

Российский физиологический журнал им. И.М. Сеченова, 2020, T. 106, № 5, стр. 539-562
Lipid Rafts and Amyloid Metabolism: Role in Pathogenesis of Alzheimer’s Disease
N. N. Nalivaeva 1, 2, *, A. J. Turner 2
1 Sechenov Institute of Evolutionary Physiology and Biochemistry RAS
St. Petersburg, Russia
2 School of Biomedical Sciences, Faculty of Biological Sciences, University of Leeds
Leeds, UK
* E-mail: n.n.nalivaeva@leeds.ac.uk
Поступила в редакцию 11.03.2020
После доработки 15.03.2020
Принята к публикации 17.03.2020
Аннотация
Brain lipids play an important role not only as ubiquitous structural membrane components providing the scaffolding and compartmentalisation outside and within the cells but also participating in various signalling processes either by facilitating them or by acting as signal molecules. Membrane lipids form highly specialised domains, called lipid rafts, which are more ordered structures than the rest of the membrane and are enriched in cholesterol and sphingolipids. These domains provide a platform for specific and targeted protein-lipid and protein-protein interactions and as such facilitate binding and/or enzymatic processes on the surface and within the membranes. These lipid-protein interactions are important for various signalling events and proper cell functioning. When normal structure and functions of lipid rafts is disturbed due to the changes in lipid metabolism, caused by various internal and environmental factors, it results in a cascade of pathological events. Among proteins whose metabolic pathways depend on the lipid raft structure and integrity is amyloid precursor protein (APP) – the protein highly implicated in the pathogenesis of Alzheimer’s disease (AD). Proteolytic processing of APP by a aspartic proteinase called β-secretase (BACE1) and a multiprotein complex called γ‑secretase results in production of the amyloid β peptide (Aβ) - one of the key molecules leading to development of AD. These events take place in the lipid rafts. Some lipid components of the rafts, including ganglioside GM1, facilitate Aβ aggregation and formation of its toxic oligomers. Understanding the mechanisms regulating lipid-protein interactions in the rafts might result in new therapeutic strategies and treatments. In this review we discuss the implications of lipids in APP processing and Aβ metabolism and possible therapeutic avenues derived from studying lipid raft structure and functions in normal and AD brain.
The amyloid cascade hypothesis of Alzheimer’s disease (AD) [1] has, since its formulation, shaped the course of research into the molecular mechanisms of the disease during almost three decades accumulating a substantial amount of data which, unfortunately, has failed to result in the design of any effective treatment. Although the hypothesis has proven itself correct based on the genetic early-onset forms of the disease [2], the late-onset forms of AD appears to be multifaceted and more difficult to tackle [3].
From the clinical point of view, the disease is characterised by global cognitive decline, associated with brain pathology involving accumulation of extracellular amyloid aggregates (also known as senile plaques) of amyloid β peptide (Aβ) and intracellular neurofibrillary tangles of hyper-phosphorylated tau protein (for review see [4]). According to the amyloid cascade hypothesis, it is the Aβ which is principally responsible for many of the pathological features of the disease [1, 5] with Aβ oligomers representing the most toxic species [6, 7]. A-ccumulation of amyloid plaques is accompanied by astrogliosis and microgliosis [8, 9] and the most affected brain areas are the neocortex and hippocampus [10]. Although there are strong genetic links, including amyloid precursor protein (APP) and presenilin mutations in familiar early-onset form on AD [11], the majority of AD cases are sporadic, late-onset forms [12]. From this point of view the predominance of AD research based on the mechanisms of early-onset disease versus the broader spectrum of the factors leading to the sporadic form might be one of the reasons for the failure of the majority of therapeutic trials and lack of any preventive measures 20 years since the amyloid hypothesis has been proposed.
One of the important cellular events underlying production of the Aβ peptide is proteolytic processing of APP initiated by its cleavage by β-secretase (BACE1) (for the most recent review see [13]) with the subsequent cleavage of the formed membrane-bound C-terminal APP fragment by a protein complex called γ-secretase [14]. This process, called the amyloidogenic APP pathway, is believed to take place in a specific membrane compartment – the lipid rafts [15, 16]. Only about 10% of all APP is cleaved by this pathway and the major part of APP molecules is processed in the non-raft compartments of cellular membranes starting with APP cleavage by a proteinase from the ADAM family called α-secretase [17]. To understand how APP is targeted to the lipid rafts for its amyloidogenic processing and how lipid composition of the rafts can affect Aβ production is very important for understanding not only the physiological significance of the whole cellular machinery of amyloidogenic APP processing in neuronal cells but also how and why it leads to pathogenic events underlying development of AD.
This review is mainly focused on the links between lipid rafts and AD pathology not only because AD-related amyloid precursor protein and production of Aβ peptide depends on the lipid rafts but also because Aβ signalling itself involves interactions of proteins resident to lipid rafts. Since we published an extensive review on this topic in 2012 [18] there have been important developments in this area of research which we shall cover in this paper. One of the aspects of the studies is related to modulation of lipid rafts with the aim to halt amyloidogenic processing of APP and Aβ production which still failed to translate into any successful drug [19]. While statin treatment is still considered beneficial [20]. there are some meta-analysis studies raising concerns on their effectiveness [21]. In assessing the literature data it is important to bear in mind that the pathology and aetiology of AD is complex and the involvement of lipids in its pathogenesis has to be analysed in a more generic way, taking into account various aspects of lipid metabolism and its changes with ageing and under pathological conditions predisposing to AD.
LIPID RAFT COMPOSITION AND PROPERTIES
With more than 20 years since the concept of the proteolytic shedding of integral membrane proteins such as APP [22] and the establishment of the “lipid raft” concept by Simons and Ikonen [23], research of these membrane-clustering compartments has attracted significant amount of efforts as extensively reviewed in [18, 24–26]. Despite the initial controversy regarding lipid raft size and precise protein and lipid composition due to the methodologies used for their isolation and analysis, it is now clear that cholesterol, sphingolipids and glycolipids represent the central components defining the structural and functional properties of the lipid rafts [25]. The lipid raft model postulates that lipids and proteins in the cellular membranes are segregated in different phases: liquid ordered (Lo) and disordered (Ld) phases. While cholesterol and sphingolipids are promoting Lo, non-saturated phospholipids maintain the Ld phases. This allows certain proteins to cluster together in the Lo phase to fulfil their functions [27].
Caveolae were the first described type of organised cholesterol-enriched microdomains which contained oligomerized caveolin protein whose composition was determined in the early 1990’s [28]. However, apart from cholesterol being the major determinant of the caveolae integrity, the role of other lipids in these domains is still unknown. While the concept of lipids rafts has been developed from lipid-enriched microdomains [29] and detergent-resistant membranes (DRMs) [30] to the currently established cholesterol-sphingomyelin rafts with the Lo phase and the Ld non-raft regions of cellular membranes, a third type of microdomains termed ceramide-rich platforms (CRPs) with a gel-like structure has also been identified [31]. It is also important to mention other cholesterol and ganglioside enriched membrane domains, which share some similarity but are physically and functionally distinct from the lipid rafts, the tetraspanin-enriched microdomains (TEMs). They represent functional platforms for the regulation of key cellular processes such as release of surface protein ectodomains (“shedding”) or regulated intramembrane proteolysis (“RIPping”) [32] and are involved in a whole range of cellular processes, from cell division and motility to gene regulation and signalling pathways [33], as well as dendritic spine maturation [34].
In relation to AD, another intracellular lipid raft-like structure involved in cholesterol and phospholipid lipid metabolism, calcium homeostasis and mitochondrial function, so-called mitochondria-associated ER membranes (MAM), are of particular significance [35]. They contain PS1 and PS2 with γ-secretase activity and process APP generating Aβ [36]. MAM and their communication with the mitochondria are important for phospholipid metabolism and transport which are altered in AD [37].
One of the important roles of lipid rafts is synaptic vesicle release [38] although more recently they have been shown to participate in intracellular lipid and protein trafficking from the endoplasmic reticulum, Golgi bodies and endosomes to the plasma membrane and production of extracellular vesicles (EVs) [39]. These nanovesicles (30–150 nm) contain proteins, RNAs and lipids, and their internalization by neighbouring cells can change their normal functions [40]. Lipid rafts may also represent the platforms for inclusion/exclusion of membrane lipids and proteins into microvesicles (MVs) which could originate from distinct domains during physiological processes and disease evolution [41].
Among the best characterised raft proteins are lipid-modified proteins, such as GPI-anchored proteins [42, 43], doubly acylated proteins, such as Src family kinases and the α subunits of heterotrimeric G proteins [44]. The 31 kDa integral membrane protein stomatin and GPI-anchored proteins were shown to be linked through lipid rafts and undergo the same cell-sorting events [45]. Another lipid raft marker, flotillin, can function as a molecular link between lipid rafts and a multimeric signaling complex at the actin cytoskeleton [46]. The flotillins are palmitoylated and myristoylated proteins, anchored to the plasma membrane [47]. They belong to a larger class of integral membrane proteins that have an evolutionarily conserved domain called the prohibitin homology domain (PHB) which determines the affinity of proteins to the lipid rafts [48].
Many other physiologically important proteins have been investigated for their possible raft localisations, including key proteins involved in AD pathogenesis, such as APP [49] and its processing enzymes: β-site APP cleaving enzyme (BACE1) [15, 50], the γ-secretase complex [51, 52] and ADAM10 – a disentegrin and metalloprotease possessing α-secretase activity [53]. Another member of the ADAMs family – the tumor necrosis factor-alpha (TNF) converting enzyme (TACE, ADAM17) responsible for the cleavage of several biologically active transmembrane proteins, including α-secretase cleavage of APP, was also shown to be targeted to lipid rafts [54]. The prion protein (PrPc) conversion to its infectious form (PrPSc) takes place in the lipid rafts [55].
Lipid rafts serve as a signaling platform for nicotinic acetylcholine receptors [56] and functioning of α7 nicotinic acetylcholine receptor depends on the lipid composition of the plasma membrane [57]. Moreover, it was shown that soluble form of Aβ peptide potentiates the activity of the presynaptic α7 nicotinic acetylcholine receptor and this event depends on the lipid raft integrity [58]. It has also been demonstrated that acetylcholinesterase (AChE) is targeted to lipid rafts [59] and in neurones this involved the proline rich membrane anchor (PRiMA) [60].
Lipid rafts play a critical role in viral infections facilitating virus entry, replication, assembly and budding [61]. Raft components are involved in cholera toxin entry via GM1 ganglioside [62]. The human immunodeficiency virus type 1 (HIV-1) entry into the cells also requires GM1 and gp120 protein localised in the lipid rafts [63]. The entry of severe acute respiratory syndrome coronavirus (SARS-CoV) to the cells via its receptor angiotensin-converting enzyme-2 (ACE2) also depends on the integrity of lipid rafts in the infected cell membrane and is disrupted by methyl-β-cyclodextrin [64]. It is likely that the newly described coronavirus (SARS-CoV-2) causing the serious epidemic COVID-19, which also uses ACE2 as its receptor, also requires lipid rafts for cell entry [65].
The mature form of the major Aβ-degrading enzyme, neprilysin (NEP) was also found to be localised in lipid rafts [66, 67]. Although the list of lipid raft-associated proteins is fast growing, it is still far from being completed, and future research will undoubtedly reveal new members of this family.
In general, lipid rafts represent signalling platforms that bring together various components of the biological membranes determining cell specificity and functions. They include receptors, channels, recognition molecules, coupling factors and enzymes, facilitating their interaction and supporting cell signalling. Lipid rafts have been implicated in a variety of physiological and pathological processes. Among the beneficial processes in the nervous system it is important to name axonal growth and branching [68], which underlie neuronal plasticity. As demonstrated in cell models, raft disruption impedes axonogenesis [69]. Lipid rafts are also involved in the stabilisation and normal functioning of synapses [70]. Disruption of these important processes are observed in AD pathogenesis and normalization of brain lipid homeostasis with formation of functional lipid raft domains might represent a therapeutic avenue in AD prevention.
AMYLOID PRECURSOR PROTEIN PROCESSING
APP is a type I integral membrane protein which exists in three isoforms (APP695, APP751 and APP770), generated from differential splicing of exons 7 and 8 of the APP gene (for review see [71]). Exon 7 is homologous to the Kunitz type protease inhibitors (KPI domain) [72], while exon 8 is related to the thymocyte MRC OX-2 antigen [73]. APP695 lacks both KPI and OX-2 domains, while APP751 only lacks the OX-2 domain. In terms of distribution, APP mRNA is expressed in almost every tissue, where only the isoform ratio differs. The ratio of different APP isoform mRNAs in human cortex is approx APP770/APP751/APP695 = 1 : 10 : 20, although there are regional differences [74] which alter in the AD brains [75, 76]. In the neurones the APP695 isoform predominates [77].
