

Российский физиологический журнал им. И.М. Сеченова, 2020, T. 106, № 2, стр. 243-253
Исследование роли нерецепторной тирозинкиназы c-Abl в регуляции р53-зависимой активации ERK1/2-зависимого каскада
Е. А. Олейник 1, А. А. Наумова 1, Д. В. Немирич 2, С. Д. Николаева 1, В. Т. Бахтеева 1, А. С. Березовская 1, Е. В. Черниговская 1, М. В. Глазова 1, *
1 Институт эволюционной физиологии и биохимии им. И.М. Сеченова РАН
Санкт-Петербург, Россия
2 Университет ИТМО, кафедра компьютерных технологий
Санкт-Петербург, Россия
* E-mail: mglazova@iephb.ru
Поступила в редакцию 28.08.2019
После доработки 02.12.2019
Принята к публикации 03.12.2019
Аннотация
Белок р53 является транскрипционным фактором, который в ответ на клеточный стресс запускает старение, остановку клеточного цикла, или апоптоз. Данные литературы свидетельствуют о том, что проапоптозный белок р53 участвует не только в апоптозе, но и в регуляции процессов нейрональной дифференцировки, а также оказывает активирующее влияние на ERK1/2 сигнальный каскад. Однако до сих пор нет точного ответа на вопрос, касающийся характера и механизмов влияния р53 на ERK1/2 сигнальный каскад. В данной работе мы провели анализ внутриклеточных механизмов, опосредующих активирующее влияние р53 на ERK1/2. Исследование было проведено на недифференцированных клетках линии РС12. Для кратковременной активации TrkA-рецепторов в среду добавляли фактор роста нервов NGF. Результаты показали, что активация p53 введением Nutlin-3 привела к повышению активности TrkA/cRaf/ERK каскада и нерецепторной киназы c-Abl. Ингибирование c-Abl на фоне активации р53 не изменило активность TrkA-рецепторов, но при этом сохранялась повышенная активность cRaf и ERK1/2. Однако сочетанное введение Nutlin-3 и ингибиторов c-Abl и TrkA-рецепторов привело к значительному снижению активности ERK1/2. Полученные нами данные свидетельствуют о том, что р53 оказывает активирующее влияние на ERK1/2 каскад как опосредованно, в составе комплекса р53/c-Abl и активируя Trk-рецепторы, так и независимо от c-Abl, влияя на ERK1/2 каскад на уровне или выше cRaf.
Изучению роли р53 посвящены тысячи исследований, но до сих пор этот белок вызывает огромный интерес. Показано, что р53 регулирует стабильность генома, апоптоз, клеточный цикл, дифференцировку, развитие, метаболизм и т.д. [1–3]. Известно, что р53 является транскрипционным фактором и контролирует экспрессию множества генов. С другой стороны, р53 также работает и как цитоплазматический белок. При этом его транскрипционная и цитоплазматическая активность зависит не только от посттрансляционных изменений, но и от прямых белковых взаимодействий, и одним из белков, связывающихся с р53 и регулирующих его активность является c-Abl (Abelson murine leukemia viral oncogene homolog 1). Так, показано, что c-Abl повышает транскрипционную активность р53 [4–7]. В цитоплазме белковый комплекс р53/c-Abl связывается с тирозинкиназным доменом Trk-рецепторов, защищает р53 от MDM2-зависимой деградации и стабилизирует р53 [6, 8].
