

Российский физиологический журнал им. И.М. Сеченова, 2021, T. 107, № 11, стр. 1385-1394
Роль NO-синтазных путей в реализации влияния провоспалительных цитокинов на паттерн дыхания и вентиляционный ответ на гипоксию
А. А. Клинникова 1, *, Г. А. Данилова 1, Н. П. Александрова 1
1 Институт физиологии им. И.П. Павлова РАН
Санкт-Петербург, Россия
* E-mail: Klinnikova.an@gmail.com
Поступила в редакцию 21.08.2021
После доработки 12.09.2021
Принята к публикации 12.09.2021
Аннотация
Интерлейкин-1β (IL-1β) и фактор некроза опухоли-α (TNF-α) являются основными провоспалительными цитокинами. Их рецепторы экспрессируются в областях ствола мозга, участвующих в контроле дыхания, а также в каротидных телах, которые контролируют содержание O2 в артериальной крови. Мы предположили, что циркулирующие провоспалительные цитокины могут влиять на вентиляцию легких и модулировать респираторный ответ на гипоксию посредством активации NO-зависимых путей. Целью нашего исследования было сравнение респираторных эффектов IL-1β и TNF-α до и после предварительной обработки L-NAME – неселективным ингибитором NO-синтазы (NOS). Вентиляционный ответ на гипоксию измеряли у анестезированных крыс линии Вистар с использованием метода возвратного дыхания до и после внутривенного введения IL-1β (2 мкг/кг) и TNF-α (40 мкг/кг). В результате было обнаружено, что повышение системного уровня провоспалительных цитокинов увеличивает вентиляцию легких при нормоксии, в то же время снижая респираторную чувствительность к гипоксии. Предварительное интраперитонеальное введение L-NAME снижало обнаруженные респираторные эффекты как IL-1β, так и TNF-α. Мы полагаем, что активация NO-синтазных путей и усиление синтеза NO при взаимодействии цитокинов с соответствующими рецепторами, опосредует респираторные эффекты провоспалительных цитокинов и лежит в основе воздействия воспаления на дыхательную функцию.
ВВЕДЕНИЕ
Гипоксический и гиперкапнический дыхательные хеморефлексы являются важнейшими элементами контроля дыхания. Эти рефлексы участвуют в поддержании газового гомеостаза артериальной крови. Хеморефлексы осуществляются при участии хеморецепторов каротидных телец, расположенных в бифуркации сонной артерии, которые возбуждаются при снижении напряжения кислорода, повышении напряжения углекислого газа и уменьшении pH артериальной крови. При гипоксии гломусные клетки каротидных телец деполяризуются в ответ на недостаток кислорода и выделяют нейромедиаторы, которые активируют сенсорные нервные волокна, передающие афферентную информацию в дыхательный центр ствола мозга.
Установлено, что гломусные клетки экспрессируют рецепторы воспалительных цитокинов, включая рецепторы TNF-α (TNF-R1 и TNF-R2), IL-1β (IL-1R1) и рецепторы IL-6 [1–4]. IL-1β и TNF-α являются основными провоспалительными цитокинами, продуцируемыми во время острой фазы иммунного ответа на инфекцию и воспаление. Сообщается, что системный уровень этих цитокинов повышается при многих респираторных заболеваниях, таких как астма, хроническая обструктивная болезнь легких и апноэ во сне [5–7]. Было обнаружено, что системное воспаление вызывает морфологические изменения в каротидном теле сонной артерии [3]. Эти изменения, связанные с повышением уровня провоспалительных цитокинов, снижают чувствительность каротидного тела к гипоксии [4]. Известно, что TNF-α может провоцировать высвобождение гломусными клетками медиатора дофамина [8, 9]. В совокупности, эти данные показывают, что функция каротидных телец при гипоксии может снижаться во время воспаления.