The amyloidogenic APP processing (Fig. 1) which results in production of Aβ peptides of different length involves sequential cleavage of APP by β- and γ-secretases (for review see [78]). APP cleavage by β-secretase at two alternative sites (at Asp1 or at Glu11 of the Aβ sequence) leads to formation of either a 99 amino acid C-terminal fragment (CTF, C99) or a 89 amino acid CTF (C89) which can further be processed by γ-secretase. It also releases a large soluble ectodomain, sAPPβ, which possesses important physiological properties [79]. The predominant, non-amyloidogenic, pathway regulated by α-secretase cleavage within the Aβ domain of the APP molecule prevents formation of Aβ but produces the large, soluble ectodomain sAPPα possessing neuroprotective properties [79, 80] and a 83 amino acid CTF (C83) which in turn undergoes proteolytic cleavage within the plasma membrane by the γ-secretase complex. Cleavage of CTFs by γ-secretase releases the APP C-terminal domain, AICD, and either Aβ (in the amyloidogenic pathway) or a shorter 3 kDa p3 fragment (in the non-amyloidogenic pathway). These p3 peptides are non-amyloidogenic but were found within diffuse plaques in AD brains [81]. Whether p3 has any functional role is still unclear. However, it was demonstrated that γ-secretase produces Aβ at a higher ratio than p3 peptides suggesting that β-CTFs and α-CTFs are differentially processed by γ-secretase [82]. APP can also undergo ε-cleavage producing a large N-terminal fragment which behaves as a type I protein and undergoes α-, β- and γ-secretase cleavages and forms the APP intracellular domain C50 [83]. This ε-cleavage resembles the S3 Notch cleavage generating Notch intracellular domain NICD which has important gene regulatory functions [84]. Lipid raft composition plays an important role in regulating APP processing by all secretases, including the ε-cleavage [85].
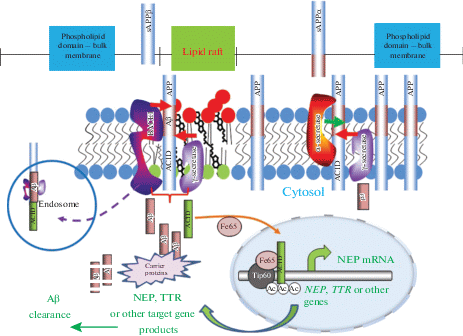
Fig. 1.
Schematic representation of APP processing in the cell membrane and lipid rafts. The proteolytic processing of APP occurs in two distinct membrane domains. The non-amyloidogenic pathway initiated by α-secretase within the Aβ sequence of APP takes place in the phospholipid domain (light blue) while the amyloidogenic pathway (the Aβ region in APP is shown in maroon) takes place in lipid rafts which are enriched in glycosphingolipids, sphingomyelin (red) and cholesterol (black). The lipid raft is shown as part of the plasma membrane. While α-secretase is not raft-associated, the β- and γ- secretases predominate in rafts. The cell surface is shown for clarity, although β-cleavage predominantly occurs in endosomes. In the amyloidogenic pathway cleavage of APP by β-secretase releases a soluble neuroprotective ectodomain (sAPPβ) and the C-terminal fragment CTF99. This, in turn, is cleaved by the γ-secretase complex, generating the transcriptional regulator APP intracellular domain (AICD), and the Aβ peptide. In the non-amyloidogenic pathway α-secretase cleavage produces a soluble ectodomain sAPPα and the C-terminal fragment CTF83. Proteolytic cleavage of CTF83 by γ‑secretase releases AICD and the p3 fragment whose functions are still unknown. The AICD fragment produced in the amyloidogenic pathway can act as a transcription factor regulating expression of a variety of genes, including the Aβ-degrading enzyme neprilysin (NEP) and a transport protein, transthyretin (TTR), which are involved in Aβ clearance and are neuroprotective. AICD produced in the non-amyloidogenic pathways is likely to be enzymatically cleared in the cytoplasm.
APP LOCALISATION IN THE RAFTS
The data suggesting that APP processing is linked to the lipid rafts have been available for many years and were previously reviewed [86, 87]. Although APP itself is not generally a raft protein, a small proportion can be detected in lipid rafts [49]. Regulation of APP raft localisation has been suggested to involve an interaction between the C-terminus of APP and flotillin-1 [88] and flotillin-2 [47] although APP can also directly bind cholesterol in the rafts [89]. It has even been proposed that one of the physiological functions of APP and Aβ is to control cholesterol transport [90]. There are data suggesting that the adaptor protein Disabled1 (Dab1) regulates APP processing together with the glycoprotein reelin which promotes interaction of APP and Dab1 in lipid rafts and that this process involves phosphorylation by Fyn kinase [91]. On the contrary, binding of the F-box and leucine rich repeat protein2 (FBL2) – a component of the E3 ubiquitin ligase complex which regulates APP metabolism through APP ubiquitination, specifically to the C-terminal fragment of APP, decreased APP protein localization in lipid rafts [92].
APP trafficking to the lipid rafts can also be regulated by the low-density lipoprotein receptor-related protein (LRP) which promotes the BACE1-APP interaction [93]. Another member of the low-density lipoprotein receptor family (LDL-R), namely ApoR2, stimulates Aβ production by shifting the proportion of APP from the non-rafts to the raft membrane domains, promoting its amyloidogenic processing [94]. It was also reported that 8–10% of APP can be palmitoylated at Cys186 and Cys187 in the E1-ectodomain which increases its preference for β-cleavage in lipid rafts [19]. Moreover, APP palmitoylation promotes its dimerization resulting in elevated BACE1-mediated cleavage [95].
Several cytoskeletal components and their binding partners together with the enzymes that regulate the cytoskeleton are localized to the lipid rafts promoting lateral diffusion of membrane lipids and proteins, including APP, in response to various extracellular signals (receptor activation, electrical conductance, oxygen and nutrient deprivation etc.) (for review see [96]).
LOCALISATION OF APP SECRETASES IN THE LIPID RAFTS
Taking into account the existence of two different pathways of APP processing (amyloidogenic and non-amyloidogenic) it was logical to suggest that APP can have two distinct pools in plasma membranes and that amyloidogenic processing might take place in the lipid rafts [15, 89]. Indeed, it was shown that the integrity and composition of lipid rafts, especially the presence of gangliosides, are crucial for Aβ production, aggregation and oligomerization [97–99]. Furthermore, the precise protein/lipid composition of the rafts was shown to influence the Aβ40/ Aβ42 ratio [100].
With regard to the enzymes processing APP via the amyloidogenic pathway it was shown that BACE1 can be palmitoylated at four C-terminal cysteine residues, which facilitates its raft localisation [101, 102]. On the contrary, treatment of cells with the BACE1 inhibitor KMI-574 resulted in relocation of BACE1 from lipid rafts to the non-raft membrane area. Moreover, proteolytically inactive mutants of BACE1 defective in glycosylation were also shifted from the lipid rafts to the non-raft areas, while a double mutant (D93A/D289A) localized exclusively in the non-raft membranes [103]. When BACE1 was specifically targeted to lipid rafts via GPI-anchoring the production of Aβ was increased which testified to enhanced amyloidogenic APP processing [16]. Later it was confirmed that wild type BACE1 cleaves APP at two sites (β and β` sites) producing full length and N-terminally truncated Aβ whereas GPI-anchorage of the enzyme increases production of the full length Aβ [104].
It is important to mention that one of the lipid-raft associated proteins, namely the GPI-anchored prion protein (PrPc) plays an important role in APP processing via inhibiting BACE1 activity [105]. Interaction of the N-terminal region of PrPC with glycosaminoglycans in the rafts results in decreased BACE1 presence at the cell surface and in endosomes where it preferentially cleaves wild type APP but increased in the Golgi where it preferentially cleaves APP with the Swedish mutation (APPSwe) responsible for the early-onset AD [105]. Because there was no effect of PrPc deficit on processing of APPSwe it was suggested that PrPc may be a key protective player against sporadic AD [106].
Apart from β-secretase, lipid rafts also harbour the main subunits of the γ-secretase complex, namely PS1-derived fragments, mature nicastrin, APH-1 and PEN-2 [51, 107]. S-palmitoylation of nicastrin and APH-1 plays an important role in localization and stability of the γ-secretase complex within the rafts although not modulating its activity [108]. On the contrary, caveolin-1 was shown to be an important regulator of γ-secretase distribution and activity [109]. However, γ-secretase activity and Aβ generation in the membranes to a great extent depends on the cholesterol levels in rafts since treating cells with AY9944, which blocks the last step of cholesterol biosynthesis, reduced γ-secretase activity and Aβ generation [110]. Interestingly, PS1 was shown to induce lipid raft formation since brain membranes from mice expressing human wild-type PS1 were less fluid and contained higher cholesterol and sphingomyelin levels [111]. Genome-wide functional analysis has identified a novel modulator of γ-secretase, named OCIAD2 (ovarian cancer immunoreactive antigen domain containing 2), that stimulates Aβ production via facilitating the formation of an active γ-secretase complex and enhancing its localization to lipid rafts [112]. Although the functional role of such regulatory molecules in APP homeostasis is as yet unknown, it clearly demonstrates that cells might possess a variety of mechanisms affecting γ-secretase activity in relation to lipid rafts.
Unlike β- and γ-secretases which are clearly linked to lipid rafts, the enzymes possessing α-secretase activity, mainly ADAM10 and ADAM17, are believed to be located in non-raft membrane compartments where they participate in shedding of various membrane proteins [17]. However, targeting ADAM10 to lipid rafts by expressing its GPI-anchored variant was found to reduce APP processing by β-secretases clearly indicating that the two opposing enzymes might compete for the APP molecules within the rafts [53]. On the other hand, ADAM10 is regulated by tetraspanins. In particular, TSPAN12 is associated with ADAM10 but not with ADAM17 and overexpression of TSPAN12 in SH-SY5Y cells was shown to enhance non-amyloidogenic processing of APP [113]. More recently it was confirmed that ADAM10 interacts directly with all members of the TspanC8 subgroup of tetraspanins which all regulate ADAM10 exit from the endoplasmic reticulum, but differentially regulate its subsequent trafficking and activity [114]. However, tetraspanins also regulate γ‑secretase activity [115] and as such the fate of APP metabolism might depend on a very intricate balance in the localisation of the enzymes and substrate in the cellular membranes.
APP processing in the lipid rafts not only facilitates interactions between APP and BACE1 but also promotes APP endocytosis. This process is APP isoform-dependent and the neuronal APP695 isoform is mostly processed via the β-secretase pathway while APP751 and APP770 mainly undergo α-secretase cleavage [116]. There is an alternative trafficking route by which APP can bypass endosomes and be transported directly to the lysosomes. This, though, does not occur with either APPSwe or APPLondon variants suggesting that these mutations increase the chance of the molecules being processed in the lipid rafts [117]. Regulation of APP intracellular trafficking also depends on its interaction with sortilin both at the N- and C-terminal regions. In sortilin knockout mice APP has decreased lysosomal distribution and is increased in lipid rafts [118]. Interestingly, defects in phosphatidylinositol-3-phosphate (PI3P) synthesis due to disruption of its synthesizing enzyme PI 3-kinase Vps34 impairs not only autophagy, lysosomal degradation and lipid metabolism but also results in secretion of unique exosomes enriched with APP-CTFs, specific sphingolipids and phospholipid bis(monoacylglycero)phosphate, which normally reside in endolysosomes [119]. Moreover, it was shown that in AD-mice purified exosomes were enriched in APP-CTFs as compared to control brains. Furthermore, γ-secretase inhibition resulted in APP-CTF oligomerization by preventing C99 proteolysis and led to accumulation of exosomes containing oligomeric APP-CTFs [120]. Future studies should evaluate whether these disease-associated APP-CTFs- or Aβ-enriched exosomes can be related to the progression and spreading of AD pathology [121].
LIPID RAFTS AND THE AMYLOID PRECURSOR PROTEIN INTRACELLULAR DOMAIN (AICD)
In recent years studies of the physiological role of the C-terminal fragments of APP have received significant attention due to the fact that the peptide released from APP by γ-secretase, namely called AICD, can act as a transcription factor (for review see [122]). The initial controversy in AICD studies, which was due to the utilisation of different cell types expressing different APP isoforms and to the very short half-life of the peptide, have been overcome when several groups have demonstrated that AICD can upregulate expression of the amyloid-degrading enzyme neprilysin (NEP) [123, 124]. This process is APP isoform- and neuronal cell-specific and depends on the integrity of lipid rafts. Moreover, transcriptionally active AICD is formed only in the amyloidogenic APP processing pathway which suggests an intrinsic feed-back mechanism for Aβ removal [116]. It was later shown that AICD regulates transcription of several target genes [125, 126] including a transport protein transthyretin [127]. Moreover, there are data that AICD can also regulate APP and BACE 1 [128] and lipid metabolism [129]. In particular, AICD regulates expression of serine-palmitoyl transferase and sphingolipid synthesis and, in turn, controls lipid raft composition and APP processing [130]. AICD suppresses the expression of the major lipoprotein receptor LRP1 gene and as such affects apoE/cholesterol metabolism [131].