Нерецепторная тирозинкиназа с-Abl экспрессируется практически во всех типах клеток млекопитающих с максимальным уровнем в мозге, селезенке и тимусе [9]. В нейронах киназа с-Abl преимущественно локализована в цитоплазме, где связывается с мембранами, актиновыми филаментами и различными белками. Ядерная локализация с-Abl определяет ее транскрипционную активность, основанную на ее прямом связывании с С-терминальным доменом РНК-полимеразы II типа, или, как уже было сказано выше, на образовании комплекса с р53. Более того, c-Abl участвует в регуляции репарации ДНК и контроле клеточного цикла [10]. Также показана ее роль в процессах апоптоза, где она может вести себя как про- так и анти-апоптозный фактор. Нокаутирование с-Abl летально для организма (приводит к гибели на ранних сроках эмбрионального развития) [9]. В нейронах c-Abl регулирует формирование синапсов [11]. Последние исследования свидетельствуют о связи активации с-Abl с нейродегенеративными процессами, в частности, с болезнями Паркинсона и Альцгеймера. Было показано, что с-Abl может фосфорилировать белок Parkin, что приводит к его инактивации и, соответственно, к гибели дофаминергических нейронов [12]. На основании этих данных с-Abl рассматривается как потенциальная мишень в разработке новых подходов в лечении болезни Паркинсона [13, 14]. С другой стороны, показано, что с-Abl и ERK1/2- киназа (extracellular signal related protein kinase 1 and 2) функционально связаны [15]. Известно, что ERK1/2 киназа также является одним из факторов, вовлеченным в патогенез болезни Паркинсона [16, 17]. В частности, показано, что гибель дофаминергических нейронов черной субстанции непосредственно связана с повышением активности ERK1/2 в тельцах Леви на ранних стадиях развития болезни Паркинсона [16]. При этом гибель дофаминергических нейронов опосредована активацией р53-зависимого пути апоптоза [18]. В свою очередь опубликованные данные свидетельствуют об активирующем влиянии р53 на ERK1/2-сигнальный каскад. Было показано, что в клетках меланомы, экспрессирующих мутантную, неактивную, форму р53 отмечается снижение активности ERK1/2 [19]. На различных клеточных линиях также были получены аналогичные результаты, при этом было показано, что функционально активные Ras и cRaf являются необходимыми посредниками р53-зависимой активации каскада [20–23]. С другой стороны, активирующее влияние р53 на ERK1/2 каскад может быть опосредовано c-Abl. На культуре клеток РС12 было показано, что при NGF-стимулированной дифференцировке белок р53 является необходимым фактором для стимуляции ERK1/2 [24]. Таким образом, в разных типах клеток влияние р53 на активность ERK1/2 одинаково, однако механизм выявленного эффекта до конца не выяснен. Предполагается, что эти эффекты опосредованы транскрипционной активностью р53, поскольку мутантная форма р53, не обладающая транскрипционной активностью, снижает активность ERK1/2, а ингибирование транскрипционной активности р53 введением специфического блокатора Pifithrin-alfa снижает уровень фосфорилирования ERK1/2 in vivo [20, 25–27]. Важно подчеркнуть, что c-Abl, так же, как и р53, принимает участие в регуляции активности ERK1/2 каскада как в норме, так и при различных патологических состояниях клетки [28, 29].
Таким образом, наличие функциональных связей между р53 и cAbl, р53 и ERK1/2, и cAbl и ERK1/2 объединяет их в тесно связанное функциональное трио, однако это третичное взаимодействие до сих пор остается не изученным. В данной работе было проведено исследование внутриклеточных механизмов, опосредующих влияние р53 на активность ERK1/2 в экспериментах с использованием различных сочетаний блокаторов и/или активаторов р53, c-Abl и членов ERK1/2 каскада. В качестве модели исследования мы выбрали клетки линии РС12, которые являются широко применяемым объектом для изучения регуляции функциональной активности дофаминергических нейронов. Полученные результаты показали, что р53-зависимая активация ERK каскада опосредована взаимодействием р53, T-rkA и протеинкиназы с-Abl.
МЕТОДЫ ИССЛЕДОВАНИЯ
Роль протеинкиназы c-Abl в опосредовании влияния p53 на активацию ERK каскада исследовалась на клеточной линии PC12 (Sigma-Aldrich). Данную клеточную линию получают из феохромацитомы крыс – опухоли, представленной хромаффинными клетками мозгового вещества надпочечников, они секретируют дофамин и широко используются как модель дофаминергических нейронов, а также широко применяются в исследованиях механизмов внутриклеточной сигнализации. Культуры клеток PC12 инкубировали в среде DMEM (#D5796, Sigma-Aldrich), с добавлением 10% лошадиной сыворотки (HS) (#H1138, Sigma-Aldrich) и 5% сыворотки плодов коровы (FBS) (#F9665, Sigma-Aldrich). Клетки содержались в инкубаторе с подачей 5% СО2 при температуре 37°С с 95%-ной влажностью. Клетки инкубировались на планшетах, покрытых коллагеном IV типа. Смена среды производилась каждые два дня. Исследуемые вещества добавляли в среду на 5-й день инкубации по достижению 70% конфлюентности культуры. Каждая серия экспериментов включала следующие экспериментальные группы:
1. Контроль – введение DMSO;
2. Введение селективного активатора р53 Nutlin-3 (20 мкМ; #3984/10, Tocris);
3. Сочетанное введение Nutlin-3 (20 мкМ) и селективного аллостерического ингибитора c-Abl GNF-5 (10 мкМ; #4908/10, Tocris);
4. Сочетанное введение Nutlin-3 (20 мкМ) и селективного ингибитора TrkA-рецепторов GW 441756 (50 мкМ; 2238/10, Tocris);
5. Сочетанное введение Nutlin-3 (20 мкМ), GNF-5 (10 мкМ) и GW 441756 (50 мкМ).
