of the prominent features of AD is the early loss of cholinergic neurons (for review, see [86]). In AD modeling APPSw mice, it was shown that high Aβ load damages cholinergic axons in the cortex with subsequent retrograde-induced cell death of cholinergic neurons [87]. This degenerative process might be due to the Aβ binding to the α7 nicotinic acetylcholine receptors, which explicitly affects the proteome in cholinergic neurons [88], leading to aberrant neuritic sprouting and cholinergic dysfunction [89]. Recent studies have also shown that streptozotocin-induced late-onset AD phenotype is accompanied by a decrease in brain ACh, increased AChE activity, and reduced NEP protein levels [90]. Since cholinergic neurons do not express NEP, they might lack an intrinsic mechanism of Aβ removal, which makes them vulnerable to Aβ-related cell death.
Because one of the early symptoms of AD is impaired olfaction [91], and early accumulation of Aβ peptide in the olfactory areas was reported in AD mice [92], it is reasonable to expect some NEP deficit in these brain areas. However, such data to our knowledge are still missing. Our data suggest that, in the rat brain, NEP mRNA levels decline with aging in the olfactory bulbs and more significantly in the entorhinal cortex and hippocampus (unpublished data, manuscript in preparation). Importantly, in the anterior olfactory nucleus levels of somatostatin – one of the NEP substrates – were reported to be 50% lower in AD brains compared to controls [93]. Although decreased somatostatin levels might be caused by increased degradation of this peptide, which plays an essential role in olfactory signaling, it might also lead to reduced NEP levels since somatostatin induces its expression [94]. Anyway, the involvement of the olfactory system in AD pathology and the role of NEP in the olfactory signaling in health and disease deserves further investigation.
REGULATION OF NEP IN THE BRAIN
Due to the critical role which NEP plays in brain functions and AD pathogenesis, numerous approaches have been developed to up-regulate its levels. Recently, we have extensively reviewed current pharmacological and epigenetic approaches for modulating NEP expression and activity [21]. Below we shall be mostly focusing on our works using the model of prenatal hypoxia as an in vivo tool for testing various biologically active compounds and highlighting some other therapeutic avenues which aim at up-regulation of NEP levels and activity (Table 1).
Table 1.
Summary of the effects of various treatments on the memory, synaptic plasticity, and NEP activity in rats
| Conditions | Short-term memory | Number of synaptopodin-positive dendritic spines | NEP activity | |||
|---|---|---|---|---|---|---|
| radial 8-arm maze | novel object recognition test (10 min after initial testing) | parietal cortex | hippocampus | parietal cortex | hippocampus | |
| Prenatal hypoxia on E14 | Impaired compared to controls | Impaired compared to controls | Decreased compared to controls | Decreased compared to controls | Decreased compared to controls | Decreased compared to controls |
| Normal aging (14 months old) | Declines compared to normal adult rats (3-4 months old) | No statistically significant changes | Decreased compared to normal adult rats (3-4 months old) | Decreased compared to normal adult rats (3-4 months old) | Decreased compared to normal adult rats (3-4 months old) | Decreased compared to normal adult rats (3-4 months old) |
| Phosphoramidon (cortical injections to adult rats) | Impaired compared to controls | Impaired compared to controls | Decreased compared to controls | No statistically significant changes | Decreased compared to controls | Decreased compared to controls |
| Sodium valproate administration to adult rats subjected to prenatal hypoxia on E14 | Improved compared to rats treated with saline | Improved compared to rats treated with saline | Increased compared to rats treated with saline | No statistically significant changes | Increased compared to rats treated with saline | Increased compared to rats treated with saline |
| EGCG administration to adult rats subjected to prenatal hypoxia on E14 | Improved compared to rats treated with saline | Improved compared to rats treated with saline | No statistically significant changes | Increased compared to rats treated with saline | Increased compared to rats treated with saline | Increased compared to rats treated with saline |
In neuronal cell culture studies it was found that NEP gene expression is controlled by competitive binding to its promoter of histone deacetylases HDAC1 and HDAC3 and the amyloid-precursor protein (APP) intracellular domain AICD which is formed in amyloidogenic APP processing alongside Aβ [10, 95] (Fig. 2). Such regulation provides an endogenous feedback mechanism allowing cells to control levels of Aβ produced by APP cleavage, as initially suggested in [96]. Also, the NEP-dependent mechanism of controlling Aβ levels indicates that Aβ is a physiologically relevant neuropeptide, as suggested by various authors [35, 97, 98] with particular relevance to activity-dependent regulation of synaptic vesicle release [99] and memory consolidation [100].
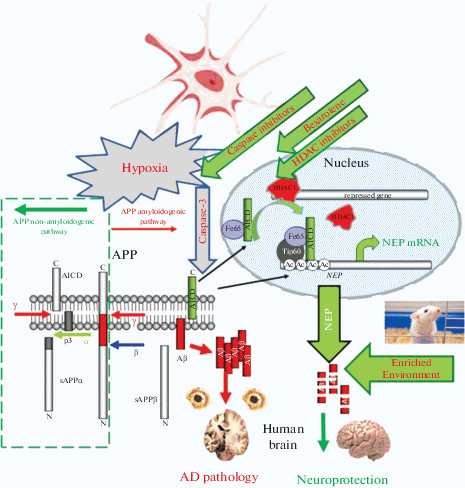
Fig. 2.
Pathways for the regulation of NEP expression. In neuronal cells, NEP regulation involves the binding of the amyloid precursor protein (APP) intracellular domain (AICD) to its gene promoter. AICD is formed alongside Aβ amyloid peptide in the amyloidogenic pathway, which includes proteolytic cleavage of APP by β- and γ-secretases. AICD with a stabilizing protein Fe65 is transported to the nucleus, where it engages a histone acetylase Tip60. This complex binds to the NEP gene promoter, replacing HDAC and increasing histone acetylation, which allows NEP gene expression [10]. This provides neuronal cells with a feedback mechanism that should control Aβ levels. Treatment of cells or animals with the HDAC inhibitor sodium valproate leads to increased binding of AICD to the NEP gene promoter, which up-regulates NEP mRNA and protein levels and enzyme activity. Bexarotene also activates the expression of NEP via reducing HDAC occupancy at the NEP gene promoter [109]. Hypoxia leads to an increased caspase activity, which reduces AICD levels and NEP expression while caspase inhibitors rescue functionally active AICD both in the cells and rat brain [105, 107]. Maintenance of animals under enriched environment also stimulates NEP expression leading to clearance of amyloid peptide and improving their memory [114]. In the human brain, the accumulation of Aβ peptide leads to AD pathology, which can be prevented by the upregulation of NEP expression and activity.