Importantly, NEP can also be associated with lipid rafts, e.g. in ectopeptidase-rich membrane microdomains in human synoviocytes [66]. Sato and colleagues have suggested that cholesterol and other lipids regulate translocation of NEP to the rafts where its substrate Aβ accumulates. However, this does not modulate the protease activity of NEP itself [67]. However, only the mature, fully glycosylated form of NEP preferentially in dimerised form and in association with phosphatidylserine was observed in the lipid rafts [132]. Studying subcellular distribution and amyloid-degrading activity of NEP these authors also suggested that localisation of the enzyme in different intracellular compartments may provide overall neuronal clearance of Aβ. Another Aβ-degrading enzyme, namely insulin-degrading enzyme (IDE), although primarily a cytosolic protein, can also be associated with lipid rafts increasing the capacity of the cells to catabolize Aβ [133].
LIPIDS RAFTS IN THE AD BRAIN
Apart from Aβ production and clearance, lipid rafts underlie many cellular processes which can be disrupted due to impaired lipid metabolism. Lipid composition and properties are changing in response to various factors including environmental conditions (temperature, air pressure, oxygen content), nutritional changes, infections, gut microbiota, etc. In this regard it is important to commemorate works of the late academician Eugene M. Kreps (1899–1985) who was the pioneer and one of the founders of lipid research in Russia and who, using an evolutionary and comparative approach, has significantly enriched our knowledge of brain lipids and their functions especially the changes they undergo under pathological conditions (for review see [134]).
The data on the role of lipid rafts in AD pathogenesis accumulated over more than two decades provide strong indication that changes in lipid metabolism have an underlying role in the disease pathogenesis. A recent mathematical modelling study has shown a correlation between the changes in lipid composition and physicochemical properties of raft-like membranes. The authors have indicated that the size of the rafts and lipid mobility in the non-raft membrane increases during age and these are accelerated in the transgenic mouse model of AD [135]. These changes in lipid composition can lead to aberrant protein aggregation resulting in accumulation of toxic peptide species, including Aβ, and neurodegeneration (for review see [136]). Recent studies have demonstrated that perturbations in sphingolipid metabolism are consistently associated with preclinical and prodromal forms of AD and that sphingolipids might serve as biomarkers for early AD [137]. Below we discuss how the changes in the major lipid raft components can result in AD pathogenesis.
Sphingomyelin
Sphingomyelin (SM) is one the major components of lipid rafts with SM-SM hydrogen bonds contributing to the formation of ordered membrane regions, which stabilize the temporal clusters of signalling proteins and their signalling pathways [138]. SM with its metabolites themselves act as important second messengers in various signal transduction events during cell differentiation, development, the immune response and adaptation of the organism [139–141]. SM is important for the activity of various types of receptors, e.g. α7 nicotinic receptor, NMDA receptors, neurotrophic tyrosine kinase receptor type 2, serotonin1A receptor, the urokinase receptor (uPAR) (for review see [18]). Moreover, Aβ and PrP have a common SM-recognition site which supports an important role of lipid rafts in the pathogenesis of AD, HIV-1 and prion disease [142].
There are well documented studies that lipid metabolism, and in particular that of sphingolipids, undergoes significant changes both in the normally ageing brain and especially in AD pathology, By comparing levels of over 800 lipid species in AD brains by shotgun lipidomics it was demonstrated that levels of SM were decreased, and ceramide levels increased, in the affected brains which might be explained by the AD-related activation of SMase which results in increased SM hydrolysis and ceramide production [143]. Activation of SMase was shown to depend on accumulation of Aβ, and especially of its most toxic form, Aβ42 [144], or its fibrils [145]. Because SMase-related changes in SM metabolism takes please early in AD it can lead to disruption of protein-lipid interaction and downstream signalling pathways [146]. In particular, changes in the balance of the activity of the major enzymes of SM synthesis and degradation can result in perturbation of the levels of ceramide, which is involved in the processes of cell proliferation and apoptosis and as such lead to the compromised homeostasis in the brain which is observed in AD [147]. Elevated levels of SM synthase 1 and SM in AD brains have been reported to increase BACE1 activity while inhibition or knockdown of this enzyme resulted in lysosomal degradation of BACE1 attenuating AD-like pathology in mice [148]. Another enzyme of SM metabolism, namely neutral sphingomyelinase smpd3, which has mostly neuronal expression in the brain but still not with clear function, was shown to play an important role in lipid bilayer remodelling. Its deficiency results in accumulation of the neurotoxic Aβ and phosphorylated tau with accompanying neuropathology and cognitive deficit characteristic to the AD pathology [149].
The contents of ceramide, SM and sulfatide were found to correlate with age in the hippocampus of males, while sphingosine and sphingosine-1-phosphate correlated inversely with age in females. This supports the existence of gender-specific differences in sphingolipid metabolism in the ageing human brain and explains the increased risk of AD development in women since S1P levels contribute significantly to neurodegeneration in the ageing brain [150].
Gangliosides
The role of gangliosides, sialic acid containing glycosphingolipids, in lipid rafts and brain functions have been recently extensively reviewed in [26]. Knockout of various classes of gangliosides was shown to lead to accelerated neurodegeneration and AD-like pathology [151]. With ageing, and especially in AD, the content of ganglioside decreases significantly in different brain structures, and particularly in the areas of AD pathology [152, 153]. With regard to the ganglioside composition, the aged brain had lower levels of gangliosides of the ganglio-series (GT1b, GD1b, GD1a, GM1) with the statistically significant decrease mainly in the frontal cortex, temporal cortex and white matter [154]. However, in the aged frontal and parietal cortex elevated levels of simple gangliosides (GM2, GM3, GM4, GD3) have been reported [154]. In the AD brain the decreased content of gangliosides of the ganglio-series correlated with degeneration of cortical neurons while elevation of simple gangliosides in the frontal and parietal cortex might be related to accelerated degradation of gangliosides during neuronal death [154]. More recent analysis of lipid rafts from the frontal cortex (representing early stages) and the temporal cortex (representing later stages of AD pathology) has revealed that AD brains contained a significantly higher level of GM1 and GM2 in lipid rafts than matching age controls [155]. Gangliosides were also shown to accumulate in the senile plaques in AD brains [156]. Lipidomic analysis by chromatography-mass spectrometry has revealed elevated levels of diacylglycerol and sphingolipids in the prefrontal cortex of AD patients. Moreover, in the AD entorhinal cortex there were increased levels of lysobisphosphatidic acid, sphingomyelin, GM3 and cholesterol esters [157]. Recently, fluorescent probes allowing analysis of the dynamic behaviour of gangliosides in living cells have been developed and their use has revealed that GPI-anchored receptors and gangliosides interact in a cholesterol-dependent manner [158]. This approach will allow researchers to extend the analysis of ganglioside dynamics in AD-affected membranes.
The link of gangliosides with AD pathology has been mostly related to their role in GM1-induced aggregation of Aβ peptide (for review see [159]). Aβ shows high affinity for GM1 and its N-terminal region interacts with GM1 membrane clusters through hydrogen bonds and electrostatic interactions [160]. It was also suggested that cholesterol may facilitate GM1 clustering in the membranes [161] although depletion of cholesterol by MβCD did not have an effect on Aβ oligomerisation [110]. Analysis of the role of lipid raft proteins in Aβ aggregation has confirmed that they do not play a significant role in this process since treatment with proteinase K or SDS did not affect the capacity of lipid rafts to facilitate aggregation of Aβ [110]. However, lipid rafts from ganglioside-rich cells were able to induce Aβ aggregation more potently than ganglioside-poor cells [110] (Fig. 2).
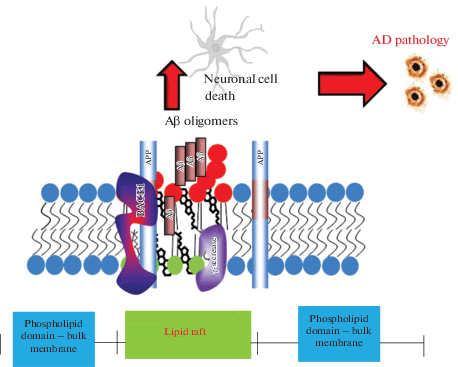
Fig. 2.
Role of ganglioside GM1 and lipid rafts in Aβ oligomer formation. Due to its very high ability for aggregation, Aβ forms dimers, trimers and oligomers of higher levels which are toxic to cells and cause neuronal death leading to AD pathology. Formation of amyloid plaques from Aβ aggregates in complex with other proteins is a hallmark of AD. Formation of toxic amyloid oligomers is initiated in the plasma membrane and depends on the ratio of Aβ/GM1.
GM1 also affects APP processing in cell cultures by inhibiting α-secretase cleavage [162]. Moreover, Aβ1-40 oligomers were shown to activate APP amyloidogenic processing by complexing with GM1 ganglioside and stimulating their own production [163]. Age-and AD-dependent increase in GM1 in the lipid rafts was shown to be more pronounced in the brain of transgenic mice overexpressing ApoE4 [164].
The mechanisms linking gangliosides with AD was shown to involve inhibition of GD3-synthase (GD3S, the key-enzyme converting a-series to major brain b-series gangliosides) by Aβ and down-regulation of GD3S expression by AICD [165]. The authors suggested that changes in the properties of lipid rafts containing different amounts of GM3 or GD3 gangliosides might modulate activity of the amyloidogenic β- and γ-secretases leading to increased production of Aβ.
Because different species of gangliosides have different impact on the integrity and properties of neuronal membranes, designing any strategy to prevent AD by altering their metabolism needs to be treated with a certain degree of caution. Despite the established role in Aβ toxicity, treatment of AD patients with GM1 was shown to halt progression of cognitive decline and improve motor performance [166]. Various studies have demonstrated a potential of GM1 administration in reducing Aβ load in AD patients [153] either by activation of its clearance by autophagy [167] or by promoting Aβ elimination by microglia via intracerebrally administered exosomes carrying surface gangliosides [168]. Peripheral administration of GM1 for prevention of Aβ aggregation in the brain via reducing peripheral/brain dynamics of Aβ was also shown to be effective [169]. However, such strategies need to take into account the antigenic properties of gangliosides which might provoke some side-effects of GM1 administration [170]. Although clinical application of gangliosides, and especially of GM1, in AD (for review see [171]) is still far from being well substantiated further studies in the field might find some appropriate therapeutic venues for these multifunctional natural compounds.
Cholesterol
The role of cholesterol and its metabolism in lipid rafts and AD has been extensively studied and reviewed in [136, 172]. The levels of cholesterol and its precursors were found to be elevated in AD patients although other groups reported lower levels of cholesterol in AD brains [173]. Familial AD-associated PS1 ΔE9 mutation was shown to upregulate total cholesterol levels and increase localization of APP in lipid rafts [174]. However, quantification of global cholesterol levels in the brain does not necessarily correlate with the amount and distribution of lipid rafts in neuronal cells [175].
High cholesterol levels were found to increase lipid raft abundance and Aβ formation [15] while low cholesterol levels resulted in up-regulation of the α-secretase activity [176]. Because cholesterol is integral for lipid raft formation, its depletion subsequently leads to raft disruption and reduction in Aβ production [16, 110]. Cholesterol has been shown to bind the C99 fragment of APP promoting Aβ production [89] and increased levels of Aβ, in turn, can change cholesterol homeostasis in the Golgi and plasma membrane [177]. And, as already discussed above, Aβ aggregation was shown to be cholesterol dependent [161]. From the biophysical point of view, increased cholesterol content in the membrane facilitates insertion of Aβ into the plasma membrane distorting membrane integrity [178]. Aβ can, as a component of lipoprotein complexes, affect cholesterol transport [179]. It was also shown to inhibit the key enzyme of cholesterol biosynthesis, hydroxymethylglutaryl-CoA reductase [146]. Following this, the APP intracellular domain AICD was found to regulate cholesterol levels via LRP1 [180].
Despite a significant amount of data on the links between cholesterol levels and AD pathogenesis they are still rather inconclusive for using cholesterol as a marker of cognitive decline in AD [181].
LIPID-RAFT RELATED THERAPEUTIC APPROACHES IN AD
The most developed drugs based on cholesterol metabolism are statins which inhibit HMG CoA reductase, a key enzyme of cholesterol biosynthesis. The current state of the field and problems have been recently discussed and the role of statins has been reviewed [21]. The authors point out that despite high scientific quality of the studies the overall results did not reveal significant efficacy between statins and placebo treatment for AD. This might be because physiological effects of statins are not solely related to inhibition of cholesterol biosynthesis but include perturbation of other mevalonate-dependent pathways such as protein prenylation which plays a certain role in AD pathogenesis [182]. On the other hand, cholesterol metabolic pathways in the brain are autonomous from the systemic ones and the blood-brain barrier penetration of different statins might vary [183]. Unlike cholesterol, oxidized cholesterol metabolites, namely oxysterols, can cross the blood brain barrier from the circulation. These oxysterols and their metabolising enzymes are altered in AD brains which opens new avenues for designing disease modifying therapeutic approaches [20].