Известно, что клетки РС12 как в дифференцированном, так и в недифференцированном состоянии экспрессируют не только катехоламины, c-Abl, p53 и белки ERK1/2 MAPK каскада, но также TrkA-рецепторы фактора роста нервов NGF. При этом связывание TrkA-рецепторов с NGF приводит к быстрому фосфорилированию тирозинкиназного домена TrkA-рецептора и к последующей активации ERK1/2. При длительном введении NGF постоянная активация TrkA/ERK1/2 каскада вызывает остановку клеточного цикла и дифференцировку клеток РС12 [30]. Для активации TrkA-рецепторов в недифференцированных клетках NGF (100 нг/мл; #01-125, Millipore) добавляли в среду за 15 мин перед добавлением исследуемых веществ. Клетки инкубировали 3 ч и затем собирали в лизатном буфере для дальнейшего Вестерн-блот анализа. Было проведено 4 серии экспериментов с двойным повтором каждой экспериментальной группы.
Вестерн-блот анализ
Белки в пробах разделяли с помощью электрофореза в 10%-ном полиакриламидном геле по Лэммли (SDS-PAGE) и переносили на нитроцеллюлозную мембрану. Для Вестерн-блот анализа были использованы следующие антитела против: cRaf1 (1 : 1000; Cell Signaling, #9422), p-cRaf1 (Ser3381 : 1000; Cell Signaling, #2330), ERK1/2 (1 : 10 000; Cell Signaling, #9102), pERK1/2 (Thr202/Thr204; 1 : 10 000; Cell Si-gnaling, #4376); p53 (1 : 1000; Cell Signaling, #2524), p-p53 (Ser392; 1 : 500; Santa Cruz, #sc-51960), p-TrkA (Tyr496; 1 : 500, Santa Cruz, #sc-8058), TrkA (1 : 500, Santa Cruz, #sc-7268), p-c-Abl (Tyr393/412; 1 : 1000, Sigma, #SAB4504330) и Actin (1 : 2000, abcam, # ab3280). Визуализация результатов проводилась при помощи SuperSignal@West Dura Extended Duration Substrate (#34075, ThermoFisher Scientific).
После промывок в TBST-T мембраны помещали в раствор вторых антител против иммуноглобулинa G кролика, конъюгированные с биотином (1 : 3000, Sigma) или против иммуноглобулина G мыши, конъюгированные с биотином (1 : 3000, Sigma) в TBS-T. Затем мембраны инкубировали с раствором стрептавидин-пероксидазы (Sigma). Визуализация результатов проводилась при помощи ECL plus системы (Amersham Biosciences).
Денситометрический анализ количества белка осуществлялся в программе I-mageJ после сканирования пленок, полученных в результате как минимум трех экспериментов. Уровень серого специфических бендов был скорректирован по фоновому сигналу и нормирован по уровню GAPDH, используемого для определения общего количества белка в пробе. Также в анализе учитывалась площадь специфических бендов.
Статистическая обработка результатов
Обработка полученных данных производилась при помощи непараметрического теста ANOVA (критерий Краскела–Уоллиса) и U-критерия Манна–Уитни в коммерческой программе GraphPad Prism7. Данные представлены в виде медианы и интерквартильного размаха. Достоверными считались отличия при уровне значимости p < 0.05.
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
Анализ уровня активности р53
Полученные данные показали, что при введении Nutlin-3, как и ожидалось, наблюдалось достоверное увеличение уровня фосфорилирования р53 (рис. 1). В группах с сочетанным введением активатора р53 и ингибиторов c-Abl и TrkA также наблюдалось достоверное повышение уровня активности белка р53 относительно контроля (рис. 1). Мы полагаем, что активация р53 в группах с введением ингибитора TrkA-рецепторов связана с активацией NGF нейротрофинового рецептора р75NT, что, как известно, приводит к активации р53.