Однако механизмы, с помощью которых провоспалительные цитокины влияют на вентиляцию легких и гипоксическую хеморецепцию, до сих пор не изучены и могут включать множество воспалительных молекул, которые влияют не только на центральные и периферические хеморецепторы, но и на дыхательные нейроны [10]. Влияние цитокинов на дыхательную систему может быть опосредовано несколькими способами: высвобождением простагландинов, норадреналина, рилизинг-фактора кортикотропина, оксида азота (NO) [11–15].
Целью настоящего исследования было изучение влияния провоспалительных цитокинов IL-1β и TNF-α на паттерн дыхания и гипоксическую хеморецепцию, а также выяснение роли NO-синтазных путей в реализации респираторных эффектов данных цитокинов.
МЕТОДЫ ИССЛЕДОВАНИЯ
Исследование было выполнено на 48 наркотизированных трахеостомированных спонтанно дышащих крысах самцах линии Вистар массой 270 ± 20 г (ЦКП Биоколлекция Института физиологии им. И.П. Павлова РАН). Все животные находились под общей анестезией (уретан, 1400 мг/кг, интраперитонеально). Эксперименты выполнены с соблюдением основных норм и правил биомедицинской этики (Europian Community Council Directives 86/609/EEC).
Для регистрации объемно-временных параметров внешнего дыхания использовалась пневмотахографическая методика. К трахеостомической канюле подключалась пневмометрическая трубка MLT-1L (AD Instruments, Австралия). По кривой пневмотахограммы измерялась средняя скорость инспираторного потока (Vинсп) и частота дыхательных движений (ЧДД). При интеграции пневмотахографической кривой автоматически получали кривую дыхательных объемов – спирограмму и вычисляли дыхательный объем (ДО). Минутный объем дыхания (МОД) рассчитывали, как произведение ДО на ЧДД. Парциальное давление кислорода и углекислого газа (PEТO2 и PEТСO2) в конечной порции выдыхаемого воздуха измерялось с помощью респираторного газоанализатора (Gemini, США).
Животные были разделены на 6 экспериментальных групп. Животные первой группы (n = 8) использовались для выяснения собственных респираторных эффектов IL-1β, этим животным вводили только IL-1β (2 мкг/кг, “Беталейкин”, ФГУП ОЧБ ФМБА) в хвостовую вену. Вторая группа (n = 8) была предназначена для выяснения респираторных эффектов TNF-α, этой группе внутривенно вводили TNF-α (40 мкг/кг, Sigma). Третья и четвертая группы (n = 8 каждая) были созданы для выяснения роли нитрергических механизмов, участвующих в реализации респираторных эффектов цитокинов. Животным этих групп за 20 мин до введения IL-1β или TNF-α производилась внутрибрюшинная инъекция неселективного ингибитора NO-синтаз (L-NAME) (10 мг/кг, Sigma). Пятая группа (n = 8) использовалась для выявления возможного собственного влияния L-NAME на вентиляцию легких и гипоксическую хеморецепцию; этой группе вводили только ингибитор, внутрибрюшинно. Помимо этого, была создана группа животных (n = 8), которая являлась контрольной. Этим животным вводили внутривенно 0.25 мл физиологического раствора.
Чувствительность к гипоксическому стимулу исследовали классическим методом возвратного дыхания, адаптированным нами для использования на мелких лабораторных животных. Дыхание производилось в замкнутом контуре, заполненном азотно-кислородной гипоксической газовой смесью (80% N2, 15% О2, 5% СО2). СО2 добавлялся в дыхательную смесь в концентрации, соответствующей нормальному содержанию СО2 в организме для предотвращения гипокапнии, развивающейся при гипоксической гипервентиляции.