Since NEP expression is controlled by HDACs, it was reasonable to suggest that HDAC inhibitors are capable of up-regulating its levels. Indeed, treatment of SH-SY5Y cells expressing low levels of NEP with an HDAC inhibitor sodium valproate (VA) was shown to increase NEP mRNA, protein, and activity in these cells [10]. Our further work in rats has confirmed that VA administration up-regulated NEP expression and activity in rat brain structures, in particular in the hippocampus where sodium VA treatment led to increased binding of AICD to the NEP gene promoter [57]. This effect was brain region- and age-specific. In rats subjected to prenatal hypoxia, we also observed that VA administration restored NEP activity and memory deficit, as well as increased the number of dendritic spines [67]. Later it was demonstrated that VA reduced Aβ-related neuropathology in AD mice subjected to prenatal hypoxia by preventing the decrease of H3-acetylation in the NEP gene promoter region and increasing levels of NEP expression [101]. This suggests that the application of VA or more specific HDAC inhibitors is a viable strategy for the up-regulation of NEP expression and activity and improvement of cognitive functions of pathologically developing or aging brain. Indeed, recently it was shown that administration of a specific HDAC3 inhibitor RGFP-966 to the AD mice (3xTg-AD) was able to reverse tau pathology and decrease levels of Aβ42 in the brain and periphery by increasing NEP levels, which correlated with improved spatial learning memory [102]. Our study using a green tea catechin, epigallocatechin gallate (EGCG), which also acts as an HDAC inhibitor [103], demonstrated that intracortical or oral administration of this compound to adult rats was able to restore levels of NEP in the cortex and hippocampus reduced after prenatal hypoxia. Moreover, EGCG administration resulted in an increased number of dendritic spines in the hippocampal CA1 area, which correlated with memory enhancement [58]. In AD mice, EGCG administration was also shown to attenuate cognitive deterioration by up-regulating NEP expression [104].
Analyzing the reasons for reduced NEP expression under hypoxic conditions, we have found that it correlates with the up-regulation of caspases, which degrade AICD [105]. Incubation of cells under hypoxic conditions with a caspase-3 inhibitor Z-DEVD-FMK rescued AICD and allowed its normal binding to the NEP promoter. Taking this paradigm to the rat model, we have found that prenatal hypoxia led to a significance increase in caspase levels and activity in the brain of rat offspring, which correlated with reduced levels of AICD and NEP [106, 107]. Administration of a caspase inhibitor Ac-DEVD-CHO to developing rats subjected to prenatal hypoxia increased levels of AICD and NEP expression in their cortex, which correlated with the improved cognitive performance [107, 108].
Among other pharmacological compounds capable of up-regulating NEP expression and activity are bexarotene, an agonist of the nuclear retinoid X receptor, which was shown to up-regulate NEP expression affecting HDAC1 binding to the NEP gene promoter [109] and a tyrosine kinase inhibitor Gleevec (imatinib) whose administration to the SH-SY5Y cells increased levels of “functional” AICD and up-regulated NEP leading to lower levels of cellular Aβ [105]. Intraventricular injection of bexarotene to young rats subjected to prenatal hypoxia was shown to increase the number of synaptopodin-positive dendritic spines in the СА1 field of the hippocampus and improve their memory [110].
Because somatostatin can up-regulate the expression of its degrading peptidase NEP [94], it was also suggested that agonists of somatostatin receptors might have a potential therapeutic effect. Indeed, i.c.v. administration of the somatostatin receptor subtype-4 agonist, NNC 26-9100, to 12-month SAMP8 mice, increased NEP activity in the cortical tissue associated with increased protein levels in the extracellular and decreased in the intracellular fractions and improved learning and memory [111].
Serotoninergic activation was also suggested to be beneficial in AD models, and it was shown that 5-hydroxyindolacetic acid (5-HIAA), the final metabolite of serotonin, significantly reduces brain Aβ load in the transgenic APPSWE mice by enhancing NEP levels. 5‑HIAA treatment also improved memory in mice with phosphoramidon-induced cerebral NEP inhibition [112].
Supporting the role of NEP in synaptic plasticity are data on overexpression of a postsynaptic density protein 93 (PSD-93) in APP/PS1 mice which resulted not only in improved animal performance in the open field, Morris water maze and fear condition tests but also facilitated LTP which was accompanied by increased expression of somatostatin receptor 4 (SSTR4) and NEP [113].
There are reports that NEP levels in postnatal life are dependent on the environment in which animals have been raised and that levels of brain NEP activity were higher in mice raised in an “enriched environment”, which also correlated inversely with the Aβ levels [114]. The enriched environment was also shown to improve synaptic plasticity and cognition and neonatal rats subjected to hypoxia/ischemia [115], which correlated with the increased levels of BDNF. As suggested previously, exercising modulates expression and protein content both of NEP and BDNF, leading to improved memory [116]. In aged wild type mice, environmental enrichment enhanced neuronal plasticity by upwardly shifting the cortical excitation/inhibition balance, increased NEP protein levels, and reduced brain Aβ oligomers [117].
Although decreased NEP expression and activity in the brain observed with normal aging can lead to accumulation of Aβ and, related to it, neurodegeneration, it is important to bear in mind that NEP has multiples substrates and is involved in many critical brain processes. While in aged AD mice decreased NEP expression and activity is always accompanied by accumulation of Aβ and impaired memory [76], in NEP-knockout mice with unchanged APP expression immunohistochemical analysis did not reveal Aβ deposits even in old age. Moreover, learning abilities in aged animals with this genotype were enhanced compared to the wild type suggesting that some compensatory mechanisms might develop during the development of these animals to substitute for the absence of NEP [118].
CONCLUSIONS
Over the years since its discovery, NEP has attracted a significant amount of interest and research in various areas of physiology and medicine. Current knowledge about this enzyme made it an attractive therapeutic target in such areas as cardiovascular medicine, cancer, diabetes, and neurodegeneration. While for treatment of some diseases development of NEP inhibitors is paramount (heart failure, hypertension), in other areas, there is a need to up-regulate NEP expression and activity (cancer and AD). Although numerous pharmacological and epigenetic approaches have been developed to up-regulate NEP expression and activity (for review, see [21]), treatment of different types of pathology that involve NEP requires a more profound understanding of the role of this peptidase and specificity of its regulation in various cell types. Since the age-dependent decline in NEP expression and activity in certain brain structures is a natural process, it might have a defined intrinsic physiological role for maintaining age-dependent brain homeostasis and regulating the balance of excitatory and inhibitory processes [35]. However, it also underlies the age-dependent elevation of Aβ concentrations in the brain, which is a natural process that precedes AD pathology [119]. This requires more studies on developmental profiles of NEP expression in animal and human brain in relation to the functions of its primary substrates, including Aβ, which will open new avenues for designing strategies for manipulating NEP expression and activity for therapeutic benefits.