Рис. 1.
Анализ активности белка р53 в клетках линии РС12. Относительное содержание фосфорилированного р53 по Ser292 (р-р53) оценивалось методом Вестерн-блот анализа. Нормировка количества белка в пробе производилась по общей форме p53. По оси ординат – оптическая плотность, измеренная в условных единицах; по оси абсцисс – исследуемые группы: С – контроль, Nutlin – введение Nutlin-3 20 мкМ, Nutlin/GNF – сочетанное введение Nutlin 3 (20 мкМ) и GNF-5 (10 мкМ), Nutlin/GW – сочетанное введение Nutlin 3 (20 мкМ) и GW 441746 (50 мкМ), Nutlin/GNF/GW – сочетанное введение Nutlin 3 (20 мкМ), GNF-5 (10 мкМ) и GW 441746 (50 мкМ). Полученные данные представлены в виде медиан и интерквартильных размахов. * p < 0.05, ** p < 0.01, *** р < 0.001 по сравнению с контрольной группой. Fig. 1. Analysis of p53 activity in the PC12 cells p53 phosphorylation at Ser292 (p-p53) was estimated using the Western blot analysis and calculated as p-p53 to p53 ratio. Y axis – optical density in arbitrary units. X axis – experimental groups: С – control, Nutlin – treatment with Nutlin-3 (20 мкМ), Nutlin/GNF – treatment with Nutlin 3 (20 μМ) and GNF-5 (10 μМ), Nutlin/GW – treatment with Nutlin 3 (20 μМ) and GW 441746 (50 μМ), Nutlin/GNF/GW - treatment with Nutlin 3 (20 μМ), GNF-5 (10 μМ) and GW 441746 (50 μМ). Data are shown as box-plots. * p ˂ 0.05, ** p < 0.01, *** р < 0.001 versus control.

Активация р53 повышает активность c-Abl
При введении активатора р53 происходит значительное увеличение активности протеинкиназы с-Abl (рис. 2). При этом введение ингибитора c-Abl GNF-5 на фоне активации р53 не привело к изменению активности c-Abl (рис. 2). Также активность c-Abl не изменялась и при сочетанном введении Nutlin-3, GNF-5 и ингибитора TrkA-рецепторов GW 441756 (рис. 2). Однако введение и ингибитора TrkA-рецепторов GW 441756 и ингибитора c-Abl GNF-5 на фоне активации р53 привело к достоверному снижению активности c-Abl (рис. 2).
Рис. 2.
Оценка уровня фосфорилирования нерецепторной тирозинкиназы c-Abl. Содержание фосфорилированной киназы c-Abl по Tyr393/412 (p-c-Abl) в клетках РС12 было исследовано с помощью Вестерн-блот анализа. В качестве контроля количества белка в пробах использовался actin. По оси ординат – оптическая плотность, измеренная в условных единицах; по оси абсцисс – исследуемые группы: С – контроль, Nutlin – введение Nutlin-3 (20 мкМ), Nutlin/GNF – сочетанное введение Nutlin 3 (20 мкМ) и GNF-5 (10 мкМ), Nutlin/GW – сочетанное введение Nutlin 3 (20 мкМ) и GW 441746 (50 мкМ), Nutlin/GNF/GW – сочетанное введение Nutlin 3 (20 мкМ), GNF-5 (10 мкМ) и GW 441746 (50 мкМ). Данные представлены в виде медиан и интерквартильных размахов. *p < 0.05 по сравнению с контрольной группой. Fig. 2. Estimation of the non-receptor tyrosine kinase c-Abl phosphorylation c-Abl at Tyr393/412 (p-c-Abl) phosphorylation was estimated using the Western blot analysis and calculated as p-c-Abl to actin ratio. Y axis – optical density in arbitrary units. X axis – experimental groups: С – control, Nutlin – treatment with Nutlin-3 (20 мкМ), Nutlin/GNF – treatment with Nutlin 3 (20 μМ) and GNF-5 (10 μМ), Nutlin/GW – treatment with Nutlin 3 (20 μМ) and GW 441746 (50 μМ), Nutlin/GNF/GW – treatment with Nutlin 3 (20 μМ), GNF-5 (10 μМ) and GW 441746 (50 μМ). Data are shown as box-plots. * p < 0.05, versus control. # p <0.05, versus Nutlin.