Изокапния поддерживалась за счет удаления с помощью адсорбента (натронная известь) из выдыхаемого воздуха, поступающего в мешок (объем 50 мл) для возвратного дыхания, углекислого газа, образующегося в организме. По мере потребления кислорода при дыхании из мешка происходило постепенное убывание содержания О2 в дыхательной смеси, нарастала стимуляция периферических хеморецепторов и происходило соответствующее увеличение легочной вентиляции. Содержание СО2 в дыхательной смеси не изменялось. Продолжительность проведения пробы с возвратным дыханием составляла 4 мин. Вентиляционный ответ на гипоксию тестировался в диапазоне снижения парциального давления кислорода в выдыхаемом воздухе от 80 до 40 мм рт. ст., т.к. в этом диапазоне зависимость величины вентиляции от интенсивности гипоксического стимула практически линейна.
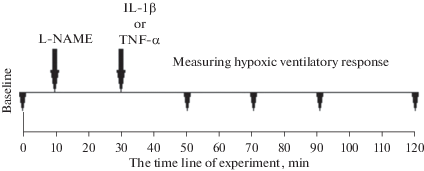
Для количественной оценки вентиляционного ответа на гипоксию производилось вычисление приростов ДО, МОД и Vинсп при снижении парциального давления кислорода в конечной порции выдыхаемого воздуха на 1 мм рт. ст. Кроме того, производилось графическое построение зависимости роста вентиляции и ее составляющих от содержания О2 в выдыхаемом воздухе. Гипоксический вентиляционный ответ тестировался до введения препаратов, а затем на 20-ой, 40-ой, 60-ой и 90-ой мин после их введения (рис. 1).
Рис. 1.
Схема эксперимента. Эксперимент длился 120 мин, на тридцатой минуте вводили IL-1β или TNF-α. L-NAME вводили на десятой минуте эксперимента (т.е. за 20 мин до введения цитокина). Треугольники – моменты измерения дыхательной реакции при гипоксии. Стрелки – моменты введения препаратов.
Для статистической обработки экспериментальных данных использовался программный пакет STATISTICA 7.0. Все значения представлены как среднее ± стандартная ошибка. Для проверки нормальности распределения данных применялись критерии Колмогорова–Смирнова и Шапиро–Уилка. Значения до и после введения препаратов оценивали с помощью парного теста Стьюдента и однофакторного дисперсионного анализа (ANOVA). Достоверными считали различия при p < 0.05.
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
Паттерн дыхания. Эксперименты показали, что экзогенное повышение системного уровня как IL-1β, так и TNF-α оказывает активирующее влияние на систему внешнего дыхания, вызывая увеличение средней скорости инспираторного потока, дыхательного объема и минутного объема дыхания (табл. 1, 2). Достоверные изменения минутного объема дыхания отмечались уже на 20-ой мин после введения цитокинов, средней скорости инспираторного потока и дыхательного объема – на 40-ой мин. Значимое увеличение частоты дыхательных движений наблюдалось лишь через 90 мин после введения IL-1β или TNF-α. При проведении серии контрольных экспериментов с внутривенным введением физиологического раствора, а также с внутрибрюшинным введением раствора L-NAME не было выявлено достоверных изменений дыхательных параметров.
Таблица 1.
Изменение параметров дыхания при повышении системного уровня IL-1β и IL-1β на фоне действия L-NAME
| Параметр | Физиологический раствор (n = 8) | IL-1β (n = 8) |
IL-1β +L-NAME (n = 8) |
||||||
|---|---|---|---|---|---|---|---|---|---|
| фон | 40 мин | 60 мин | фон | 40 мин | 60 мин | фон | 40 мин | 60 мин | |
| МОД, мл/мин | 100 ± 5.2 | 97 ± 4.0 | 96 ± 9.5 | 117 ± 9.6 | 143 ± 12.8* | 146 ± 12.0* | 210 ± 8.3 | 237 ± 10.1 | 244 ± 9.3* |
| ДО, мл | 1.0 ± 0.02 | 0.9 ± 0.05 | 1.0 ± 0.08 | 1.0 ± 0.08 | 1.36 ± 0.07* | 1.4 ± 0.07* | 1.8±0.08 | 1.9 ± 0.07# | 1.9 ± 0.07# |
| ЧДД, мин–1 | 107 ± 2.0 | 105 ± 4.0 | 105 ± 2.6 | 113 ± 7.0 | 106 ± 9.0 | 105 ± 8.0 | 114 ± 3.3 | 118 ± 2.6 | 122 ± 2.7# |
| Vинсп, мл/с–1 | 3.8 ± 0.3 | 3.9 ± 0.2 | 3.9 ± 0.1 | 3.8 ± 0.5 | 4.2 ± 0.4* | 4.3 ± 0.4* | 7.8 ± 0.3 | 8.7 ± 0.3 | 8.8 ± 0.3 |
Таблица 2.