Список литературы
Fulcher I.S., Kenny A.J. Proteins of the kidney microvillar membrane. The amphipathic forms of endopeptidase purified from pig kidneys. Biochem. J. 211: 743–753. 1983. https://doi.org/10.1042/bj2110743
Spillantini M.G., Sicuteri F., Salmon S., Malfroy B. Characterization of endopeptidase 3.4.24.11 (“enkephalinase”) activity in human plasma and cerebrospinal fluid. Biochem. Pharmacol. 39: 1353–1356. 1990. https://doi.org/10.1016/0006-2952(90)90012-a
Kuruppu S., Rajapakse N.W., Minond D., Smith A.I. Production of soluble Neprilysin by endothelial cells. Biochem. Biophys. Res. Commun. 446(2): 423–427. 2014. https://doi.org/10.1016/j.bbrc.2014.01.158
Kerr M.A., Kenny A.J. The purification and specificity of a neutral endopeptidase from rabbit kidney brush border. Biochem. J. 137: 477–488. 1974. https://doi.org/10.1042/bj1370477
Nalivaeva N.N., Turner A.J. Neprilysin. In: Neil D. Rawlings and Guy S. Salvesen, editors, Handbook of Proteolytic Enzymes. Oxford: Academic Press. 612–619. 2013.
Matsas R., Kenny A.J., Turner A.J. An immunohistochemical study of endopeptidase-24.11 (“enkephalinase”) in the pig nervous system. Neuroscience. 18: 991–1012. 1986. https://doi.org/10.1016/0306-4522(86)90113-2
Letarte M., Vera S., Tran R., Addis J.B.L., Onizuka R.J., Quackenbush E.J., Jongeneel C.V., McInnes R.R. Common acute lymphocytic leukemia antigen is identical to neutral endopeptidase. J. Exp. Med. 168: 1247–1253. 1988. https://doi.org/10.1084/jem.168.4.1247
Jongeneel C.V., Quackenbush E.J., Ronco P., Verroust P., Carrel S., Letarte M. Common Acute Lymphoblastic Leukemia Antigen Expressed on Leukemia and Melanoma Cell Lines Has Neutral Endopeptidase Activity. J. Clin. Invest. 83: 713–717. 1989. https://doi.org/10.1172/JCI113936
D'Adamio L., Shipp M.A., Masteller E.L., Reinherz E.L. Organization of the gene encoding common acute lymphoblastic leukemia antigen (neutral endopeptidase 24.11): multiple miniexons and separate 5' untranslated regions. Proc. Natl. Acad. Sci. USA. 86: 7103–7107. 1989. https://doi.org/10.1073/pnas.86.18.7103
Belyaev N.D., Nalivaeva N.N., Makova N.Z., Turner A.J. Neprilysin gene expression requires binding of the amyloid precursor protein intracellular domain to its promoter: implications for Alzheimer disease. EMBO Rep. 10: 94–100. 2009. https://doi.org/10.1038/embor.2008.222
Li C., Booze R.M., Hersh L.B. Tissue-specific expression of rat neutral endopeptidase (neprilysin) mRNAs. J. Biol. Chem. 270: 5723–5728. 1995. https://doi.org/10.1074/jbc.270.11.5723
Turner A.J., Isaac R.E., Coates D. The neprilysin (NEP) family of zinc metalloendopeptidases: genomics and function. Bioessays. 23: 261–269. 2001. https://doi.org/10.1002/1521-1878(200103)23:3<261::AID-BIES1036>3.0.CO;2-K
Zappulla J.P., Wickham. L., Bawab. W., Yang. X.F., Storozhuk. M.V., Castellucci. V.F., DesGroseillers. L. Cloning and characterization of Aplysia neutral endopeptidase, a metallo-endopeptidase involved in the extracellular metabolism of neuropeptides in Aplysia californica. J. Neurosci. 19: 4280–4292. 1999. https://doi.org/10.1523/JNEUROSCI.19-11-04280.1999
Shipp M.A., Richardson N.E., Sayre P.H., Brown N.R., Masteller E.L., Clayton L.K., Ritz J., Reinherz E.L. Molecular cloning of the common acute lymphoblastic leukemia antigen (CALLA) identifies a type II integral membrane protein. Proc. Natl. Acad. Sci. USA. 85: 4819–23. 1988. https://doi.org/10.1073/pnas.85.13.4819
Lorkowski G., Zijderhand-Bleekemolen J.E., Erdös E.G., von Figura K., Hasilik A. Neutral endopeptidase-24.11 (enkephalinase). Biosynthesis and localization in human fibroblasts. Biochem. J. 248: 345–350. 1987. https://doi.org/10.1042/bj2480345
Zheng R., Horiguchi A., Iida K., Lee J., Shen R., Goodman O.B. Jr., Nanus D.M. Neutral endopeptidase is a myristoylated protein. Mol. Cell. Biochem. 335: 173–180. 2010. https://doi.org/10.1007/s11010-009-0253-8
Ganju R.K., Shpektor R.G., Brenner D.G., Shipp M.A. CD10/neutral endopeptidase 24.11 is phosphorylated by casein kinase II and coassociates with other phosphoproteins including the lyn src-related kinase. Blood 88: 4159–4165. 1996. https://doi.org/10.1182/blood.V88.11.4159.bloodjournal88114159
Siepmann M., Kumar S., Mayer G., Walter J. Casein kinase 2 dependent phosphorylation of neprilysin regulates receptor tyrosine kinase signaling to Akt. PLoS One 5: e13134. 2010. https://doi.org/10.1371/journal.pone.0013134
Bayes-Genis A., Barallat J., Richards A.M. A Test in Context: Neprilysin: Function, Inhibition, and Biomarker. J. Am. Coll. Cardiol. 68: 639–653. 2016. https://doi.org/10.1016/j.jacc.2016.04.060
Nalivaeva N.N., Belyaev N.D., Zhuravin I.A., Turner A.J. The Alzheimer’s amyloid-degrading peptidase, neprilysin: can we control it? Int. J. Alzheimers Dis. 2012: 383796. 2012. https://doi.org/10.1155/2012/383796
Nalivaeva N.N., Turner A.J. Targeting amyloid clearance in Alzheimer’s disease as a therapeutic strategy. Br. J. Pharmacol. 176: 3447–3463. 2019. https://doi.org/10.1111/bph.14593
Kerr M.A., Kenny A.J. The purification and specificity of a neutral endopeptidase from rabbit kidney brush border. Biochem. J. 137: 477–488. 1974. https://doi.org/10.1042/bj1370477
Roques B.P., Fournié-Zaluski M.C., Florentin D., Waksman G., Sassi A., Chaillet P., Collado H., Costentin J. New enkephalinase inhibitors as probes to differentiate “enkephalinase” and angiotensin-converting-enzyme active sites. Life Sci. 31: 1749–1752. 1982. https://doi.org/10.1016/0024-3205(82)90201-6