р53 повышает активность TrkA-рецепторов и членов ERK1/2-зависимого сигнального каскада
Анализ уровня активности TrkA-рецепторов показал, что активация р53 введением Nutlin-3 значительно повышала уровень фосфорилирования TrkA (рис. 3А). Однако ингибирование c-Abl на фоне активации р53 не изменило активность Tr-kA-рецепторов по сравнению с контрольными значениямия, но была достоверно ниже, чем при введении Nutlin-3 (рис. 3А). При этом ингибирование TrkA как на фоне активации р53, так и при сочетанном введении Nutli-3 и GNF-5, как и ожидалось, привело к значительному снижению уровня фосфорилирования TrkA (рис. 3А).
Рис. 3.
Анализ активности TrkA-рецепторов и членов ERK1/2 сигнального каскада. Оценка содержания фосфорилированных форм рецепторов TrkA (p-TrkA по Tyr490) (A), c-Raf (p-c-Raf по Ser 338) (B) и ERK1/2-киназ (p-ERK1/2 по Thr202/Thr204) (C) в клетках РС12 проводилась с применением Вестерн-блот анализа. Для нормировки количества белка в пробе были использованы общие формы TrkA (А), c‑Raf (B) и ERK1/2 (C). По оси ординат – оптическая плотность, измеренная в условных единицах; по оси абсцисс – исследуемые группы: С – контроль, Nutlin – введение Nutlin-3 (20 мкМ), Nutlin/GNF – сочетанное введение Nutlin 3 (20 мкМ) и GNF-5 (10 мкМ), Nutlin/GW – сочетанное введение Nutlin 3 (20 мкМ) и GW 441746 (50 мкМ), Nutlin/GNF/GW – сочетанное введение Nutlin 3 (20 мкМ), GNF-5 (10 мкМ) и GW 441746 (50 мкМ). Данные представлены в виде медиан и интерквартильных размахов. * p < 0.05 по сравнению с контрольной группой. & p < 0.05 при сравнении уровня активности TrkA между группами с введением Nutlin-3 и группой с сочетанным введением Nutlin-3 и GW 441746 (Манн–Уитни U-тест). Fig. 3. Analysis of the TrkA receptor and members of ERK1/2 cascade activity Estimation of phosphorylated TrkA (p-TrkA Tyr490) (A), c-Raf (p-c-Raf Ser 338) (B) and ERK1/2 (p-ERK1/2 Thr202/Thr204) was performed using the Western blot analysis. TrkA, c-Raf and ERK1/2 phosphorylation was calculated as a ratio between phosphorylated and total studied proteins. Y axis – optical density in arbitrary units. X axis – experimental groups: С – control, Nutlin – treatment with Nutlin-3 (20 μМ), Nutlin/GNF – treatment with Nutlin 3 (20 μМ) and GNF-5 (10 μМ), Nutlin/GW – treatment with Nutlin 3 (20 μМ) and GW 441746 (50 μМ), Nutlin/GNF/GW – treatment with Nutlin 3 (20 μМ), GNF-5 (10 μМ) and GW 441746 (50 μМ). Data are shown as box-plots. * p < 0.05, ** p <0.01, *** р < 0.001 versus control. & p < 0.05 Nutlin-3 vs Nutlin-3/GW 441746 (by Mann-Whitney test).

Под влиянием повышения активированного p53 происходило увеличение фосфо-c-Raf. Также увеличение уровня активности cRaf наблюдалось в группе с сочетанным введением Nutlin-3 и GNF-5 (рис. 3B). В группах с введением GW 441756 наблюдалась тенденция к снижению уровня фосфорилирования сRaf относительно контрольной группы, однако это снижение было не достоверным (рис. 3B).
Далее мы проанализировали уровень активности ERK1/2. Результаты показали, что введение блокатора TrkA привело к достоверному снижению активных форм ERK1/2 (рис. 3C). При этом в группе с введением активатора р53, а также в группе с сочетанным введением Nutlin-3 и GNF-5 наблюдалось достоверное увеличение уровня активности ERK1/2 (рис. 3C).