Nevertheless, a significant number of in vitro and in vivo studies have shown positive effects of statins on AD pathogenetic mechanisms. Thus, treatment of brain capillary endothelial cells with simvastatin markedly reduced Aβ uptake and cell-associated Aβ oligomers [184]. Fluvastatin was shown to increase lysosomal degradation of APP C-terminal fragments and facilitate Aβ clearance [185], while lovastatin increased α-secretase cleavage of APP [176]. In AD transgenic mice bryostatin-1 was shown to significantly increase levels of sAPPα and reduce Aβ peptide levels resulting in improved cognitive behaviour [186, 187]. Another statin, rosuvastatin, was found to alter gene expression of the γ-secretase complex without affecting its enzyme activity [188]. These experimental data clearly demonstrate a potential therapeutic value of statins in preventing AD pathogenesis, but further studies are required to ratify their efficacy in humans.
With regard to designing therapeutic strategies based on utilisation of sphingolipid compounds, the application of nanoparticles based on GM1-rHDL, which have high binding affinity to Aβ, was shown to facilitate Aβ degradation by microglia with subsequent increased Aβ efflux across the blood-brain barrier [189].
Taking into account that lipid rafts are an integral part of the plasma membrane consisting of a phospholipid moiety, it is important to bear in mind that this class of lipids have significant variability of molecular species allowing them to maintain membrane fluidity due to the changeable fatty acid composition [190]. Manipulating lipid membrane components with diet, including unsaturated fatty acids, is one of the approaches to prevent AD pathology [191]. There is convincing evidence that supplementation of docosahexaenoic acid could prevent disturbances in membrane structure with ageing and prevent cognitive decline [192]. An alternative approach for maintaining healthy cholesterol levels and reducing Aβ generation might be physical exercise as shown in treadmill experiments in AD transgenic mice [193].
Concluding remarks
Future progress in lipid research especially with development of clinical lipidomic approaches and technologies [143] will bring new extension to lipid raft research by characterizing lipid profiles, pathways and networks in isolated cells, tissue biopsy or body fluids of patients [194]. Combined with clinical proteomic and genomic data this will reveal further the role of lipid metabolism in AD pathogenesis in relation to amyloid-induced membrane damage [195]. The discovery of the role of lipid rafts in production of the functionally active transcriptional regulator AICD, which regulates expression of a variety of neuronal genes [124, 127, 165], suggests that any new therapeutic approaches aimed at modification of lipid raft components should be treated with caution. The multifunctional role of lipids, their structural variability and adaptive potential are of great importance for normal functions of cells and organisms and any advance in their research will be beneficial not only for clinical application but mostly for general science. The view on the fundamental role of lipids in cellular membranes and brain functions advocated by academician Eugene M. Kreps in the second half of the last century has reached new horizons and opened new perspectives in our current research.
Список литературы
Hardy J.A., Higgins G.A. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 256: 184–185. 1992.https://doi.org/10.1126/science.1566067
Selkoe D.J., Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 8: 595–608. 2016. https://doi.org/10.15252/emmm.201606210
Armstrong R.A. Risk factors for Alzheimer’s disease. Folia Neuropathol. 57: 87–105. 2019. https://doi.org/10.5114/fn.2019.85929
Nelson P.T., Alafuzoff I., Bigio E.H., Bouras C., Braak H., Cairns N.J., Castellani R.J., Crain B.J., Davies P., Del Tredici K., Duyckaerts C., Frosch M.P., Haroutunian V., Hof P.R., Hulette C.M., Hyman B.T., Iwatsubo T., Jellinger K.A., Jicha G.A., Kövari E., Kukull W.A., Leverenz J.B., Love S., Mackenzie I.R., Mann D.M., Masliah E., McKee A.C., Montine T.J., Morris J.C., Schneider J.A., Sonnen J.A., Thal D.R., Trojanowski J.Q., Troncoso J.C., Wisniewski T., Woltjer R.L., Beach T.G. Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J. Neuropathol. Exp. Neurol. 71: 362–381. 2012. https://doi.org/10.1097/NEN.0b013e31825018f7
Sakono M., Zako T. Amyloid oligomers: formation and toxicity of Aβ oligomers. FEBS J. 277: 1348–1358. 2010. https://doi.org/10.1111/j.1742-4658.2010.07568.x
Walsh D.M., Selkoe D.J. A β oligomers – a decade of discovery. J. Neurochem. 101: 1172–1184. 2007. https://doi.org/10.1111/j.1471-4159.2006.04426.x
Yang T., Li S., Xu H., Walsh D.M., Selkoe D.J. Large Soluble Oligomers of Amyloid β-Protein from Alzheimer Brain Are Far Less Neuroactive Than the Smaller Oligomers to Which They Dissociate. J. Neurosci. 37: 152–163. 2017. https://doi.org/10.1523/JNEUROSCI.1698-16.2016
Condello C., Yuan P., Schain A., Grutzendler J. Microglia constitute a barrier that prevents neurotoxic protofibrillar Aβ42 hotspots around plaques. Nat. Commun. 6: 6176. 2015. https://doi.org/10.1038/ncomms7176
Rodríguez-Arellano J.J., Parpura V., Zorec R., Verkhratsky A. Astrocytes in physiological aging and Alzheimer’s disease. Neuroscience. 323: 170–182. 2016. https://doi.org/10.1016/j.neuroscience.2015.01.007
Desikan R.S., Sabuncu M.R., Schansky N.J., Reuter M., Cabral H.J., Hess C.P., Weiner M.W., Biffi A., Anderson C.D., Rosand J., Salat D.H., Kemper T.L., Dale A.M., Sperling R.A., Fischl B. Alzheimer’s Disease Neuroimaging Initiative. Selective disruption of the cerebral neocortex in Alzheimer’s disease. PLoS One. 5:e12853. 2010. https://doi.org/10.1371/journal.pone.0012853
Carmona S., Hardy J., Guerreiro R. The genetic landscape of Alzheimer disease. Handb. Clin. Neurol. 148: 395–408. 2018. https://doi.org/10.1016/B978-0-444-64076-5.00026-0
Guerreiro R.J., Gustafson D.R., Hardy J. The genetic architecture of Alzheimer’s disease: beyond APP, PSENs and APOE. Neurobiol. Aging. 33: 437–456. 2012. https://doi.org/10.1016/j.neurobiolaging.2010.03.025
Hampel H., Vassar R., De Strooper B., Hardy J., Willem M., Singh N., Zhou J., Yan R., Vanmechelen E., De Vos A., Nisticò R., Corbo M., Imbimbo B.P., Streffer J., Voytyuk I., Timmers M., Tahami Monfared A.A., Irizarry M., Albala B., Koyama A., Watanabe N., Kimura T., Yarenis L., Lista S., Kramer L., Vergallo A. The β-secretase BACE1 in Alzheimer’s disease. Biol. Psychiatry. 2020. https://doi.org/10.1016/j.biopsych.2020.02.001
Haass C., Kaether C., Thinakaran G., Sisodia S. Trafficking and proteolytic processing of APP. Cold Spring Harb Perspect Med. 2: a006270. 2012. https://doi.org/10.1101/cshperspect.a006270
Ehehalt R., Keller P., Haass C., Thiele C., Simons K. Amyloidogenic processing of the Alzheimer β-amyloid precursor protein depends on lipid rafts. J. Cell Biol. 160: 113–123. 2003. https://doi.org/10.1083/jcb.200207113
Cordy J.M., Hussain I., Dingwall C., Hooper N.M., Turner A.J. Exclusively targeting β-secretase to lipid rafts by GPI-anchor addition up-regulates β-site processing of the amyloid precursor protein. Proc. Natl. Acad. Sci. USA. 100: 11735–11740. 2003. https://doi.org/10.1073/pnas.1635130100
Allinson T.M., Parkin E.T., Turner A.J., Hooper N.M. ADAMs family members as amyloid precursor protein alpha-secretases. J. Neurosci. Res. 74: 342–352. 2003. https://doi.org/10.1002/jnr.10737
Hicks D.A., Nalivaeva N.N., Turner A.J. Lipid rafts and Alzheimer’s disease: protein-lipid interactions and perturbation of signaling. Front Physiol. 3: 189. 2012. https://doi.org/10.3389/fphys.2012.00189
Bhattacharyya R., Barren C., Kovacs D.M. Palmitoylation of amyloid precursor protein regulates amyloidogenic processing in lipid rafts. J. Neurosci. 33: 11169–11183. 2013. https://doi.org/10.1523/JNEUROSCI.4704-12.2013
Loera-Valencia R., Goikolea J., Parrado-Fernandez C., Merino-Serrais P., Maioli S. Alterations in cholesterol metabolism as a risk factor for developing Alzheimer’s disease: Potential novel targets for treatment. J. Steroid. Biochem. Mol. Biol. 190: 104–114. 2019. https://doi.org/10.1016/j.jsbmb.2019.03.003
Mejías-Trueba M., Pérez-Moreno M.A., Fernández-Arche M.Á. Systematic review of the efficacy of statins for the treatment of Alzheimer’s disease. Clin. Med. (Lond). 18: 54–61. 2018. https://doi.org/10.7861/clinmedicine.18-1-54
Hooper N.M., Karran E.H., Turner A.J. Membrane protein secretases. Biochem. J. 321: 265–279. 1997. https://doi.org/10.1042/bj3210265
Simons K., Ikonen E. Functional rafts in cell membranes. Nature. 387: 569–572. 1997. https://doi.org/10.1038/42408
Sonnino S., Prinetti A. Membrane domains and the “lipid raft” concept. Curr. Med. Chem. 20: 4–21. 2013. https://doi.org/10.2174/0929867311320010003
Bieberich E. Sphingolipids and lipid rafts: Novel concepts and methods of analysis. Chem. Phys. Lipids. 216: 114–131. 1918. https://doi.org/10.1016/j.chemphyslip.2018.08.003
Grassi S., Giussani P., Mauri L., Prioni S., Sonnino S., Prinetti A. Lipid rafts and neurodegeneration: Structural and functional roles in physiologic aging and neurodegenerative diseases. J. Lipid. Res. pii: jlr.TR119000427. 2019. https://doi.org/10.1194/jlr.TR119000427
Morigaki K., Tanimoto Y. Evolution and development of model membranes for physicochemical and functional studies of the membrane lateral heterogeneity. Biochim. Biophys. Acta Biomembr. 1860: 2012–2017. 2018. https://doi.org/10.1016/j.bbamem.2018.03.010
Rothberg K.G., Heuser J.E., Donzell W.C., Ying Y.S., Glenney J.R., Anderson R.G. Caveolin, a protein component of caveolae membrane coats. Cell. 68: 673–682. 1992. https://doi.org/10.1016/0092-8674(92)90143-z
van Meer G., Simons K. Lipid polarity and sorting in epithelial cells. J. Cell Biochem. 36:51-58. 1988. https://doi.org/10.1002/jcb.240360106
Brown D.A., London E. Structure of detergent-resistant membrane domains: does phase separation occur in biological membranes? Biochem. Biophys. Res. Commun. 240: 1–7. 1997. https://doi.org/10.1006/bbrc.1997.7575
Jiang X., Zhu Z., Qin H., Tripathi P., Zhong L., Elsherbini A., Karki S., Crivelli S.M., Zhi W., Wang G., Spassieva S.D., Bieberich E. Visualization of Ceramide-Associated Proteins in Ceramide-Rich Platforms Using a Cross-Linkable Ceramide Analog and Proximity Ligation Assays With Anti-ceramide Antibody. Front Cell Dev. Biol. 7: 166. 2019. https://doi.org/10.3389/fcell.2019.00166
Yáñez-Mó M., Gutiérrez-López M.D., Cabañas C. Functional interplay between tetraspanins and proteases. Cell Mol. Life Sci. 68: 3323–3335. 2011. https://doi.org/10.1007/s00018-011-0746-y
Hemler M.E. Tetraspanin functions and associated microdomains. Nat. Rev. Mol. Cell Biol. 6, 801–811. 2005. https://doi.org/10.1038/nrm1736
Moretto E., Longatti A., Murru L., Chamma I., Sessa A., Zapata J., Hosy E., Sainlos M., Saint-Pol J., Rubinstein E., Choquet D., Broccoli V., Schiavo G., Thoumine O., Passafaro M. TSPAN5 Enriched Microdomains Provide a Platform for Dendritic Spine Maturation through Neuroligin-1 Clustering. Cell Rep. 29: 1130–1146.e8. 2019. https://doi.org/10.1016/j.celrep.2019.09.051