Изменение параметров дыхания при повышении системного уровня TNF-α и TNF-α на фоне действия L-NAME
| Параметр | Физиологический раствор (n = 8) |
TNF-α (n = 8) |
TNF-α + L-NAME (n = 8) |
||||||
|---|---|---|---|---|---|---|---|---|---|
| фон | 40 мин | 60 мин | Фон | 40 мин | 60мин | фон | 40 мин | 60 мин | |
| МОД, мл/мин | 100 ± 5.2 | 97 ± 4.0 | 96 ± 9.5 | 119 ± 15.6 | 172 ± 13.8* | 179 ± 11.5* | 132 ± 8.8 | 138 ± 7.0# | 145.2 ± 7.0*# |
| ДО, мл | 1.0 ± 0.02 | 0.9 ± 0.05 | 1.0 ± 0.08 | 1.1 ± 0.07 | 1.3 ± 0.06* | 1.4 ± 0.05* | 1.3 ± 0.06 | 1.2 ± 0.07# | 1.2 ± 0.06# |
| ЧДД, мин–1 | 107 ± 2.4 | 105 ± 3 | 105 ± 2.6 | 111 ± 5.1 | 114 ± 5.3 | 119 ± 6.1 | 108 ± 5.6 | 112 ± 4.8 | 110 ± 6.0 |
| Vинсп, мл/с–1 | 3.8 ± 0.3 | 3.9 ± 0.2 | 3.9 ± 0.1 | 4.2 ± 0.4 | 5.4 ± 0.5* | 5.8 ± 0.3* | 4.2 ± 0.3 | 4.3 ± 0.2# | 4.4 ± 0.4# |
Обозначения такие же, как в табл. 1.
После предварительной обработки L-NAME влияние IL-1β и TNF-α на паттерн дыхания было ослаблено. Достоверное увеличение минутного объема дыхания наблюдалось только через 60 мин после повышения системного уровня данных цитокинов (табл. 1, 2). Величина прироста этого параметра составляла 25 и 50% при введении IL-1β и TNF-α соответственно. При действии IL-1β и TNF-α на фоне предварительного введения L-NAME прирост минутного объема дыхания через 60 мин после введения цитокинов составлял 16 и 10%.
Вентиляционный ответ на гипоксию. При возвратном дыхании гипоксической газовой смесью наблюдалась значительная корреляция между увеличением минутной вентиляции, дыхательного объема, средней скорости нспираторного потока и снижением PETO2 как до, так и после внутривенных инъекций IL-1β или TNF-α. Однако внутривенное введение как IL-1β, так и TNF-α вызывало уменьшение угла наклона линий тренда, усредняющих вентиляционные кривые, зарегистрированные в нескольких экспериментах. Линии тренда становились более пологими, что свидетельствует о снижении вентиляционной чувствительности к гипоксической стимуляции (рис. 2a, b; 3a, b).
Рис. 2.
Вентиляционный ответ на гипоксию до (сплошная линия) и через 40 мин после (пунктирная линия) внутривенного введения IL-1β (a, b) и IL-1β на фоне действия L-NAME (c, d).