Komiyama T., Aoyagi T., Takeuchi T., Umezawa H. Inhibitory effects of phosphoramidon on neutral metalloendopeptidases and its application on affinity chromatography. Biochem. Biophys. Res. Commun. 65: 352–357. 1975. https://doi.org/10.1016/s0006-291x(75)80100-8
Campbell D.J. Long-term neprilysin inhibition – implications for ARNIs. Nat. Rev. Cardiol. 14: 171–186. 2017. https://doi.org/10.1038/nrcardio.2016.200
Rougeot C., Messaoudi M., Hermitte V., Rigault A.G., Blisnick T., Dugave C., Desor D., Rougeon F. Sialorphin, a natural inhibitor of rat membrane-bound neutral endopeptidase that displays analgesic activity. Proc. Natl. Acad. Sci. USA. 100: 8549–8554. 2003. https://doi.org/10.1073/pnas.1431850100
Wisner A., Dufour E., Messaoudi M., Nejdi A., Marcel A., Ungeheuer M.N., Rougeot C. Human Opiorphin, a natural antinociceptive modulator of opioid-dependent pathways. Proc. Natl. Acad. Sci. USA. 103: 17979–17984. 2006. https://doi.org/10.1073/pnas.0605865103
Barnes K., Matsas R., Hooper N.M., Turner A.J., Kenny A.J. Endopeptidase-24.11 is striosomally ordered in pig brain and, in contrast to aminopeptidase N and peptidyl dipeptidase A ('angiotensin converting enzyme') is a marker for a set of striatal efferent fibres. Neuroscience. 27: 799–817. 1988. https://doi.org/10.1016/0306-4522(88)90184-4
Bourne A., Kenny A.J. The hydrolysis of brain and atrial natriuretic peptides by porcine choroid plexus is attributable to endopeptidase-24.11. Biochem J. 271: 381–385. 1990. https://doi.org/10.1042/bj2710381
Nalivaeva N.N., Fisk L., Kochkina E.G., Plesneva S.A., Zhuravin I.A., Babusikova E., Dobrota D., Turner A.J. Effect of hypoxia/ischemia and hypoxic preconditioning/reperfusion on expression of some amyloid-degrading enzymes. Ann. N.Y. Acad. Sci. 1035: 21–33. 2004. https://doi.org/10.1196/annals.1332.002
Barnes K., Doherty S., Turner A.J. Endopeptidase-24.11 is the integral membrane peptidase initiating degradation of somatostatin in the hippocampus. J. Neurochem. 64: 1826–1832. 1995. https://doi.org/10.1046/j.1471-4159.1995.64041826.x
Waksman G., Hamel E., Delay-Goyet P., Roques BP. Neuronal localization of the neutral endopeptidase 'enkephalinase' in rat brain revealed by lesions and autoradiography. EMBO J. 5: 3163–3166. 1986. PMID: .3545813
Iijima-Ando K., Hearn S.A., Granger L., Shenton C., Gatt A., Chiang H.C., Hakker I., Zhong Y., Iijima K. Overexpression of neprilysin reduces alzheimer amyloid-beta42 (Abeta42)-induced neuron loss and intraneuronal Abeta42 deposits but causes a reduction in cAMP-responsive element-binding protein-mediated transcription, age-dependent axon pathology, and premature death in Drosophila. J. Biol. Chem. 283: 19066–19076. 2008. https://doi.org/10.1074/jbc.M710509200
Fukami S., Watanabe K., Iwata N., Haraoka J., Lu B., Gerard N.P., Gerard C., Fraser P., Westaway D., St George-Hyslop P., Saido T.C. Aβ-degrading endopeptidase, neprilysin, in mouse brain: synaptic and axonal localization inversely correlating with Aβ pathology. Neurosci. Res. 43: 39–56. 2002. https://doi.org/10.1016/s0168-0102(02)00015-9
Pacheco-Quinto J., Eckman C.B., Eckman E.A. Major amyloid-β-degrading enzymes, endothelin-converting enzyme-2 and neprilysin, are expressed by distinct populations of GABAergic interneurons in hippocampus and neocortex. Neurobiol. Aging. 48: 83–92. 2016. https://doi.org/10.1016/j.neurobiolaging.2016.08.011
Fisk L., Nalivaeva N.N., Boyle J.P., Peers C.S., Turner A.J. Effects of hypoxia and oxidative stress on expression of neprilysin in human neuroblastoma cells and rat cortical neurones and astrocytes. Neurochem. Res. 32: 1741–1748. 2007. https://doi.org/10.1007/s11064-007-9349-2
Shimizu E., Kawahara K., Kajizono M., Sawada M., Nakayama H. IL-4-induced selective clearance of oligomeric β-amyloid peptide1–42 by rat primary type 2 microglia. J. Immunol. 181: 6503–6513. 2008. https://doi.org/10.4049/jimmunol.181.9.6503
Ries M., Sastre M. Mechanisms of Aβ Clearance and Degradation by Glial Cells. Front. Aging Neurosci. 8: 160. 2016. https://doi.org/10.3389/fnagi.2016.00160
Waksman G., Hamel E., Fournié-Zaluski M.C., Roques B.P. Autoradiographic Comparison of the Distribution of the Neutral Endopeptidase “Enkephalinase” and of Mu and Delta Opioid Receptors in Rat Brain. Proc. Natl. Acad. Sci. USA. 83: 1523–1527. 1986. https://doi.org/10.1073/pnas.83.5.1523
Saria A., Hauser K.F., Traurig H.H., Turbek C.S., Hersh L., Gerard C. Opioid-related changes in nociceptive threshold and in tissue levels of enkephalins after target disruption of the gene for neutral endopeptidase (EC 3.4.24.11) in mice. Neurosci. Lett. 234: 27–30. 1997. https://doi.org/10.1016/s0304-3940(97)00660-5
Barnes K., Turner A.J., Kenny A.J. An immunoelectron microscopic study of pig substantia nigra shows colocalization of endopeptidase-24.11 with substance P. Neuroscience. 53: 1073–1082. 1993. https://doi.org/10.1016/0306-4522(93)90490-7
Feindt J., Krisch B., Lucius R., Mentlein R. Meningeal cells are targets and inactivation sites for the neuropeptide somatostatin. Brain Res. Mol. Brain. Res. 44: 293–300. 1997. https://doi.org/10.1016/s0169-328x(96)00229-x