Schon E.A., Area-Gomez E. Mitochondria-associated ER membranes in Alzheimer disease. Mol. Cell Neurosci. 55: 26–36. 2013. https://doi.org/10.1016/j.mcn.2012.07.011
Pera M., Larrea D., Guardia-Laguarta C., Montesinos J., Velasco K.R., Agrawal R.R., Xu Y., Chan R.B., Di Paolo G., Mehler M.F., Perumal G.S., Macaluso F.P., Freyberg Z.Z., Acin-Perez R., Enriquez J.A., Schon E.A., Area-Gomez E. Increased localization of APP-C99 in mitochondria-associated ER membranes causes mitochondrial dysfunction in Alzheimer disease. EMB-O J. 36: 3356–3371. 2017. https://doi.org/10.15252/embj.201796797
Hayashi T., Rizzuto R., Hajnoczky G., Su T.P. MAM: more than just a housekeeper. Trends Cell Biol. 19: 81–88. 2009. https://doi.org/10.1016/j.tcb.2008.12.002
Teixeira G., Vieira L.B., Gomez M.V., Guatimosim C. Cholesterol as a key player in the balance of evoked and spontaneous glutamate release in rat brain cortical synaptosomes. Neurochem. Int. 61: 1151–1159. 2012. https://doi.org/10.1016/j.neuint.2012.08.008
Ouweneel A.B., Thomas M.J., Sorci-Thomas M.G. The ins and outs of lipid rafts: Functions in intracellular cholesterol homeostasis, microparticles, and cell membranes. J. Lipid Res. pii: jlr.TR119000383. 2019. https://doi.org/10.1194/jlr.TR119000383
Pascual M., Ibáñez F., Guerri C. Exosomes as mediators of neuron-glia communication in neuroinflammation. Neural. Regen. Res. 15: 796–801. 2020. https://doi.org/10.4103/1673-5374.268893
Pollet H., Conrard L., Cloos A.S., Tyteca D. Plasma Membrane Lipid Domains as Platforms for Vesicle Biogenesis and Shedding. Biomolecules. 8: pii: E94. 2018. https://doi.org/10.3390/biom8030094
Turner A.J. PIG-tailed membrane proteins. Essays Biochem. 28: 113–127. 1994. PMID: 7925314
Kinoshita T., Fujita M. Biosynthesis of GPI-anchored proteins: special emphasis on GPI lipid remodeling. J. Lipid. Res. 571: 6–24. 2016. https://doi.org/10.1194/jlr.R063313
Oh P., Schnitzer J.E. Segregation of heterotrimeric G proteins in cell surface microdomains. G(q) binds caveolin to concentrate in caveolae, whereas G(i) and G(s) target lipid rafts by default. Mol. Biol. Cell. 123: 685–698. 2001. https://doi.org/10.1091/mbc.12.3.685
Snyers L., Umlauf E., Prohaska R. Association of stomatin with lipid-protein complexes in the plasma membrane and the endocytic compartment. Eur. J. Cell Biol. 7811: 802–812. 1999. https://doi.org/10.1016/S0171-9335(99)80031-4
Liu J., Deyoung S.M., Zhang M., Dold L.H., Saltiel A.R. The stomatin/prohibitin/flotillin/HflK/C domain of flotillin-1 contains distinct sequences that direct plasma membrane localization and protein interactions in 3T3-L1 adipocytes. J. Biol. Chem. 28016: 16125–16134. 2005. https://doi.org/10.1074/jbc.M500940200
Schneider A., Rajendran L., Honsho M., Gralle M., Donnert G., Wouters F., Hell S.W., Simons M. Flotillin-dependent clustering of the amyloid precursor protein regulates its endocytosis and amyloidogenic processing in neurons. J. Neurosci. 28: 2874–2882. 2008. https://doi.org/10.1523/JNEUROSCI.5345-07.2008
Morrow I.C., Parton R.G. Flotillins and the PHB domain protein family: rafts, worms and anaesthetics. Traffic. 6: 725–740. 2005. https://doi.org/10.1111/j.1600-0854.2005.00318.x
Parkin E.T., Turner A.J., Hooper N.M. (1999). Amyloid precursor protein, although partially detergent-insoluble in mouse cerebral cortex, behaves as an atypical lipid raft protein. Biochem. J. 344: 23–30. 1999. PMCID: PMC1220609.
Kalvodova L., Kahya N., Schwille P., Ehehalt R., Verkade P., Drechsel D., Simons K. Lipids as modulators of proteolytic activity of BACE: involvement of cholesterol, glycosphingolipids, and anionic phospholipids in vitro. J. Biol. Chem. 280: 36815–36823. 2005. https://doi.org/10.1074/jbc.M504484200
Hur J.Y., Welander H., Behbahani H., Aoki M., Franberg J., Winblad B., Frykman S., Tjernberg L.O. Active γ-secretase is localized to detergent-resistant membranes in human brain. FEBS J. 275: 1174–1187. 2008. https://doi.org/10.1111/j.1742-4658.2008.06278.x
Matsumura N., Takami M., Okochi M., Wada-Kakuda S., Fujiwara H., Tagami S., Funamoto S., Ihara Y., Morishima-Kawashima M. γ-Secretase associated with lipid rafts: multiple interactive pathways in the stepwise processing of β-carboxyl-terminal fragment. J. Biol. Chem. 289: 5109–5121. 2014. https://doi.org/10.1074/jbc.M113.510131
Harris B., Pereira I., Parkin E. Targeting ADAM10 to lipid rafts in neuroblastoma SH-SY5Y cells impairs amyloidogenic processing of the amyloid precursor protein. Brain Res. 1296: 203–215. 2009. https://doi.org/10.1016/j.brainres.2009.07.105
Tellier E., Canault M., Rebsomen L., Bonardo B., Juhan-Vague I., Nalbone G., Peiretti F. The shedding activity of ADAM17 is sequestered in lipid rafts. Exp. Cell. Res. 312: 3969–3980. 2006. https://doi.org/10.1016/j.yexcr.2006.08.027
Pinheiro T.J. The role of rafts in the fibrillization and aggregation of prions. Chem. Phys. Lipids. 141: 66–71. 2006. https://doi.org/10.1016/j.chemphyslip.2006.02.022
Zhu D., Xiong W.C., Mei L. Lipid rafts serve as a signaling platform for nicotinic acetylcholine receptor clustering. J. Neurosci. 26: 4841–4851. 2006. https://doi.org/10.1523/JNEUROSCI.2807-05.2006
Colón-Sáez J.O., Yakel J.L. The α7 nicotinic acetylcholine receptor function in hippocampal neurons is regulated by the lipid composition of the plasma membrane. J. Physiol. 589: 3163–3174. 2011. https://doi.org/10.1113/jphysiol.2011.209494
Khan G.M., Tong M., Jhun M., Arora K., Nichols R.A. β-Amyloid activates presynaptic α7 nicotinic acetylcholine receptors reconstituted into a model nerve cell system: involvement of lipid rafts. Eur. J. Neurosci. 31: 788–796. 2010. https://doi.org/10.1111/j.1460-9568.2010.07116.x
Moral-Naranjo M.T., Montenegro M.F., Muñoz-Delgado E., Campoy F.J., Vidal C.J. Targeting of acetylcholinesterase to lipid rafts of muscle. Chem. Biol. Interact. 175: 312–317. 2008. https://doi.org/10.1016/j.cbi.2008.04.018
Xie H.Q., Liang D., Leung K.W., Chen V.P., Zhu K.Y., Chan W.K., Choi R.C., Massoulie J., Tsim K.W. Targeting acetylcholinesterase (AChE) to membrane rafts: A function mediated by the proline rich membrane anchor (PRiMA) in neurons. J. Biol. Chem 285: 11537–11546. 2010. https://doi.org/10.1074/jbc.M109.038711
Suzuki T., Suzuki Y. Virus infection and lipid rafts. Biol. Pharm. Bull. 29: 1538–1541. 2006. https://doi.org/10.1248/bpb.29.1538
Margheri G., D’Agostino R., Trigari S., Sottini S., Del Rosso M. The β-subunit of cholera toxin has a high affinity for ganglioside GM1 embedded into solid supported lipid membranes with a lipid raft-like composition. Lipids. 49: 203–206. 2014. https://doi.org/10.1007/s11745-013-3845-8
Popik W., Alce T.M., Au W.C. Human immunodeficiency virus type 1 uses lipid raft-colocalized CD4 and chemokine receptors for productive entry into CD4(+) T cells. J. Virol. 76: 4709–4722. 2002. https://doi.org/10.1128/jvi.76.10.4709-4722.2002
Lu Y., Liu D.X., Tam J.P. Lipid rafts are involved in SARS-CoV entry into Vero E6 cells. Biochem. Biophys. Res. Commun. 369: 344–349. 2008. https://doi.org/10.1016/j.bbrc.2008.02.023
Yan R., Zhang Y., Li Y., Xia L., Guo Y., Zhou Q. Structural basis for the recognition of the SARS-CoV-2 by full-length human ACE2. Science. pii: eabb2762. 2020. https://doi.org/10.1126/science.abb2762
Riemann D., Hansen G.H., Niels-Christiansen L., Thorsen E., Immerdal L., Santos A.N., Kehlen A., Langner J., Danielsen E.M. Caveolae/lipid rafts in fibroblast-like synoviocytes: ectopeptidase-rich membrane microdomains. Biochem. J. 354: 47–55. 2001. https://doi.org/10.1042/0264-6021:3540047
Sato K., Tanabe C., Yonemura Y., Watahiki H., Zhao Y., Yagishita S., Ebina M., Suo S., Futai E., Murata M., Ishiura S. Localization of mature neprilysin in lipid rafts. J. Neurosci. Res. 90: 870–877. 2012. https://doi.org/10.1002/jnr.22796
Grider M.H., Park D., Spencer D.M., Shine H.D. Lipid raft-targeted Akt promotes axonal branching and growth cone expansion via mTOR and Rac1, respectively. J. Neurosci. Res. 87: 3033–3042. 2009. https://doi.org/10.1002/jnr.22140
Petro K.A., Schengrund C.L. Membrane raft disruption promotes axonogenesis in n2a neuroblastoma cells. Neurochem. Res 34: 29–37. 2009. https://doi.org/10.1007/s11064-008-9625-9
Willmann R., Pun S., Stallmach L., Sadasivam G., Santos A.F., Caroni P., Fuhrer C. Cholesterol and lipid microdomains stabilize the postsynapse at the neuromuscular junction. EMBO J. 25: 4050–4060. 2006. https://doi.org/10.1038/sj.emboj.7601288
Nalivaeva N.N., Turner A.J. The amyloid precursor protein: a biochemical enigma in brain development, function and disease. FEBS Lett. 587: 2046–2054. 2013. https://doi.org/10.1016/j.febslet.2013.05.010
Kitaguchi N., Takahashi Y., Tokushima Y., Shiojiri S., Ito H. (1988). Novel precursor of Alzheimer’s disease amyloid protein shows protease inhibitory activity. Nature. 331: 530–532. https://doi.org/10.1038/331530a0
Sandbrink R., Masters C.L., Beyreuther K. APP gene family. Alternative splicing generates functionally related isoforms. Ann. NY Acad. Sci. 777: 281–287. 1996. https://doi.org/10.1111/j.1749-6632.1996.tb34433.x
Tanaka S., Shiojiri S., Takahashi Y., Kitaguchi N., Ito H., Kameyama M., Kimura J., Nakamura S., Ued-a K. Tissue-specific expression of three types of β-protein precursor mRNA: enhancement of protease inhibitor-harboring types in Alzheimer’s disease brain. Biochem. Biophys. Res. Commun. 165: 1406–1414. 1989. https://doi.org/10.1016/0006-291x(89)92760-5
Golde T.E., Estus S., Usiak M., Younkin L.H., Younkin S.G. Expression of β amyloid protein precursor mRNAs: recognition of a novel alternatively spliced form and quantitation in Alzheimer’s disease using PCR. Neuron. 4: 253–267. 1990. https://doi.org/10.1016/0896-6273(90)90100-t
Moir R.D., Lynch T., Bush A.I., Whyte S., Henry A., Portbury S., Multhaup G., Small D.H., Tanzi R.E., Beyreuther K., Masters C.L. Relative increase in Alzheimer’s disease of soluble forms of cerebral Ab amyloid protein precursor containing the Kunitz protease inhibitory domain. J. Biol. Chem. 273: 5013–5019. 1998. https://doi.org/10.1074/jbc.273.9.5013
Yamada T., Araki E., Izumi R., Goto I., Sasaki H., Sakaki Y. Expression of Alzheimer amyloid β-protein precursor gene in neuronal cells. Gerontology. 37(Suppl 1): 24–30. 1991. https://doi.org/10.1159/000213294
Zhang H., Ma Q., Zhang Y.W., Xu H. Proteolytic processing of Alzheimer’s β-amyloid precursor protein. J. Neurochem. 120(Suppl 1) :9–21. https://doi.org/10.1111/j.1471-4159.2011.07519.x
Chasseigneaux S., Allinquant B. Functions of Aβ, sAPPα and sAPPβ : similarities and differences. J. Neurochem. 120(Suppl 1): 99–108. https://doi.org/10.1111/j.1471-4159.2011.07584.x
Octave J.N., Pierrot N., Ferao Santos S., Nalivaeva N.N., Turner A.J. From synaptic spines to nuclear signaling: nuclear and synaptic actions of the amyloid precursor protein. J. Neurochem. 126: 183–190. 2013. https://doi.org/10.1111/jnc.12239
Higgins L.S., Murphy G.M., Jr., Forno L.S., Catalano R., Cordell B. P3 β-amyloid peptide has a unique and potentially pathogenic immunohistochemical profile in Alzheimer’s disease brain. Am. J. Pathol. 149: 585–596. 1996. PMCID: PMC1865300.