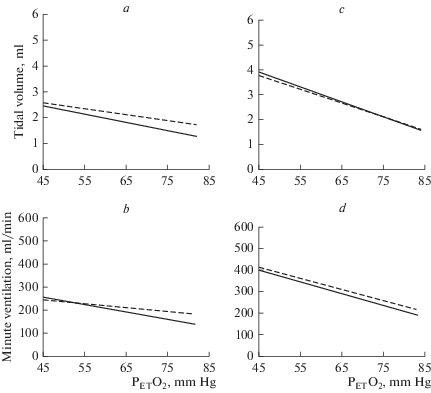
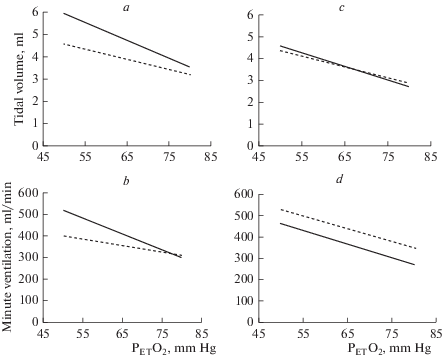
Рис. 3.
Вентиляционный ответ на гипоксию до (сплошная линия) и через 40 мин после (пунктирная линия) внутривенного введения TNF-α (a, b) и TNF-α на фоне действия L-NAME (c, d).
Проведение количественных расчетов подтвердило достоверность снижения прироста респираторных параметров в ответ на гипоксическую стимуляцию на фоне действия IL-1β. Максимальное снижение приростов дыхательных параметров наблюдалось на 40-ой мин действия цитокина. Так, расчет величины прироста параметров при снижении РETО2 на 1 мм рт. ст. показал, что через 40 мин после введения IL-1β прирост МОД уменьшался с 3.34 ± 0.23 до 2.02 ± 0.19 мл/мин/мм рт. ст. (–41%, p < 0.05), ДО – с 0.031 ± 0.005 в контроле до 0.022 ± 0.004 мл/мм рт. ст. (–29%, p < 0.05) и скорости инспираторного потока с 0.114 ± 0.017 до 0.06 ± 0.012 мл/с/мм рт. ст. (–47%, p < 0.05) по сравнению с фоновыми величинами (рис. 4а).
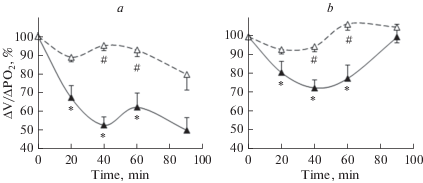
Рис. 4.
Динамика вентиляционного ответа на гипоксию на протяжении всего эксперимента, т.е. на исходном уровне и через 20, 40, 60 и 90 мин после введения IL-1β (a) или TNF-α (b), до (сплошная линия) и после (пунктирная линия) предварительного введения L-NAME. * p < 0.05 – достоверные отличия от фоновых значений, # p < 0.05 – достоверные отличия от IL-1β или TNF-α. Представлены средние значения ± SEM.
Максимальное снижение чувствительности респираторной системы к гипоксии было обнаружено и через 40 мин после введения TNF-α. Количественные расчеты показали, что МОД снизился с 6.06 ± 0.91 до 3.48 ± 0.38 мл/мин/мм рт. ст. (–40%) через 40 мин после введения TNF-α (p < 0.05). ДО и Vинсп. снизились с 0.06 ± 0.01 до 0.04 ± 0.01 мл/мм рт. ст. (–27%, p < 0.05) и с 0.23 ± 0.02 в контроле до 0.15 ± ± 0.01 мл/с/мм рт. ст. (–27%, p < 0.05) через 40 мин после введения TNF-α соответственно (рис. 4b). В то же время, внутривенное введение физиологического раствора не изменило вентиляционный ответ на гипоксию.
В совокупности полученные данные показывают, что повышенный уровень провоспалительных цитокинов в крови снижает чувствительность дыхательной системы к гипоксии и подавляет гипоксический хеморецепторный контроль дыхания.