Nocera S., Simon A., Fiquet O., Chen Y., Gascuel J., Datiche F., Schneider N., Epelbaum J., Viollet C. Somatostatin Serves a Modulatory Role in the Mouse Olfactory Bulb: Neuroanatomical and Behavioral Evidence. Front. Behav. Neurosci. 13: 61. 2019. https://doi.org/10.3389/fnbeh.2019.00061
Dutriez I., Salès N., Fournié-Zaluski M.C., Roques B.P. Pre- and post-natal ontogeny of neutral endopeptidase 24-11 ('enkephalinase') studied by in vitro autoradiography in the rat. Experientia. 48: 290–300. 1992. https://doi.org/10.1007/bf01930479
Dauch P., Masuo Y., Vincent J.P., Checler F. A survey of the cerebral regionalization and ontogeny of eight exo- and endopeptidases in murines. Peptides. 14: 593–599. 1993. https://doi.org/10.1016/0196-9781(93)90150-f
Kioussi C., Crine P., Matsas R. Endopeptidase-24.11 is suppressed in myelin-forming but not in non-myelin-forming Schwann cells during development of the rat sciatic nerve. Neuroscience. 50: 69–83. 1992. https://doi.org/10.1016/0306-4522(92)90382-c
Kioussi C., Mamalaki A., Jessen K., Mirsky R., Hersh L.B., Matsas R. Expression of endopeptidase-24.11 (common acute lymphoblastic leukaemia antigen CD10) in the sciatic nerve of the adult rat after lesion and during regeneration. Eur. J. Neurosci. 7: 951–961. 1995. https://doi.org/10.1111/j.1460-9568.1995.tb01083.x
Higuchi Y., Hashiguchi A., Yuan J., Yoshimura A., Mitsui J., Ishiura H., Tanaka M., Ishihara S., Tanabe H., Nozuma S., Okamoto Y., Matsuura E., Ohkubo R., Inamizu S., Shiraishi W., Yamasaki R., Ohyagi Y., Kira J., Oya Y., Yabe H., Nishikawa N., Tobisawa S., Matsuda N., Masuda M., Kugimoto C., Fukushima K., Yano S., Yoshimura J., Doi K., Nakagawa M., Morishita S., Tsuji S., Takashima H. Mutations in MME cause an autosomal-recessive Charcot-Marie-Tooth disease type 2. Ann. Neurol. 79: 659–672. 2016. https://doi.org/10.1002/ana.24612
Maguer-Satta V., Besançon R., Bachelard-Cascales E. Concise review: neutral endopeptidase (CD10): a multifaceted environment actor in stem cells, physiological mechanisms, and cancer. Stem Cells 29, 389–396. 2011. https://doi.org/10.1002/stem.592
Lu B., Gerard N.P., Kolakowski L.F. Jr., Bozza M., Zurakowski D., Finco O., Carroll M.C., Gerard C. Neutral endopeptidase modulation of septic shock. J. Exp. Med. 181: 2271–2275. 1995. https://doi.org/10.1084/jem.181.6.2271
Saria A., Hauser K.F., Traurig H.H., Turbek C.S., Hersh L., Gerard C. Opioid-related changes in nociceptive threshold and in tissue levels of enkephalins after target disruption of the gene for neutral endopeptidase (EC 3.4.24.11) in mice. Neurosci. Lett. 234, 27–30. 1997. https://doi.org/10.1016/s0304-3940(97)00660-5
Fischer H.S., Zernig G., Schuligoi R., Miczek K.A., Hauser K.F., Gerard C., Saria A. Alterations within the endogenous opioid system in mice with targeted deletion of the neutral endopeptidase ('enkephalinase') gene. Regul. Pept. 96: 53–58. 2000. https://doi.org/10.1016/s0167-0115(00)00200-7
Siems W., Maul B., Krause W., Gerard C., Hauser K.F., Hersh L.B., Fischer H.S., Zernig G., Saria A. Neutral endopeptidase and alcohol consumption, experiments in neutral endopeptidase-deficient mice. Eur. J. Pharmacol. 397: 327–334. 2000. https://doi.org/10.1016/s0014-2999(00)00222-3
Krämer H.H., He L., Lu B., Birklein F., Sommer C. Increased pain and neurogenic inflammation in mice deficient of neutral endopeptidase. Neurobiol. Dis. 35: 177–183. 2009. https://doi.org/10.1016/j.nbd.2008.11.002
Zhuravin I.A., Dubrovskaya N.M., Tumanova N.L., Vasilev D.S., Nalivaeva N.N. Ontogenetic and phylogenetic approaches for studying the mechanisms of cognitive dysfunctions. In: Evolutionary Physiology and Biochemistry: Advances and Perspectives / ed. V.F. Levchenko. InTech: Rijeka, Croatia. 2018. Chapter 15, 205–224. http://www.intechopen.com/books/evolutionary-physiology-andbiochemistry-advances-and-perspectives. https://doi.org/10.5772/intechopen.73666
Dubrovskaya N.M., Zhuravin I.A. Ontogenetic characteristics of behavior in rats subjected to hypoxia on day 14 or day 18 of embryogenesis. Neurosci. Behav. Physiol. 40: 231–238. 2010. https://doi.org/10.1007/s11055-009-9235-2
Nalivaeva N.N., Belyaev N.D., Lewis D.I., Pickles A.R., Makova N.Z., Bagrova D.I., Dubrovskaya N.M., Plesneva S.A., Zhuravin I.A., Turner A.J. Effect of sodium valproate administration on brain neprilysin expression and memory in rats. J. Mol. Neurosci. 46: 569–577. 2012. https://doi.org/10.1007/s12031-011-9644-x
Zhuravin I.A., Dubrovskaya N.M., Vasilev D.S., Kozlova D.I., Kochkina E.G., Tumanova N.L., Nalivaeva N.N. Regulation of Neprilysin Activity and Cognitive Functions in Rats After Prenatal Hypoxia. Neurochem. Res. 44: 1387–1398. 2019. https://doi.org/10.1007/s11064-019-02796-3
Vasilev D.S., Dubrovskaya N.M., Tumanova N.L., Zhuravin I.A. Prenatal Hypoxia in Different Periods of Embryogenesis Differentially Affects Cell Migration, Neuronal Plasticity, and Rat Behavior in Postnatal Ontogenesis. Front. Neurosci. 10: 126. 2016. https://doi.org/10.3389/fnins.2016.00126
Deller T., Bas Orth C., Vlachos A., Merten T., Del Turco D., Dehn D., Mundel P., Frotscher M. Plasticity of synaptopodin and the spine apparatus organelle in the rat fascia dentata following entorhinal cortex lesion. J. Compar. Neurol. 499: 471–484. 2006. https://doi.org/10.1002/cne.21103
Vasilev D.S., Tumanova N.L., Kalinina D.S. Prenatal hypoxia disturbs the formation of pyramidal neurones in the entorhinal cortex of the rat brain. Russ. J. Physiol. 2020. In press.