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ ИССЛЕДОВАНИЙ
Полученные в ходе данного исследования данные показали, что влияние р53 на ERK1/2 каскад во многом зависит не только от активности р53, но и от уровня активности нейротрофиновых рецепторов и нерецепторной протеинкиназы c-Abl. Наши данные показали, что на фоне повышения активности р53 также происходит повышение активности c-Abl. Одним из объяснений данного влияния является прямое белок-белковое взаимодействие р53 и c-Abl, которое приводит к переходу киназного домена c-Abl в открытую активную конформацию [6]. С другой стороны, мы показали, что ингибирование активности c-Abl приводит к де-фосфорилированию р53. Однако данных литературы, свидетельствующих о c-Abl-зависимом фосфорилировании р53 мы не обнаружили. Было показано, что c-Abl повышает транскрипционную активность р53 [5, 6]. Также известно, что c-Abl фосфорилирует белок р75, относящийся к семейству белков р53 [5], а формирование комплекса р53/c-Abl стабилизирует активность р53 [6]. На основании полученных нами данных о снижении уровня фосфорилирования р53 при введении ингибитора с-Abl и данных литературы о c-Abl-зависимом фосфорилировании р75 можно предположить, что протеинкиназа c-Abl не только стабилизирует белок р53, но и принимает участие в его фосфорилировании. Таким образом, мы показали, что связь между p53 и c-Abl двухсторонняя: не только c-Abl регулирует активность р53, но и от уровня активности р53 может зависеть активность c-Abl.
Известно, что дифференцировка клеток линии PC12 по нейрональному типу происходит за счет активации ERK1/2 зависимого каскада при введении NGF [30], что также сопровождается активацией белка р53 [24]. С другой стороны, в цитоплазме белковый комплекс р53/c-Abl связывается с тирозинкиназным доменом Trk-рецепторов, приводя к активации ERK1/2 каскада [6, 8]. Кроме того, было показано, что эффект p53 на ERK1/2 каскад может быть опосредован взаимодействиями только одного белка p53 с рецептором TrkA [31, 32]. Однако имеются данные, свидетельствующие о позитивном влиянии р53 на активность ERK1/2 без участия нейротрофиновой стимуляции Trk-рецепторов [20, 23]. При этом авторы показали, что р53 активирует ERK-зависимый MAPK каскад на уровне или выше cRaf-киназы. Мы показали, что при введении ингибитора c-Abl на фоне активации р53 в группах с введением NGF наблюдается снижение уровня фосфорилирования TrkA-рецепторов по сравнению с введением только Nutlin-3. Этот факт позволяет предположить, что протеинкиназа c-Abl действительно способствует активации TrkA-рецепторов при участии р53. С другой стороны, активация c-Raf и ERK1/2 при сочетанном введении Nutlin-3 и GNF-5 свидетельствует о том, что при активации TrkA-рецепторов р53 может оказывать влияние на активность ERK1/2-сигнального каскада на уровне или выше c-Raf и независимо от c-Abl.
Важно отметить, что р53, c-Abl и ERK1/2 вовлечены в развитие нейродегенеративных болезней, в частности, болезни Хантингтона, Альцгеймера и Паркинсона [12, 18, 33, 34]. На сегодняшний день в терапии в основном раковых заболеваний уже применяются или разрабатываются препараты, основанные на точечном влиянии на р53, c-Abl и ERK1/2 [13, 35–38]. Однако действие применяемых препаратов ограничено и в ряде случаев малоэффективно, а в большинстве случаев также развивается толерантность к применяемым препаратам. Логично предположить, что малая эффективность препаратов, направленных только на одну мишень, связана с изменением активности как минимум одного или обоих составляющих данного комплекса.
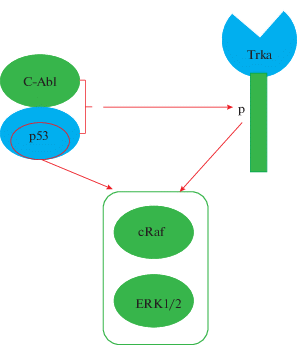
Таким образом, наши данные свидетельствуют о том, что р53 оказывает активирующее влияние на ERK1/2 каскад как опосредованно, в составе комплекса р53/c-Ab-l и активируя Trk-рецепторы, так и независимо от c-Abl, влияя на ERK1/2 каскад на уровне или выше cRaf (рис. 4 ). Также на основании полученных нами данных можно предположить, что применение различных сочетаний блокаторов и/или активаторов р53, c-Abl и членов ERK1/2 каскада может стать основой для разработки комплексной терапии для лечения нейродегенеративных состояний.