Повышение системного уровня как IL-1β, так и TNF-α на фоне действия ингибитора NO-синтаз – L-NAME не вызывало ослабления вентиляционного ответа на гипоксию: угол наклона линий тренда, характеризующий зависимость дыхательных параметров (МОД, ДО, Vинсп) от величины гипоксической стимуляции, не изменялся после введения цитокинов (рис. 2c, d; 3c, d).
Количественная оценка реакции на гипоксию после введения цитокинов на фоне ингибирования активности NO-синтаз также показала, что в течение всего эксперимента достоверного снижения приростов МОД, ДО и Vинсп в ответ на гипоксическую стимуляцию, относительно их фоновых значений, не наблюдалось. Сохранялась лишь слабо выраженная тенденция к ослаблению вентиляционного ответа на гипоксию (рис. 4).
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
В нормальных условиях уровень цитокинов в организме очень низкий. Однако при системном воспалении, стрессе, гипоксии, обструктивной болезни легких наблюдается значительный рост уровня провоспалительных цитокинов. Известно, что внутривенное введение липополисахарида вызывает повышение уровня TNF в крови с 750 до 5000 нг/мл [16].
Проведенное нами исследование показало, что при спокойном дыхании воздухом экзогенное повышение уровня IL-1β и TNF-α вызывает увеличение минутной вентиляции, дыхательного объема и средней скорости инспираторного потока. Подобное влияние провоспалительных цитокинов на вентиляцию легких было показано и в более ранних исследованиях. Было обнаружено, что вводимый внутривенно эндотоксин, который приводит к высвобождению провоспалительных цитокинов (IL-1β, IL-6 и TNF-α), вызывает усиление вентиляции легких [17]. В предыдущих исследованиях мы показали, что повышение церебрального уровня IL-1β приводит к значительному увеличению объемно-временных параметров дыхания в условиях нормоксии, но в то же время ослабляет гиперкапнический и гипоксический вентиляционные ответы [15, 18]. Результаты, представленные в данной статье, показывают, что внутривенное введение IL-1β и TNF-α имеет такие же респираторные эффекты: усиление вентиляции легких и ослабление респираторной чувствительности к гипоксии.
На основании полученных нами данных мы полагаем, что повышение системного уровня провоспалительных цитокинов снижает чувствительность периферических артериальных хеморецепторов к изменению газового состава крови. Этот вывод подтверждается результатами, полученными в других исследованиях. Установлено, что IL-1β и TNF-α могут распознаваться мембранными рецепторами, расположенными в гломусных клетках каротидных телец, а также могут способствовать выделению гломусными клетками медиатора дофамина [8, 9]. В экспериментах, проведенных in vitro, установлено, что в каротидном теле TNF-α может увеличивать базовую частоту хемосенсорных разрядов нерва, иннервирующего каротидные тела, но в тоже время – уменьшать хемосенсорные разряды, вызванные гипоксией [8]. В нашей работе, в экспериментах in vivo, было показано, что внутривенная инъекция как IL-1β, так и TNF-α увеличивает минутную вентиляцию легких при спокойном дыхании воздухом, и в то же время снижает вентиляционный ответ респираторной системы на гипоксию. Принимая во внимание представленные данные литературы, можно предположить, что обнаруженные нами респираторные эффекты провоспалительных цитокинов связаны с тем, что повышение их уровня в циркулирующей крови изменяет хемосенсорную активность каротидного тела.
Результаты представленного исследования показывают также, что респираторные эффекты провоспалительных цитокинов опосредуются активацией NO-синтазных путей. Наличие NO-синтаз было обнаружено в нервных волокнах, окружающих гломусные клетки каротидных тел, а также в самих гломусных клетках [19]. При взаимодействии цитокинов с соответствующими мембранными рецепторами в этих клетках может усиливаться синтез конститутивных форм NO-синтазы и образование молекул NO, которые оказывают как аутокринное, так и паракринное действие. Внутриклеточный механизм действия NO заключается в активизации циклического гуанозинмонофосфата (цГМФ), способного влиять на проводимость ионных каналов и, таким образом, изменять функциональное состояние клеточной мембраны.