Zhang X., Li L., Zhang X., Xie W., Li L., Yang D., Heng X., Du Y., Doody R.S., Le W. Prenatal hypoxia may aggravate the cognitive impairment and Alzheimer’s disease neuropathology in APPSwe/PS1A246E transgenic mice. Neurobiol. Aging. 34: 663–678. 2013. https://doi.org/10.1016/j.neurobiolaging.2012.06.012
Turrel O., Lampin-Saint-Amaux A., Préat T., Goguel V. Drosophila Neprilysins Are Involved in Middle-Term and Long-Term Memory. J. Neurosci. 36: 9535–9546. 2016. https://doi.org/10.1523/JNEUROSCI.3730-15
Turrel O., Rabah Y., Plaçais P.Y., Goguel V., Preat T. Drosophila Middle-Term Memory: Amnesiac is Required for PKA Activation in the Mushroom Bodies, a Function Modulated by Neprilysin 1. J. Neurosci. 40: 4219–4229. 2020. https://doi.org/10.1523/JNEUROSCI.2311-19.2020
Dubrovskaya N.M., Nalivaeva N.N., Plesneva S.A., Feponova A.A., Turner A.J., Zhuravin I.A. Changes in the Activity of Amyloid-Degrading Metallopeptidases Leads to Disruption of Memory in Rats. Neurosci. Behav. Physiol. 40: 975–980. 2010. https://doi.org/10.1007/s11055-010-9355-8
Mouri A., Zou L.B., Iwata N., Saido T.C., Wang D., Wang M.W., Noda Y., Nabeshima T. Inhibition of neprilysin by thiorphan (i.c.v.) causes an accumulation of amyloid β and impairment of learning and memory. Behav. Brain Res. 168: 83–91. 2006. https://doi.org/10.1016/j.bbr.2005.10.014
Zhuravin I.A., Dubrovskaya N.M., Vasilev D.S., Tumanova N.L., Nalivaeva N.N. Epigenetic and pharmacological regulation of the amyloid-degrading enzyme neprilysin results in modulation of cognitive functions in mammals Dokl. Biol. Sci. 438: 145–148. 2011. Epub 2011 Jul 5.https://doi.org/10.1134/S001249661103015X
Zou L.B., Mouri A., Iwata N., Saido T.C., Wang D., Wang M.W., Mizoguchi H., Noda Y., Nabeshima T. Inhibition of neprilysin by infusion of thiorphan into the hippocampus causes an accumulation of amyloid Beta and impairment of learning and memory. J. Pharmacol. Exp. Ther. 317: 334–340. 2006. https://doi.org/10.1124/jpet.105.095687
Hardy J., Selkoe D.J. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 297: 353–356. 2002. https://doi.org/10.1126/science.1072994
Selkoe D.J., Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 8: 595–608. 2016. https://doi.org/10.15252/emmm.201606210
Howell S., Nalbantoglu J., Crine P. Neutral endopeptidase can hydrolyze β-amyloid (1–40) but shows no effect on β-amyloid precursor protein metabolism. Peptides. 16: 647–652. 1995. https://doi.org/10.1016/0196-9781(95)00021-b
Iwata N., Tsubuki S., Takaki Y., Watanabe K., Seikiguchi M., Hosoki E., Kawashima-Morishima M., Lee H.-J., Hama E., Sekine-Aizawa Y., Saido T.C. Identification of the major Aβ1-42 degrading catabolic pathway in brain parenchyma: Suppression leads to biochemical and pathological deposition. Nature Med. 6: 143–150. 2000. https://doi.org/10.1038/72237
Iwata N., Tsubuki S., Takaki Y., Shirotani K., Lu B., Gerard N.P., Gerard C., Hama E., Lee H., Saido T.C. Metabolic regulation of brain Aβ by neprilysin. Science 292: 1550–1552. 2001. https://doi.org/10.1126/science.1059946
Hanson L.R., Hafez D., Svitak A.L., Burns R.B., Li X., Frey W.H. 2nd, Marr RA. Intranasal phosphoramidon increases beta-amyloid levels in wild-type and NEP/NEP2-deficient mice. J. Mol. Neurosci. 43: 424–427. 2011. https://doi.org/10.1007/s12031-010-9460-8
Nilsson P., Loganathan K., Sekiguchi M., Winblad B., Iwata N., Saido T.C., Tjernberg L.O. Loss of neprilysin alters protein expression in the brain of Alzheimer’s disease model mice. Proteomics. 15: 3349–3355. 2015. https://doi.org/10.1002/pmic.201400211
Mohajeri M.H., Wolfer D.P., Neprilysin deficiency-dependent impairment of cognitive functions in a mouse model of amyloidosis. Neurochem. Res. 34: 717–726. 2009. https://doi.org/10.1007/s11064-009-9919-6
Carpentier M., Robitaille Y., DesGroseillers L., Boileau G., Marcinkiewicz M. Declining expression of neprilysin in Alzheimer disease vasculature: possible involvement in cerebral amyloid angiopathy. J. Neuropathol. Exp. Neurol. 61: 849–856. 2002. https://doi.org/10.1093/jnen/61.10.849
Yasojima K., Akiyama H., McGeer E.G., McGeer P.L. Reduced neprilysin in high plaque areas of Alzheimer brain: a possible relationship to deficient degradation of beta-amyloid peptide. Neurosci. Lett. 297: 97–100. 2001. https://doi.org/10.1016/s0304-3940(00)01675-x
Zhang H., Liu D., Wang Y., Huang H., Zhao Y., Zhou H. Meta-analysis of expression and function of neprilysin in Alzheimer’s disease. Neurosci. Lett 657: 69–76. 2017. https://doi.org/10.1016/j.neulet.2017.07.060
Li W., Wu Y., Min F., Li Z., Huang J., Huang R. A nonhuman primate model of Alzheimer’s disease generated by intracranial injection of amyloid-β42 and thiorphan. Metab. Brain. Dis. 25: 277–284. 2010. https://doi.org/10.1007/s11011-010-9207-9
Marr R.A., Rockenstein E., Mukherjee A., Kindy M.S., Hersh L.B., Gage F.H., Verma I.M., Masliah E. Neprilysin gene transfer reduces human amyloid pathology in transgenic mice. J. Neurosci. 23: 1992–1996. 2003. PMCID: PMC6742010.