Siegel G., Gerber H., Koch P., Bruestle O., Fraering P.C., Rajendran L. The Alzheimer’s Disease γ-Secretase Generates Higher 42:40 Ratios for β-Amyloid Than for p3 Peptides. Cell. Rep. 19: 1967–1976. https://doi.org/10.1016/j.celrep.2017.05.034
Lefranc-Jullien S., Sunyach C., Checler F. APPε, the ε-secretase-derived N-terminal product of the β-amyloid precursor protein, behaves as a type I protein and undergoes α-, β-, and γ‑secretase cleavages. J. Neurochem. 97: 807–817. 2006. https://doi.org/10.1111/j.1471-4159.2006.03748.x
Haupt S., Borghese L., Brüstle O., Edenhofer F. Non-genetic modulation of Notch activity by artificial delivery of Notch intracellular domain into neural stem cells. Stem. Cell. Rev. Rep. 8: 672–684. 2012. https://doi.org/10.1007/s12015-011-9335-6
Sawamura N., Ko M., Yu W., Zou K., Hanada K., Suzuki T., Gong J.S., Yanagisawa K., Michikawa M. Modulation of amyloid precursor protein cleavage by cellular sphingolipids. J. Biol. Chem. 279:11984–11991. 2004. https://doi.org/10.1074/jbc.M309832200
Cordy J.M., Hooper N.M., Turner A.J. The involvement of lipid rafts in Alzheimer’s disease. Mol. Membr.Biol. 23: 111–122. 2006. https://doi.org/10.1080/09687860500496417
Vetrivel K.S., Thinakaran G. Membrane rafts in Alzheimer’s disease β-amyloid production. Biochim. Biophys. Acta. 1801: 860–867. 2010. https://doi.org/10.1016/j.bbalip.2010.03.007
Chen T.Y., Liu P.H., Ruan C.T., Chiu L., Kung F.L. The intracellular domain of amyloid precursor protein interacts with flotillin-1, a lipid raft protein. Biochem. Biophys. Res. Commun. 342: 266–272. 2006. https://doi.org/10.1016/j.bbrc.2006.01.156
Beel A.J., Sakakura M., Barrett P.J., Sanders C.R. Direct binding of cholesterol to the amyloid precursor protein: An important interaction in lipid-Alzheimer’s disease relationships? Biochim. Biophys. Acta. 1801: 975–982. 2010. https://doi.org/10.1016/j.bbalip.2010.03.008
Yao Z.X., Papadopoulos V. Function of β-amyloid in cholesterol transport: a lead to neurotoxicity. FASEB J. 16: 1677–1679. 2002. https://doi.org/10.1096/fj.02-0285fje
Minami S.S., Hoe H.S., Rebeck G.W. Fyn kinase regulates the association between amyloid precursor protein and Dab1 by promoting their localization to detergent-resistant membranes. J. Neurochem. 118: 879–890. 2011. https://doi.org/10.1111/j.1471-4159.2011.07296.x
Watanabe T., Hikichi Y., Willuweit A., Shintani Y., Horiguchi T. FBL2 regulates amyloid precursor protein (APP) metabolism by promoting ubiquitination-dependent APP degradation and inhibition of APP endocytosis. J. Neurosci. 32: 3352–3365. 2012. https://doi.org/10.1523/JNEUROSCI.5659-11.2012
Yoon I.S., Chen E., Busse T., Repetto E., Lakshmana M.K., Koo E.H., Kang D.E. Low-density lipoprotein receptor-related protein promotes amyloid precursor protein trafficking to lipid rafts in the endocytic pathway. FASEB J. 21: 2742–2752. 2007. https://doi.org/10.1096/fj.07-8114com
Fuentealba R.A., Barría M.I., Lee J., Cam J., Araya C., Escudero C.A., Inestrosa N.C., Bronfman F.C., Bu G., Marzolo M.P. ApoER2 expression increases Aβ production while decreasing Amyloid Precursor Protein (APP) endocytosis: Possible role in the partitioning of APP into lipid rafts and in the regulation of γ-secretase activity. Mol. Neurodegener. 2: 14. 2007. https://doi.org/10.1186/1750-1326-2-14
Bhattacharyya R., Fenn R.H., Barren C., Tanzi R.E., Kovacs D.M. Palmitoylated APP Forms Dimers, Cleaved by BACE1. PLoS One. 11: e0166400. 2016. https://doi.org/10.1371/journal.pone.0166400
Head B.P., Patel H.H., Insel P.A. Interaction of membrane/lipid rafts with the cytoskeleton: impact on signaling and function: membrane/lipid rafts, mediators of cytoskeletal arrangement and cell signaling. Biochim. Biophys. Acta. 1838: 532–545. 2014. https://doi.org/10.1016/j.bbamem.2013.07.018
Rushworth J.V., Hooper N.M. Lipid Rafts: Linking Alzheimer’s Amyloid-β Production, Aggregation, and Toxicity at Neuronal Membranes. Int. J. Alzheimers Dis. 2011:603052. 2011. https://doi.org/10.4061/2011/603052
Kim S.I., Yi J.S., Ko Y.G. Amyloid β oligomerization is induced by brain lipid rafts. J. Cell Biochem. 99: 878–889. 2006. https://doi.org/10.1002/jcb.20978
Ikeda K., Yamaguchi T., Fukunaga S., Hoshino M., Matsuzaki K. Mechanism of amyloid β-protein aggregation mediated by GM1 ganglioside clusters. Biochemistry. 50: 6433–6440. 2011. https://doi.org/10.1021/bi200771m
Ogawa M., Tsukuda M., Yamaguchi T., Ikeda K., Okada T., Yano Y., Hoshino M., Matsuzaki K. Ganglioside-mediated aggregation of amyloid β-proteins (Aβ): comparison between Aβ-(1-42) and Aβ-(1-40). J. Neurochem. 116: 851–857. 2011. https://doi.org/10.1111/j.1471-4159.2010.06997.x
Hattori C., Asai M., Onishi H., Sasagawa N., Hashimoto Y., Saido T.C., Maruyama K., Mizutani S., Ishiura S. BACE1 interacts with lipid raft proteins. J. Neurosci.Res. 84: 912–917. 2006. https://doi.org/10.1002/jnr.20981
Motoki K., Kume H., Oda A., Tamaoka A., Hosaka A., Kametani F., Araki W. Neuronal β-amyloid generation is independent of lipid raft association of β-secretase BACE1: analysis with a palmitoylation-deficient mutant. Brain Behav. 2: 270–282. 2012. https://doi.org/10.1002/brb3.52
Ebina M., Futai E., Tanabe C., Sasagawa N., Kiso Y., Ishiura S. Inhibition by KMI-574 leads to dislocalization of BACE1 from lipid rafts. J. Neurosci. Res. 87: 360–368. 2009. https://doi.org/10.1002/jnr.21858
Vetrivel K.S., Barman A., Chen Y., Nguyen P.D., Wagner S.L., Prabhakar R., Thinakaran G. Loss of cleavage at β'-site contributes to apparent increase in β-amyloid peptide (Aβ) secretion by β-secretase (BACE1)-glycosylphosphatidylinositol (GPI) processing of amyloid precursor protein. J. Biol. Chem. 286: 26166–26177. 2011. https://doi.org/10.1074/jbc.M111.260471
Parkin E.T., Watt N.T., Hussain I., Eckman E.A., Eckman C.B., Manson J.C., Baybutt H.N., Turner A.J., Hooper N.M. Cellular prion protein regulates β-secretase cleavage of the Alzheimer’s amyloid precursor protein. Proc. Natl. Acad. Sci. USA. 104: 11062–11067. 2007. https://doi.org/10.1073/pnas.0609621104
Griffiths H.H., Whitehouse I.J., Baybutt H., Brown D., Kellett K.A., Jackson C.D., Turner A.J., Piccardo P., Manson J.C., Hooper N.M. Prion protein interacts with BACE1 protein and differentially regulates its activity toward wild type and Swedish mutant amyloid precursor protein. J. Biol. Chem. 286: 33489–33500. 2011. https://doi.org/10.1074/jbc.M111.278556
Vetrivel K.S., Cheng H., Lin W., Sakurai T., Li T., Nukina N., Wong P.C., Xu H., Thinakaran G. Association of γ-secretase with lipid rafts in post-Golgi and endosome membranes. J. Biol. Chem. 279: 44945–54. 2004. https://doi.org/10.1074/jbc.M407986200
Cheng H., Vetrivel K.S., Drisdel R.C., Meckler X., Gong P., Leem J.Y., Li T., Carter M., Chen Y., Nguyen P., Iwatsubo T., Tomita T., Wong P.C., Green W.N., Kounnas M.Z., Thinakaran G. S-palmitoylation of gamma-secretase subunits nicastrin and APH-1. J. Biol. Chem. 284: 1373–84. 2009. https://doi.org/10.1074/jbc.M806380200
Kapoor A., Hsu W.M., Wang B.J., Wu G.H., Lin T.Y., Lee S.J., Yen C.T., Liang S.M., Liao Y.F. Caveolin-1 regulates γ-secretase-mediated AβPP processing by modulating spatial distribution of γ-secretase in membrane. J. Alzheimers Dis. 22: 423–242. 2010. https://doi.org/10.3233/JAD-2010-100531
Kim Y., Kim C., Jang H.Y., Mook-Jung I. Inhibition of Cholesterol Biosynthesis Reduces γ-Secretase Activity and Amyloid-β Generation. J. Alzheimers Dis. 51: 1057–1068. 2016. https://doi.org/10.3233/JAD-150982
Eckert G.P., Müller W.E. Presenilin 1 modifies lipid raft composition of neuronal membranes. Biochem. Biophys. Res. Commun. 382: 673–677. 2009. https://doi.org/10.1016/j.bbrc.2009.03.070
Han J., Jung S., Jang J., Kam T.I., Choi H., Kim B.J., Nah J., Jo D.G., Nakagawa T., Nishimura M., Jung Y.K. OCIAD2 activates γ-secretase to enhance amyloid β production by interacting with nicastrin. Cell. Mol. Life Sci. 71: 2561–2576. 2014. https://doi.org/10.1007/s00018-013-1515-x
Xu D., Sharma C., Hemler M.E. Tetraspanin12 regulates ADAM10-dependent cleavage of amyloid precursor protein. FASEB J. 23:3674-3681. 2009. https://doi.org/10.1096/fj.09-133462
Seipold L., Saftig P. The Emerging Role of Tetraspanins in the Proteolytic Processing of the Amyloid Precursor Protein. Front Mol. Neurosci. 9: 149. 2016. https://doi.org/10.3389/fnmol.2016.00149
Seipold L., Damme M., Prox J., Rabe B., Kasparek P., Sedlacek R., Altmeppen H., Willem M., Boland B., Glatzel M., Saftig P. Tetraspanin 3: A central endocytic membrane component regulating the expression of ADAM10, presenilin and the amyloid precursor protein. Biochim. Biophys. Acta Mol. Cell. Res. 1864: 217–230. 2017. https://doi.org/10.1016/j.bbamcr.2016.11.003
Belyaev N.D., Kellett K.A., Beckett C., Makova N.Z., Revett T.J., Nalivaeva N.N., Hooper N.M., Turner A.J. The transcriptionally active amyloid precursor protein (APP) intracellular domain is preferentially produced from the 695 isoform of APP in a β-secretase-dependent pathway J. Biol. Chem. 285: 41443–41454. 2010. https://doi.org/10.1074/jbc.M110.141390
Lorenzen A., Samosh J., Vandewark K., Anborgh P.H., Seah C., Magalhaes A.C., Cregan S.P., Ferguson S.S., Pasternak S.H. Rapid and direct transport of cell surface APP to the lysosome defines a novel selective pathway. Mol. Brain 3: 11. 2010. https://doi.org/10.1186/1756-6606-3-11
Yang M., Virassamy B., Vijayaraj S.L., Lim Y., Saadipour K., Wang Y.J., Han Y.C., Zhong J.H., Morales C.R., Zhou X.F. The intracellular domain of sortilin interacts with amyloid precursor protein and regulates its lysosomal and lipid raft trafficking. PLoS One. 8:e63049. 2013. https://doi.org/10.1371/journal.pone.0063049
Miranda A.M., Lasiecka Z.M., Xu Y., Neufeld J., Shahriar S., Simoes S., Chan R.B., Oliveira T.G., Small S.A., Di Paolo G. Neuronal lysosomal dysfunction releases exosomes harboring APP C-terminal fragments and unique lipid signatures. Nat. Commun. 9: 291. 2018. https://doi.org/10.1038/s41467-017-02533-w