Установлено, что оксид азота в физиологических концентрациях является тормозным модулятором хемосенсорных разрядов каротидных тел. Уменьшение эндогенной продукции NO вносит вклад в усиление хемосенсорных разрядов каротидных тел [20, 21]. Известно, что NO модулирует хеморецепторные процессы посредством различных механизмов, опосредовано через изменения тонуса сосудов в каротидных телах и доставки кислорода, и напрямую через модуляцию возбудимости гломусных клеток и сенсорных нейронов. При этом эффект NO имеет двойственное дозозависимое влияние на хеморецепцию каротидных тел. В гипоксических условиях NO является преимущественно тормозным модулятором каротидной хеморецепции, тогда как при нормоксии NO усиливает хемосенсорные разряды [20, 21]. Этот факт объясняет обнаруженный нами двойственный респираторный эффект IL-1β и TNF-α: увеличение базовой вентиляции при нормоксии, но уменьшение гипоксического вентиляционного ответа.
Полученные нами данные, указывающие на восстановление вентиляционного гипоксического ответа после ингибирования NO-синтаз, находят объяснения и в других исследованиях, которые показывают, что уменьшенная эндогенная продукция NO усиливает хеморецепторную активность каротидных тел. Так, например, было установлено, что у кроликов с сердечной недостаточностью и усиленным ответом на гипоксию наблюдается уменьшенная эндогенная активность NO-синтаз в каротидных телах по сравнению с нормальными животными. При этом было показано, что внедрение этим животным аденовируса, экспрессирующего NO-синтазы, уменьшало базовые разряды хеморецепторов каротидных тел и ослабляло ответ на гипоксию [22]. В экспериментах in vitro на перфузированных каротидных телах кошек при регистрации активности синусного нерва было показано, что обе конститутивные изоформы NO-синтазы – нейрональная (nNOS) и эндотелиальная (еNOS) – вносят вклад в действие NO на хеморецепторную активность [23].
Таким образом, результаты проведенных нами исследований и анализ данных литературы указывают на участие ключевых воспалительных цитокинов IL-1β и TNF-α в модуляции паттерна дыхания и гипоксического хеморефлекса посредством активации NO-синтаз и усиления синтеза NO в гломусных клетках каротидных тел.
Список литературы
Wang X, Wang BR, Duan X L, Zhang P, Ding YQ, Jia Y, Jiao XY, Ju G (2002) Strong expression of interleukin-1 receptor types I in the rat carotid body. J Histochem Cytochem 50 (12): 1677–1684. https://doi.org/10.1177/002215540205001213
Wang X, Zhang XJ, Xu Z, Li X, Li G L, Ju G, Wang BR (2006) Morphological evidence for existence of IL-6 receptor alpha in the glomus cells of rat carotid body. Anat Rec A Discov Mol Cell Evol Biol 288 (3): 292–296. https://doi.org/10.1002/ar.a.20310
Lam SY, Tipoe GL, Liong EC, Fung ML (2008) Chronic hypoxia upregulates the expression and function of proinflammatory cytokines in the rat carotid body. Histochem Cell Biol 130 (3): 549–559. https://doi.org/10.1007/s00418-008-0437-4
Gauda EB, Shirahata M, Masona A, Pichard LE, Kostuk EW, Chavez-Valdeza R (2013) Inflammation in the carotid body during development and its contribution to apnoea of prematurity. Respir Physiol Neurobiol 185 (1): 120–131. https://doi.org/10.1016/j.resp.2012.08.005
Vgontzas AN, Papanicolaou DA, Bixler EO (2000) Sleep apnoea and daytime sleepiness and fatigue: relation to visceral obesity, insulin resistance, and hypercytokinemia. J Clin Endocrinol Metab 85: 1151–1158.