Park M.H., Lee J.K., Choi S., Ahn J., Jin H.K., Park J.S., Bae J.S. Recombinant soluble neprilysin reduces amyloid-beta accumulation and improves memory impairment in Alzheimer’s disease mice. Brain Res. 1529: 113–124. 2013. https://doi.org/10.1016/j.brainres.2013.05.045
Hüttenrauch M., Baches S., Gerth J., Bayer T.A., Weggen S., Wirths O. Neprilysin deficiency alters the neuropathological and behavioral phenotype in the 5XFAD mouse model of Alzheimer’s disease. J. Alzheimers. Dis. 44: 1291–1302. 2015. https://doi.org/10.3233/JAD-142463
Huang S.M., Mouri A., Kokubo H., Nakajima R., Suemoto T., Higuchi M., Staufenbiel M., Noda Y., Yamaguchi H., Nabeshima T., Saido T.C., Iwata N. Neprilysin-sensitive synapse-associated amyloid-beta peptide oligomers impair neuronal plasticity and cognitive function. J. Biol. Chem. 281: 17941–17951. 2006. https://doi.org/10.1074/jbc.M601372200
Blurton-Jones M., Spencer B., Michael S., Castello N.A., Agazaryan A.A., Davis J.L., Müller F.J., Loring J.F., Masliah E., LaFerla F.M. Neural stem cells genetically-modified to express neprilysin reduce pathology in Alzheimer transgenic models. Stem. Cell Res. Ther. 5: 46. 2014. https://doi.org/10.1186/scrt440
Schliebs R., Arendt T. The cholinergic system in aging and neuronal degeneration. Behav. Brain Res. 221: 555–563. 2011. https://doi.org/10.1016/j.bbr.2010.11.058
Foidl B.M., Do-Dinh P., Hutter-Schmid B., Bliem H.R., Humpel C. Cholinergic neurodegeneration in an Alzheimer mouse model overexpressing amyloid-precursor protein with the Swedish-Dutch-Iowa mutations. Neurobiol. Learn. Mem. 136: 86–96. 2016. https://doi.org/10.1016/j.nlm.2016.09.014
Fodero L.R., Mok S.S., Losic D., Martin L.L., Aguilar M.I., Barrow C.J., Livett B.G., Small D.H. Alpha7-nicotinic acetylcholine receptors mediate an Aβ(1–42)-induced increase in the level of acetylcholinesterase in primary cortical neurones. J. Neurochem. 88: 1186–1193. https://doi.org/10.1046/j.1471-4159.2003.02296.x
Masliah E., Alford M., Adame A., Rockenstein E., Galasko D., Salmon D., Hansen L.A., Thal L.J. Aβ1-42 promotes cholinergic sprouting in patients with AD and Lewy body variant of AD. Neurology. 61: 206–211. 2003. https://doi.org/10.1212/01.WNL.0000073987.79060.4B
Sorial M.E. El Sayed NSED. Protective effect of valproic acid in streptozotocin-induced sporadic Alzheimer’s disease mouse model: possible involvement of the cholinergic system. Naunyn. Schmiedebergs Arch. Pharmacol. 390: 581–593. 2017. https://doi.org/10.1007/s00210-017-1357-4
Djordjevic J., Jones-Gotman M., De Sousa K., Chertkow H. Olfaction in patients with mild cognitive impairment and Alzheimer’s disease. Neurobiol. Aging. 29: 693–706. 2008. https://doi.org/10.1016/j.neurobiolaging.2006.11.014
Wesson D.W., Levy E., Nixon R.A., Wilson D.A. Olfactory dysfunction correlates with amyloid-β burden in an Alzheimer’s disease mouse model. J. Neurosci. 30: 505–514. 2010. https://doi.org/10.1523/JNEUROSCI.4622-09.2010
Saiz-Sanchez D., Ubeda-Bañon I., de la Rosa-Prieto C., Argandoña-Palacios L., Garcia-Muñozguren S., Insausti R., Martinez-Marcos A. Somatostatin, Tau, and β-Amyloid Within the Anterior Olfactory Nucleus in Alzheimer Disease. Exp. Neurol. 223: 347–350. 2010. https://doi.org/10.1016/j.expneurol.2009.06.010
Saito T., Iwata N., Tsubuki S., Takaki Y., Takano J., Huang S.M., Suemoto T., Higuchi M., Saido T.C. Somatostatin regulates brain amyloid β peptide Aβ42 through modulation of proteolytic degradation. Nat. Med. 11: 434–439. 2005. https://doi.org/10.1038/nm1206
Belyaev N.D., Kellett K.A., Beckett C., Makova N.Z., Revett T.J., Nalivaeva N.N., Hooper N.M., Turner A.J. The transcriptionally active amyloid precursor protein (APP) intracellular domain is preferentially produced from the 695 isoform of APP in a β-secretase-dependent pathway. J. Biol. Chem. 285: 41443–41454. 2010. https://doi.org/10.1074/jbc.M110.141390
Pardossi-Piquard R., Petit A., Kawarai T., Sunyach C., Alves da Costa C., Vincent B., Ring S., D’Adamio L., Shen J., Müller U., St George Hyslop P., Checler F. Presenilin-dependent transcriptional control of the Aβ-degrading enzyme neprilysin by intracellular domains of βAPP and APLP. Neuron. 46: 541–554. 2005. https://doi.org/10.1016/j.neuron.2005.04.008
Koudinov A.R., Berezov T.T. Alzheimer’s amyloid-β (Aβ) is an essential synaptic protein, not neurotoxic junk. Acta Neurobiol. Exp. (Wars). 64: 71–79. 2004. PMID: .15190681
Wang H., Megill A., He K., Kirkwood A., Lee H.K. Consequences of inhibiting amyloid precursor protein processing enzymes on synaptic function and plasticity. Neural Plast. 2012: 272374. 2012. https://doi.org/10.1155/2012/272374
Abramov E., Dolev I., Fogel H., Ciccotosto G.D., Ruff E., Slutsky I. Amyloid-β as a positive endogenous regulator of release probability at hippocampal synapses. Nat. Neurosci. 12: 1567–1576. 2009. https://doi.org/10.1038/nn.2433
Finnie P.S.P., Nader K. Amyloid β Secreted during Consolidation Prevents Memory Malleability. Curr. Biol. 30: 1934–1940.e4. 2020. https://doi.org/10.1016/j.cub.2020.02.083
Wang Z., Zhang X.J., Li T., Li J., Tang Y., Le W. Valproic acid reduces neuritic plaque formation and improves learning deficits in APP(Swe) /PS1(A246E) transgenic mice via preventing the prenatal hypoxia-induced down-regulation of neprilysin. CNS Neurosci. Ther. 20: 209–217. 2014. https://doi.org/10.1111/cns.12186