Lauritzen I., Bécot A., Bourgeois A., Pardossi-Piquard R., Biferi M.G., Barkats M., Checler F. Targeting γ-secretase triggers the selective enrichment of oligomeric APP-CTFs in brain extracellular vesicles from Alzheimer cell and mouse models. Transl. Neurodegener. 8:35. 2019. https://doi.org/10.1186/s40035-019-0176-6
Rosas-Hernandez H., Cuevas E., Raymick J.B., Robinson B.L., Ali S.F., Hanig J., Sarkar S. Characterization of Serum Exosomes from a Transgenic Mouse Model of Alzheimer’s Disease. Curr. Alzheimer Res. 16: 388–395. 2019. https://doi.org/10.2174/1567205016666190321155422
Beckett, Nalivaeva N.N., Belyaev N.D., Turner A.J. Nuclear signalling by membrane protein intracellular domains: the AICD enigma. Cell Signal. 24: 402–409. 2012. https://doi.org/10.1016/j.cellsig.2011.10.007
Pardossi-Piquard R., Petit A., Kawarai T., Sunyach C., Alves da Costa C., Vincent B., Ring S., D’Adamio L., Shen J., Muller U., St George Hyslop P., Checler F. Presenilin-dependent transcriptional control of the Aβ-degrading enzyme neprilysin by intracellular domains of βAPP and APLP. Neuron. 46: 541–554. 2005. https://doi.org/10.1016/j.neuron.2005.04.008
Belyaev N.D., Nalivaeva N.N., Makova N.Z., Turner A.J. Neprilysin gene expression requires binding of the amyloid precursor protein intracellular domain to its promoter: implications for Alzheimer disease. EMBO Rep. 10: 94–100. 2009. https://doi.org/10.1038/embor.2008.222
Pardossi-Piquard R., Checler F. The physiology of the β-amyloid precursor protein intracellular domain AICD. J. Neurochem. 120(Suppl 1): 109–124. 2012. https://doi.org/10.1111/j.1471-4159.2011.07475.x
Nalivaeva N.N., Belyaev N.D., Kerridge C., Turner A.J. Amyloid-clearing proteins and their epigenetic regulation as a therapeutic target in Alzheimer’s disease. Front Aging Neurosci. 6: 235. 2014. https://doi.org/10.3389/fnagi.2014.00235
Kerridge C., Belyaev N.D., Nalivaeva N.N., Turner A.J. The Aβ-clearance protein transthyretin, like neprilysin, is epigenetically regulated by the amyloid precursor protein intracellular domain. J. Neurochem. 130: 419–431. 2014. https://doi.org/10.1111/jnc.12680
von Rotz R.C., Kohli B.M., Bosset J., Meier M., Suzuki T., Nitsch R.M., Konietzko U. The APP intracellular domain forms nuclear multiprotein complexes and regulates the transcription of its own precursor. J. Cell Sci. 117: 4435–4448. 2004. https://doi.org/10.1242/jcs.01323
Grimm M.O., Rothhaar T.L., Hartmann T. The role of APP proteolytic processing in lipid metabolism. Exp. Brain Res. 217: 365–375. 2012. https://doi.org/10.1007/s00221-011-2975-6
Grimm M.O., Grösgen S., Rothhaar T.L., Burg V.K., Hundsdörfer B., Haupenthal V.J., Friess P., Müller U., Fassbender K., Riemenschneider M., Grimm H.S., Hartmann T. Intracellular APP Domain Regulates Serine-Palmitoyl-CoA Transferase Expression and Is Affected in Alzheimer’s Disease. Int. J. Alzheimers Dis. 2011: 695413. 2011. https://doi.org/10.4061/2011/695413
Liu Q., Zerbinatti C.V., Zhang J., Hoe H.S., Wang B., Cole S.L., Herz J., Muglia L., Bu G. Amyloid precursor protein regulates brain apolipoprotein E and cholesterol metabolism through lipoprotein receptor LRP1. Neuron. 56: 66–78. 2007. https://doi.org/10.1016/j.neuron.2007.08.008
Hama E., Shirotani K., Iwata N., Saido T.C. Effects of neprilysin chimeric proteins targeted to subcellular compartments on amyloid β peptide clearance in primary neurons. J. Biol. Chem. 279: 30259–30264. 2004. https://doi.org/10.1074/jbc.M401891200
Bulloj A., Leal M.C., Surace E.I., Zhang X., Xu H., Ledesma M.D., Castaño E.M., Morelli L. Detergent resistant membrane-associated IDE in brain tissue and cultured cells: Relevance to Aβ and insulin degradation. Mol. Neurodegener. 3: 22. 2008. https://doi.org/10.1186/1750-1326-3-22
Kreps E.M., Avrova N.F., Chebotarëva M.A., Chirkovskaya E.V., Levitina M.V., Pomazanskaya L.F., Pravdina N.I. Some aspects of comparative biochemistry of brain lipids in teleost and elasmobranch fish. Comp. Biochem. Physiol. B. 52: 293–299. 1975. https://doi.org/10.1016/0305-0491(75)90067-x
Santos G., Díaz M., Torres N.V. Lipid Raft Size and Lipid Mobility in Non-raft Domains Increase during Aging and Are Exacerbated in APP/PS1 Mice Model of Alzheimer’s Disease. Predictions from an Agent-Based Mathematical Model. Front Physiol. 7: 90. 2016. https://doi.org/10.3389/fphys.2016.00090
Quinto- Marin R., Fabelo N., Fernández-Echevarría C., Canerina-Amaro A., Rodríguez-Barreto D., Alemany D., Mesa-Herrera F., Díaz M. Lipid Raft Alterations in Aged-Associated Neuropathologies. Curr. Alzheimer Res. 13: 973–984. 2016. https://doi.org/10.2174/1567205013666160314150017
Varma V.R., Oommen A.M., Varma S., Casanova R., An Y., Andrews R.M., O’Brien R., Pletnikova O., Troncoso J.C., Toledo J., Baillie R., Arnold M., Kastenmueller G., Nho K., Doraiswamy P.M., Saykin A.J., Kaddurah-Daouk R., Legido-Quigley C., Thambisetty M. Brain and blood metabolite signatures of pathology and progression in Alzheimer disease: A targeted metabolomics study. PLoS Med. 15: e1002482. 2018. https://doi.org/10.1371/journal.pmed.1002482
Kinoshita M., Suzuki K.G.N., Murata M., Matsumori N. Evidence of lipid rafts based on the partition and dynamic behavior of sphingomyelins. Chem. Phys. Lipids. 215: 84–95. 2018. https://doi.org/10.1016/j.chemphyslip.2018.07.002
Herget T., Esdar C., Oehrlein S.A., Heinrich M., Schütze S., Maelicke A., van Echten-Deckert G. Production of ceramides causes apoptosis during early neural differentiation in vitro. J. Biol. Chem. 275: 30344–30354. 2000.https://doi.org/10.1074/jbc.M000714200
Nalivaeva N.N., Rybakina E.G., Pivanovich I.Yu., Kozinets I.A., Shanin S.N., Bartfai T. Activation of neutral sphingomyelinase by IL-1β requires the type 1 interleukin 1 receptor. Cytokine. 12: 229–232. 2000. https://doi.org/10.1006/cyto.1999.0547
Clement A.B., Gamerdinger M., Tamboli I.Y., Lütjohann D., Walter J., Greeve I., Gimpl G., Behl C. Adaptation of neuronal cells to chronic oxidative stress is associated with altered cholesterol and sphingolipid homeostasis and lysosomal function. J. Neurochem. 111: 669–682. 2009. https://doi.org/10.1111/j.1471-4159.2009.06360.x
Mahfoud R., Garmy N., Maresca M., Yahi N., Puigserver A., Fantini J. Identification of a common sphingolipid-binding domain in Alzheimer, prion, and HIV-1 proteins. J. Biol. Chem. 277: 11292–11296. 2002. https://doi.org/10.1074/jbc.M111679200
Han X., Rozen S., Boyle S.H., Hellegers C., Cheng H., Burke J.R., Welsh-Bohmer K.A., Doraiswamy P.M., Kaddurah-Daouk R. Metabolomics in early Alzheimer’s disease: identification of altered plasma sphingolipidome using shotgun lipidomics. PLoS One. 6: e21643. 2011. https://doi.org/10.1371/journal.pone.0021643
Grimm M.O., Grimm H.S., Pätzold A.J., Zinser E.G., Halonen R., Duering M., Tschäpe J.A., De Strooper B., Müller U., Shen J., Hartmann T. Regulation of cholesterol and sphingomyelin metabolism by amyloid-β and presenilin. Nat. Cell. Biol. 7: 1118–1123. 2005. https://doi.org/10.1038/ncb1313
Jana A., Pahan K. Fibrillar amyloid-β-activated human astroglia kill primary human neurons via neutral sphingomyelinase: implications for Alzheimer’s disease. J. Neurosci. 30: 12676–12689. 2010. https://doi.org/10.1523/JNEUROSCI.1243-10.2010
Haughey N.J., Bandaru V.V., Bae M., Mattson M.P. Roles for dysfunctional sphingolipid metabolism in Alzheimer’s disease neuropathogenesis. Biochim. Biophys. Acta. 1801:878-886. 2010. https://doi.org/10.1016/j.bbalip.2010.05.003
Bienias K., Fiedorowicz A., Sadowska A., Prokopiuk S., Car H. Regulation of sphingomyelin metabolism. Pharmacol. Rep. 68: 570–581. 2016. https://doi.org/10.1016/j.pharep.2015.12.008
Lu M.H., Ji W.L., Xu D.E., Yao P.P., Zhao X.Y., Wang Z.T., Fang L.P., Huang R., Lan L.J., Chen J.B., Wang T.H., Cheng L.H., Xu R.X., Liu C.F., Puglielli L., Ma Q.H. Inhibition of sphingomyelin synthase 1 ameliorates alzheimer-like pathology in APP/PS1 transgenic mice through promoting lysosomal degradation of BACE1. Exp. Neurol. 311: 67–79. 2019. https://doi.org/10.1016/j.expneurol.2018.09.012
Stoffel W., Jenke B., Schmidt-Soltau I., Binczek E., Brodesser S., Hammels I. SMPD3 deficiency perturbs neuronal proteostasis and causes progressive cognitive impairment. Cell. Death Dis. 9:507. 2018. https://doi.org/10.1038/s41419-018-0560-7
Couttas T.A., Kain N., Tran C., Chatterton Z., Kwok J.B., Don A.S. Age-Dependent Changes to Sphingolipid Balance in the Human Hippocampus are Gender-Specific and May Sensitize to Neurodegeneration. J. Alzheimers Dis. 63: 503–514. 2018. https://doi.org/10.3233/JAD-171054
Furukawa K., Ohmi Y., Ohkawa Y., Tokuda N., Kondo Y., Tajima O., Furukawa K. Regulatory mechanisms of nervous systems with glycosphingolipids. Neurochem. Res. 36: 1578–1586. 2011. https://doi.org/10.1007/s11064-011-0494-2
Kracun I., Rosner H., Drnovsek V., Heffer-Lauc M., Cosović C., Lauc G. Human brain gangliosides in development, aging and disease. Int. J. Dev. Biol. 35: 289–295. 1991. PMID: .1814411
Ariga T., McDonald M.P., Yu R.K. Role of ganglioside metabolism in the pathogenesis of Alzheimer’s disease – a review. J. Lipid Res. 49: 1157–1175. 2008. https://doi.org/10.1194/jlr.R800007-JLR200
Kalanj S., Kracun I., Rosner H., Cosović C. Regional distribution of brain gangliosides in Alzheimer’s disease. Neurol. Croat. 40: 269–281. 1991. PMID: 1751644
Molander-Melin M., Blennow K., Bogdanovic N., Dellheden B., Månsson J.E., Fredman P. Structural membrane alterations in Alzheimer brains found to be associated with regional disease development; increased density of gangliosides GM1 and GM2 and loss of cholesterol in detergent-resistant membrane domains. J. Neurochem. 92: 171–182. 2005. https://doi.org/10.1111/j.1471-4159.2004.02849.x
Nishinaka T., Iwata D., Shimada S., Kosaka K., Suzuki Y. Anti-ganglioside GD1a monoclonal antibody recognizes senile plaques in the brains of patients with Alzheimer-type dementia. Neurosci. Res. 17: 171–176. 1993. https://doi.org/10.1016/0168-0102(93)90093-6
Chan R.B., Oliveira T.G., Cortes E.P., Honig L.S., Duff K.E., Small S.A., Wenk M.R., Shui G., Di Paolo G.J. Comparative lipidomic analysis of mouse and human brain with Alzheimer disease. Biol. Chem. 287: 2678–2688. 2012. https://doi.org/10.1074/jbc.M111.274142