Рис. 4.
Предполагаемая схема p53 зависимой регуляции активности ERK1/2 сигнального каскада. Fig. 4. Hypothetical scheme of p53-dependent regulation of the ERK1/2 signaling.
Список литературы
Ko L.J., Prives C. p53: puzzle and paradigm. Genes Dev. 10(9): 1054–1072. 1996.
Sola S., Aranha M.M., Rodrigues C.M. Driving apoptosis-relevant proteins toward neural differentiation. Mol. Neurobiol. 46(2): 316–331. 2012.
Vousden K.H., Prives C. Blinded by the Light: The Growing Complexity of p53. Cell. 137(3): 413–431. 2009.
Jing Y., Wang M., Tang W., Qi T., Gu C., Hao S., Zeng X. c-Abl tyrosine kinase activates p21 transcription via interaction with p53. J. Biochem. 141(5): 621–626. 2007.
Kharbanda S., Yuan Z.M., Weichselbaum R., Kufe D. Determination of cell fate by c-Abl activation in the response to DNA damage. Oncogene. 17(25): 3309–3318. 1998.
Levav-Cohen Y., Goldberg Z., Zuckerman V., Grossman T., Haupt S., Haupt Y. C-Abl as a modulator of p53. Biochem. Biophys. Res. Commun. 331(3): 737–749. 2005.
Nie Y., Li H.H., Bula C.M., Liu X. Stimulation of p53 DNA binding by c-Abl requires the p53 C terminus and tetramerization. Mol. Cell Biol. 20(3): 741–748. 2000.
Brown A., Browes C., Mitchell M., Montano X. c-abl is involved in the association of p53 and trk A. Oncogene. 19(26): 3032–3040. 2000.
Tsygankov A.Y. Non-receptor protein tyrosine kinases. Front. Biosci. 8: s595–635. 2003.
Raitano A.B., Whang Y.E., Sawyers C.L. Signal transduction by wild-type and leukemogenic Abl proteins. Biochim. Biophys. Acta. 1333(3): F201–F216. 1997.
Wang J.Y. The capable ABL: what is its biological function? Mol. Cell Biol. 34(7): 1188–1197. 2014.
Ko H.S., Lee Y., Shin J.H., Karuppagounder S.S., Gadad B.S., Koleske A.J., Pletnikova O., Troncoso J.C., Dawson V.L., Dawson T.M. Phosphorylation by the c-Abl protein tyrosine kinase inhibits parkin’s ubiquitination and protective function. Proc. Natl. Acad. Sci. USA. 107(38): 16691–16696. 2010.
Abushouk A.I., Negida A., Elshenawy R.A., Zein H., Hammad A.M., Menshawy A., Mohamed W.M.Y. C-Abl Inhibition; A Novel Therapeutic Target for Parkinson’s Disease. CNS Neurol. Disord. Drug Targets. 17(1): 14–21. 2018.
Zhou H., Yamamura Y., Ogawa M., Tsuji R., Tsuchiya K., Kasahara J., Goto S. c-Abl Inhibition Exerts Symptomatic Antiparkinsonian Effects Through a Striatal Postsynaptic Mechanism. Front. Pharmacol. 9: 1311. doi: eCollection 2018https://doi.org/10.3389/fphar.2018.01311
Long J., Liao G., Yinna Wang Y., Tang D.D. Specific protein 1, c-Abl and ERK1/2 form a regulatory loop. J. Cell Science. 132. doi: 2019https://doi.org/10.1242/jcs.222380
Dzamko N., Zhou J., Huang Y., Halliday G.M. Parkinson’s disease-implicated kinases in the brain; insights into disease pathogenesis. Front. Mol. Neurosci. 7, article 57. https://doi.org/10.3389/fnmol.2014.00057
Bohush A., Niewiadomska G., Filipek A. Role of Mitogen Activated Protein Kinase Signaling in Parkinson’s Disease. Int. J. Mol. Sci. 19, 2973; doi: 2018https://doi.org/10.3390/ijms19102973
Szybińska A., Leśniak W. P53 Dysfunction in Neurodegenerative Diseases – The Cause or Effect of Pathological Changes? Aging and Disease. 8(4): 506–518. 2017.