Janczura K.J., Volmar C.H., Sartor G.C., Rao S.J., Ricciardi N.R., Lambert G., Brothers S.P., Wahlestedt C. Inhibition of HDAC3 reverses Alzheimer’s disease-related pathologies in vitro and in the 3xTg-AD mouse model. Proc. Natl. Acad. Sci. USA. 115: E11148–E11157. 2018. https://doi.org/10.1073/pnas.1805436115
Hu Q., Chang X., Yan R., Rong C., Yang C., Cheng S., Gu X., Yao H., Hou X., Mo Y., Zhao L., Chen Y., Dinlin X., Wang Q., Fang S. (−)-Epigallocatechin-3-gallate induces cancer cell apoptosis via acetylation of amyloid precursor protein. Med. Oncol. 32: 390. 2015. https://doi.org/10.1007/s12032-014-0390-0
Chang X., Rong C., Chen Y., Yang C., Hu Q., Mo Y., Zhang C., Gu X., Zhang L., He W., Cheng S., Hou X., Su R., Liu S., Dun W., Wang Q., Fang S. (-)-Epigallocatechin-3-gallate attenuates cognitive deterioration in Alzheimer’s disease model mice by upregulating neprilysin expression. Exp. Cell Res. 334: 136–145. 2015. https://doi.org/10.1016/j.yexcr.2015.04.004
Kerridge C., Kozlova D.I., Nalivaeva N.N., Turner A.J. Hypoxia Affects Neprilysin Expression Through Caspase Activation and an APP Intracellular Domain-dependent Mechanism. Front Neurosci. 9: 426. 2015. https://doi.org/10.3389/fnins.2015.00426
Vasilev D.S., Dubrovskaya N.M., Nalivaeva N.N., Zhuravin I.A. 2016. Regulation of caspase-3 content and activity in rat cortex in norm and after prenatal hypoxia. Neurochem. J. 10: 144–150. https://doi.org/10.1134/S1819712416020100
Kozlova D.I., Vasylev D.S., Dubrovskaya N.M., Nalivaeva N.N., Tumanova N.L., Zhuravin I.A. Role of caspase-3 in regulation of the amyloid-degrading neuropeptidase neprilysin level in the rat cortex after hypoxia. J. Evol. Biochem. Physiol. 51: 480–484. 2015. https://doi.org/10.1134/S0022093015060046
Dubrovskaya N.M., Tikhonravov D.L., Alekseeva O.S., Zhuravin I.A. Recovery of learning and memory impaired by prenatal hypoxic stress in rats after injection of caspase-3 inhibitor during early ontogenesis. J. Evol. Biochem. Physiol. 53(1): 66–68. 2017. https://doi.org/10.1134/S0022093017010082
Nalivaeva N.N., Belyaev N.D., Turner A.J. New Insights into Epigenetic and Pharmacological Regulation of Amyloid-Degrading Enzymes. Neurochem. Res. 41: 620–630. 2016. https://doi.org/10.1007/s11064-015-1703-1
Dubrovskaya N.M., Vasilev D.S., Nalivaeva N.N., Tumanova N.L., Alekseeva O.S., Zhuravin I.A. Bexarotene, an Agonist of Nuclear X Retinoid Receptors, Improves Cognitive Functions in Rats Subjected to Prenatal Hypoxia. Russ. J. Physiol. 105: 165–177. 2019. (Article in Russian). .https://doi.org/10.1134/S0869813919020043
Sandoval K.E., Farr S.A., Banks W.A., Crider A.M., Morley J.E., Witt K.A. Somatostatin receptor subtype-4 agonist NNC 26-9100 decreases extracellular and intracellular Aβ1−42 trimers. Eur. J. Pharmacol. 683: 116–124. 2012. https://doi.org/10.1016/j.ejphar.2012.03.020
Klein C., Roussel G., Brun S., Rusu C., Patte-Mensah C., Maitre M., Mensah-Nyagan A.G. 5-HIAA induces neprilysin to ameliorate pathophysiology and symptoms in a mouse model for Alzheimer’s disease. Acta Neuropathol. Commun. 6: 136. 2018. https://doi.org/10.1186/s40478-018-0640-z
Yu L., Liu Y., Yang H., Zhu X., Cao X., Gao J., Zhao H., Xu Y. PSD-93 Attenuates Amyloid-β-Mediated Cognitive Dysfunction by Promoting the Catabolism of Amyloid-β. J. Alzheimers Dis. 59: 913–927. 2017. https://doi.org/10.3233/JAD-170320
Lazarov O., Robinson J., Tang Y.P., Hairston I.S., Korade-Mirnics Z., Lee V.M., Hersh L.B., Sapolsky R.M., Mirnics K., Sisodia S.S. Environmental enrichment reduces Aβ levels and amyloid deposition in transgenic mice. Cell. 120: 701–713. 2005. https://doi.org/10.1016/j.cell.2005.01.015
Griva M., Lagoudaki R., Touloumi O., Nousiopoulou E., Karalis F., Georgiou T., Kokaraki G., Simeonidou C., Tata D.A., Spandou E. Long-term effects of enriched environment following neonatal hypoxia-ischemia on behavior, BDNF and synaptophysin levels in rat hippocampus: Effect of combined treatment with G-CSF. Brain Res. 1667: 55–67. 2017. Epub 2017 May 8.https://doi.org/10.1016/j.brainres.2017.05.004
Radak Z., Kumagai S., Taylor A.W., Naito H., Goto S. Effects of exercise on brain function: role of free radicals. Appl. Physiol. Nutr. Metab. 32: 942–946. 2007. https://doi.org/10.1139/H07-081
Mainardi M., Di Garbo A., Caleo M., Berardi N., Sale A., Maffei L. Environmental enrichment strengthens corticocortical interactions and reduces amyloid-β oligomers in aged mice. Front. Aging Neurosci. 6: 1. 2014. https://doi.org/10.3389/fnagi.2014.00001
Walther T., Albrecht D., Becker M., Schubert M., Kouznetsova E., Wiesner B., Maul B., Schliebs R., Grecksch G., Furkert J., Sterner-Kock A., Schultheiss H.P., Becker A., Siems W.E. Improved learning and memory in aged mice deficient in amyloid β-degrading neutral endopeptidase. PLoS One. 4: e4590. 2009. https://doi.org/10.1371/journal.pone.0004590
Saito T., Takaki Y., Iwata N., Trojanowski J., Saido T.C. Alzheimer’s disease, neuropeptides, neuropeptidase, and amyloid-β peptide metabolism. Sci. Aging Knowledge Environ. 2003: PE1. 2003. https://doi.org/10.1126/sageke.2003.3.pe1
Дополнительные материалы отсутствуют.
Инструменты
Российский физиологический журнал им. И.М. Сеченова