Suzuki K.G.N., Ando H., Komura N., Fujiwara T., Kiso M., Kusumi A. Unraveling of Lipid Raft Organization in Cell Plasma Membranes by Single-Molecule Imaging of Ganglioside Probes. Adv. Exp. Med. Biol. 1104: 41–58. 2018. https://doi.org/10.1007/978-981-13-2158-0_3
Matsuzaki K. Aβ-ganglioside interactions in the pathogenesis of Alzheimer’s disease. Biochim. Biophys. Acta Biomembr. 3:183233. 2020.https://doi.org/10.1016/j.bbamem.2020.183233
Lemkul J.A., Bevan D.R. Lipid composition influences the release of Alzheimer’s amyloid β-peptide from membranes. Protein Sci. 20: 1530–1545. 2011. https://doi.org/10.1002/pro.678
Kakio A., Nishimoto S.I., Yanagisawa K., Kozutsumi Y., Matsuzaki K. Cholesterol-dependent formation of GM1 ganglioside-bound amyloid β-protein, an endogenous seed for Alzheimer amyloid. J. Biol. Chem. 276: 24985–24990. 2001. https://doi.org/10.1074/jbc.M100252200
Zha Q., Ruan Y., Hartmann T., Beyreuther K., Zhang D. GM1 ganglioside regulates the proteolysis of amyloid precursor protein. Mol. Psychiatry. 9: 946–952. 2004. https://doi.org/10.1038/sj.mp.4001509
Peters I., Igbavboa U., Schütt T., Haidari S., Hartig U., Rosello X., Böttner S., Copanaki E., Deller T., Kögel D., Wood W.G., Müller W.E., Eckert G.P. The interaction of β-amyloid protein with cellular membranes stimulates its own production. Biochim. Biophys. Acta. 1788: 964–972. 2009. https://doi.org/10.1016/j.bbamem.2009.01.012
Yamamoto N., Igbabvoa U., Shimada Y., Ohno-Iwashita Y., Kobayashi M., Wood W.G., Fujita S.C., Yanagisawa K. Accelerated Aβ aggregation in the presence of GM1-ganglioside-accumulated synaptosomes of aged apoE4-knock-in mouse brain. FEBS Lett. 569: 135–139. 2004. https://doi.org/10.1016/j.febslet.2004.05.037
Grimm M.O., Zinser E.G., Grösgen S., Hundsdörfer B., Rothhaar T.L., Burg V.K., Kaestner L., Bayer T.A., Lipp P., Müller U., Grimm H.S., Hartmann T. Amyloid precursor protein (APP) mediated regulation of ganglioside homeostasis linking Alzheimer’s disease pathology with ganglioside metabolism. PLoS One. 7: e34095. 2012. https://doi.org/10.1371/journal.pone.0034095
Svennerholm L., Bråne G., Karlsson I., Lekman A., Ramström I., Wikkelsö C. Alzheimer disease – effect of continuous intracerebroventricular treatment with GM1 ganglioside and a systematic activation programme. Dement. Geriatr. Cogn. Disord. 14: 128–136. 2002. https://doi.org/10.1159/000063604
Dai R., Zhang S., Duan W., Wei R., Chen H., Cai W., Yang L., Wang Q. Enhanced autophagy contributes to protective effects of GM1 ganglioside against Aβ1-42-induced neurotoxicity and cognitive deficits. Neurochem. Res. 42: 2417–2426. 2017. https://doi.org/10.1007/s11064-017-2266-0
Yuyama K., Sun H., Sakai S., Mitsutake S., Okada M., Tahara H., Furukawa J., Fujitani N., Shinohara Y., Igarashi Y. Decreased amyloid-β pathologies by intracerebral loading of glycosphingolipid-enriched exosomes in Alzheimer model mice. J. Biol. Chem. 289: 24488–24498. 2014. https://doi.org/10.1074/jbc.M114.577213
Matsuoka Y., Saito M., LaFrancois J., Saito M., Gaynor K., Olm V., Wang L., Casey E., Lu Y., Shiratori C., Lemere C., Duff K. Novel therapeutic approach for the treatment of Alzheimer’s disease by peripheral administration of agents with an affinity to β-amyloid. J. Neurosci. 23: 29–33. 2003. https://doi.org/10.1523/JNEUROSCI.23-01-00029.2003
Goodfellow J.A., Willison H.J. Gangliosides and Autoimmune Peripheral Nerve Diseases. Prog Mol Biol Transl Sci. 156: 355–382. 2018. https://doi.org/10.1016/bs.pmbts.2017.12.010
Magistretti P.J., Geisler F.H., Schneider J.S., Li P.A., Fiumelli H., Sipione S. Gangliosides: Treatment Avenues in Neurodegenerative Disease. Front Neurol. 10: 859. 2019. https://doi.org/10.3389/fneur.2019.00859
Allinquant B., Clamagirand C., Potier M.C. Role of cholesterol metabolism in the pathogenesis of Alzheimer’s disease. Curr. Opin. Clin. Nutr. Metab. Care. 17: 319–323. 2014. https://doi.org/10.1097/MCO.0000000000000069
Eckert G.P., Hooff G.P., Strandjord D.M., Igbavboa U., Volmer D.A., Müller W.E., Wood W.G. Regulation of the brain isoprenoids farnesyl- and geranylgeranylpyrophosphate is altered in male Alzheimer patients. Neurobiol. Dis. 35: 251–257. 2009.https://doi.org/10.1016/j.nbd.2009.05.005
Cho Y.Y., Kwon O.H., Park M.K., Kim T.W., Chung S. Elevated cellular cholesterol in Familial Alzheimer’s presenilin 1 mutation is associated with lipid raft localization of β-amyloid precursor protein. PLoS One. 14: e0210535. 2019. https://doi.org/10.1371/journal.pone.0210535
Leduc V., Jasmin-Belanger S., Poirier J. APOE and cholesterol homeostasis in Alzheimer’s disease. Trends Mol. Med. 16: 469–477. 2010. https://doi.org/10.1016/j.molmed.2010.07.008
Kojro E., Gimpl G., Lammich S., Marz W., Fahrenholz F. Low cholesterol stimulates the nonamyloidogenic pathway by its effect on the α-secretase ADAM 10. Proc. Natl. Acad. Sci. USA. 98: 5815–5820. 2001.
Igbavboa U., Sun G.Y., Weisman G.A., He Y., Wood W.G. Amyloid β-protein stimulates trafficking of cholesterol and caveolin-1 from the plasma membrane to the Golgi complex in mouse primary astrocytes. Neuroscience. 162: 328–338. 2009. https://doi.org/10.1016/j.neuroscience.2009.04.049
Brown A.M., Bevan D.R. Influence of sequence and lipid type on membrane perturbation by human and rat amyloid β-peptide (1-42). Arch. Biochem. Biophys. 614: 1–13. 2017. https://doi.org/10.1016/j.abb.2016.11.006
Koudinov A.R., Koudinova N.V., Berezov T.T. Alzheimer’s peptides Aβ1-40 and Aβ1-28 inhibit the plasma cholesterol esterification rate. Biochem. Mol. Biol. Int. 38: 747–752. 1996. PMID: 8728104
Grosgen S., Grimm M.O., Friess P., Hartmann T. Role of amyloid β in lipid homeostasis. Biochim. Biophys. Acta 1801: 966–974. 2010. https://doi.org/10.1016/j.bbalip.2010.05.002
McFarlane O., Kędziora-Kornatowska K. Cholesterol and Dementia: A Long and Complicated Relationship. Curr. Aging Sci. 2019 Sep 17. [Epub ahead of print].https://doi.org/10.2174/1874609812666190917155400
Jeong A., Suazo K.F., Wood W.G., Distefano M.D. Isoprenoids and protein prenylation: implications in the pathogenesis and therapeutic intervention of Alzheimer’s disease. Crit. Rev. Biochem. Mol. Biol. 53: 279–310. 2018. https://doi.org/10.1080/10409238.2018.1458070
Petek B., Villa-Lopez M., Loera-Valencia R., Gerenu G., Winblad B., Kramberger M.G., Ismail M., Eriksdotter M., Garcia-Ptacek S. Connecting the brain cholesterol and renin-angiotensin systems: potential role of statins and RAS-modifying medications in dementia. J. Intern. Med. 284: 620–642. 2018. https://doi.org/10.1111/joim.12838
Zandl-Lang M., Fanaee-Danesh E., Sun Y., Albrecher N.M., Gali C.C., Čančar I., Kober A., Tam-Amersdorfer C., Stracke A., Storck S.M., Saeed A., Stefulj J., Pietrzik C.U., Wilson M.R., Björkhem I., Panzenboeck U. Regulatory effects of simvastatin and apoJ on APP processing and amyloid-β clearance in blood-brain barrier endothelial cells. Biochim. Biophys. Acta Mol. Cell Biol. Lipids. 1863: 40–60. 2018. https://doi.org/10.1016/j.bbalip.2017.09.008
Shinohara M., Sato N., Kurinami H., Takeuchi D., Takeda S., Shimamura M., Yamashita T., Uchiyama Y., Rakugi H., Morishita R. Reduction of brain Aβ by fluvastatin, an HMG-CoA reductase inhibitor, through increase in degradation of APP-CTFs and Aβ clearance. J. Biol. Chem. 285: 22091–22102. 2010. https://doi.org/10.1074/jbc.M110.102277
Etcheberrigaray R., Tan M., Dewachter I., Kuipéri C., Van der Auwera I., Wera S., Qiao L., Bank B., Nelson T.J., Kozikowski A.P., Van Leuven F., Alkon D.L. Therapeutic effects of PKC activators in Alzheimer’s disease transgenic mice. Proc. Natl. Acad. Sci. USA. 101: 11141–11146. 2004. https://doi.org/10.1073/pnas.0403921101
Sun M.K., Alkon D.L. Dual effects of bryostatin-1 on spatial memory and depression. Eur. J. Pharmacol. 512: 43–51. 2005. https://doi.org/10.1016/j.ejphar.2005.02.028
Crestini A., Piscopo P., Iazeolla M., Albani D., Rivabene R., Forloni G., Confaloni A. Rosuvastatin and thapsigargin modulate γ-secretase gene expression and APP processing in a human neuroglioma model. J. Mol. Neurosci. 43: 461–469. 2011.https://doi.org/10.1007/s12031-010-9465-3
Huang M., Hu M., Song Q., Song H., Huang J., Gu X., Wang X., Chen J., Kang T., Feng X., Jiang D., Zheng G., Chen H., Gao X. GM1-Modified Lipoprotein-like Nanoparticle: Multifunctional Nanoplatform for the Combination Therapy of Alzheimer’s Disease. ACS Nano. 9: 10801–10816. 2015. https://doi.org/10.1021/acsnano.5b03124
Frisardi V., Panza F., Seripa D., Farooqui T., Farooqui A.A. Glycerophospholipids and glycerophospholipid-derived lipid mediators: a complex meshwork in Alzheimer’s disease pathology. Prog. Lipid Res. 50: 313–30. 2011. https://doi.org/10.1016/j.plipres.2011.06.001
Díaz M., Fabelo N., Martín V., Ferrer I., Gómez T., Marín R. Biophysical alterations in lipid rafts from human cerebral cortex associate with increased BACE1/AβPP interaction in early stages of Alzheimer’s disease. J. Alzheimers Dis. 43: 1185–1198. 2015. https://doi.org/10.3233/JAD-141146
Grimm M.O.W., Michaelson D.M., Hartmann T. Omega-3 fatty acids, lipids, and apoE lipidation in Alzheimer’s disease: a rationale for multi-nutrient dementia prevention. J. Lipid Res. 58: 2083–2101. 2017. https://doi.org/10.1194/jlr.R076331
Zhang X.L., Zhao N., Xu B., Chen X.H., Li T.J. Treadmill exercise inhibits amyloid-β generation in the hippocampus of APP/PS1 transgenic mice by reducing cholesterol-mediated lipid raft formation. Neuroreport. 307: 498–503. 2019. https://doi.org/10.1097/WNR.0000000000001230
Zhang L., Han X., Wang X. Is the clinical lipidomics a potential goldmine? Cell. Biol. Toxicol. 34: 421–423. 2018. https://doi.org/10.1007/s10565-018-9441-1
Hardy J., Escott-Price V. Genes, pathways and risk prediction in Alzheimer’s disease. Hum. Mol. Genet. 28: 235–240. 2019. https://doi.org/10.1093/hmg/ddz163
Дополнительные материалы отсутствуют.
Инструменты
Российский физиологический журнал им. И.М. Сеченова