Gulati A.P., Yang Y.M., Harter D., Mukhopadhyay A., Aggarwal B.B., Benzil D.L., Whysner J., Albino A.P., Murali R., Jhanwar-Uniyal M. Mutant human tumor suppressor p53 modulates the activation of mitogen-activated protein kinase and nuclear factor-kappaB, but not c-Jun N-terminal kinase and activated protein-1. Mol. Carcinog. 45(1): 26–37. 2006.
Lee S.W., Fang L., Igarashi M., Ouchi T., Lu K.P., Aaronson S.A. Sustained activation of Ras/Raf/mitogen-activated protein kinase cascade by the tumor suppressor p53. Proc. Natl. Acad. Sci. USA. 97(15): 8302–8305. 2000.
Sablina A.A., Chumakov P.M., Levine A.J., Kopnin B.P. p53 activation in response to microtubule disruption is mediated by integrin-Erk signaling. Oncogene. 20(8): 899–909. 2001.
Singh S., Upadhyay A.K., Ajay A.K., Bhat M.K. p53 regulates ERK activation in carboplatin induced apoptosis in cervical carcinoma: a novel target of p53 in apoptosis. FEBS Lett. 581(2): 289–295. 2007.
Zosen D.V., Glazova M.V. The role of the interaction of p53 and the MAPK cascade in controlling neuronal differentiation in the PC12 cell line. Neurosci. Behav. Physiol. 46(5): 559–565. 2016
Zhang J., Yan W., Chen X. p53 is required for nerve growth factor-mediated differentiation of PC12 cells via regulation of TrkA levels. Cell Death Differ. 13(12): 2118–2128. 2006.
Wu G.S. The functional interactions between the p53 and MAPK signaling pathways. Cancer Biol. Ther. 3(2): 156–161. 2004.
Dorofeeva N.A., Chernigovskaya E.V., Nikitina L.S., Glazova M.V. Effect of p53 inhibition by pifithrin-alpha on functional activity of vasopressin neurones in rat hypothalamus. Рос. физиол. журн. им. И.М. Сеченова. 99(8): 901–916. 2013.
Nikitina L.S., Dorofeeva N.A., Kirillova O.D., Korotkov A.A., Glazova M., Chernigovskaya E.V. Role of the ERK signaling pathway in regulating vasopressin secretion in dehydrated rats. Biotech. Histochem. 89(3): 199–208. 2014.
Cilloni D., Saglio G. Molecular pathways: BCR-ABL. Clin. Cancer Res. 18(4): 930–937. 2012.
Jain A., Tripathi R., Turpin C.P., Wang C., Plattner R. Abl kinase regulation by BRAF/ERK and cooperation with Akt in melanoma. Oncogene. 36(32): 4585–4596. 2017.
Vaudry D., Stork P.J., Lazarovici P., Eiden L.E. Signaling pathways for PC12 cell differentiation: making the right connections. Science. 296(5573): 1648–1649. 2002.
Montano X. P53 associates with trk tyrosine kinase. Oncogene. 15(3): 245–256. 1997.
Browes C., Rowe J., Brown A., Montano X. Analysis of trk A and p53 association. FEBS Lett. 497(1): 20–25. 2001.
Bowles K.R., Jones L. Kinase signalling in Huntington’s disease. J. Huntingtons Dis. 3, 89–123. 2014.
Schapira A.H., Olanow C.W., Greenamyre J.T., Bezard E. Slowing of neurodegeneration in Parkinson’s disease and Huntington’s disease: future therapeutic perspectives. Lancet. 384. 545–555. 2014.
Burgess A., Chia K.M., Haupt S., Thomas D., Haupt Y., Lim E. Clinical Overview of MDM2/X-Targeted Therapies. Front Oncol. 2016. https://doi.org/10.3389/fonc.2016.00007
Liu F., Yang X., Geng M., Huang M. Targeting ERK, an Achilles’ Heel of the MAPK pathway, in cancer therapy. Acta Pharm. Sin. B. 8: 552–562. 2018.
Rossari F., Minutolo F., Orciuolo E. Past, present, and future of Bcr-Abl inhibitors: from chemical development to clinical efficacy. J. Hematol. Oncol. 11(1): 84. 2018.
Khorasanizadeh, M., Eskian, M., Gelfand, E.W., Rezaei, N. Mitogen-activated protein kinases as therapeutic targets for asthma. Pharmacol. Ther. 174: 112–126. 2017.
Дополнительные материалы отсутствуют.
Инструменты
Российский физиологический журнал им. И.М. Сеченова