Vernooy JH, Kucukaycan M, Jacobs JA, Chavannes NH, Buurman WA, Dentener MA, Wouters EF (2002) Local and systemic inflammation in patients with chronic obstructive pulmonary disease: soluble tumor necrosis factor receptors are increased in sputum. Am J Respir Crit Care Med 166: 1218–1224.
Bucchioni E, Kharitonov SA, Allegra L, Barnes PJ (2003) High levels of interleukin-6 in the exhaled breath condensate of patients with COPD. Respir Med 97: 1299–1302.
Fernández R, González S, Rey S, Cortés PP, Maisey KR, Reyes EP, Larraín C, Zapata P (2008) Lipopolysaccharide-induced carotid body inflammation in cats: functional manifestations, histopathology and involvement of tumour necrosis factor-alpha. Exp Physiol 93 (7): 892–907. https://doi.org/10.1113/expphysiol.2008.041152
Zapata P, Larrain C, Reyes P, Fernández R (2011) Immunosensory signaling by carotid body chemoreceptors. Respir Physiol Neurobiol 178 (3): 370–374. https://doi.org/doi:10.1016/j. resp.2011.03.025
Huxtable AG, Vinit S, Windelborn JA, Crader SM, Guenther CH, Watters JJ, Mitchell GS (2011) Systemic inflammation impairs respiratory chemoreflexes and plasticity. Respir Physiol Neurobiol 178(3): 482–489. https://doi.org/10.1016/j.resp.2011.06.017
Nakamori T, Morimoto A, Murakami N (1993) Effect of a central CRF antagonist on cardiovascular and thermoregulatory responses induced by stress or IL-1β. Am J Physiol 265(4): 834–839.
Watanabe T, Tan N, Saiki Y, Makisumi T, Nakamura S (1996) Possible involvement of glucocorticoids in the modulation of interleukin-1-induced cardiovascular responses in rats. J Physiol 491(1): 231–239. https://doi.org/10.1113/jphysiol.1996.sp021211
Graff GR, Gozal D (1999) Cardiorespiratory responses to interleukin-1beta in adult rats: role of nitric oxide, eicosanoids and glucocorticoids. Arch Physiol Biochem 107(2): 97–112.
Herlenius E (2011) An inflammatory pathway to apnea and autonomic dysregulation. Respir Physiol Neurobiol 178: 449–457. https://doi.org/10.1016/j.resp.2011.06.026
Aleksandrova NP, Danilova GA, Aleksandrov VG (2015) Cyclooxygenase pathway in modulation of the ventilatory response to hypercapnia by interleukin-1β in rats. Respir Physiol Neurobiol 209: 85–90. https://doi.org/10.1016/j.resp.2014.12.006
Foster SJ, McCormick LM, Ntolosi BA, Campbell D (1993) Production of TNF alpha by LPS-stimulated murine, rat and human blood and its pharmacological modulation. Agents Actions 38: 77–79. https://doi.org/10.1007/BF01991143
Preas HL 2nd, Jubran A, Vandivier RW, Reda D, Godin PJ, Banks SM, Tobin MJ, Suffredini AF (2001) Effect of endotoxin on ventilation and breath variability: role of cyclooxygenase pathway. Am J Respir Crit Care Med 164(4): 620–626.
Aleksandrova NP, Danilova GA, Aleksandrov VG (2017) Interleukin-1beta suppresses the ventilatory hypoxic response in rats via prostaglandin dependent pathways. Canad J Physiol Pharmacol 95(6): 681–685. https://doi.org/10.1139/cjpp-2016-0419
Tanaka K, Chiba T (1994) Nitric oxide synthase containing neurons in the carotid body and sinus of the guinea pig. Microscopy Res Techniq J 29(2): 90–93. https://doi.org/10.1002/jemt.1070290205
Iturriaga R (2001) Nitric oxide and carotid body chemoreception. Biol Res 34(2): 135–139. https://doi.org/10.4067/s0716-97602001000200019
Moya EA, Alcayaga J, Iturriaga R (2012) NO modulation of carotid body chemoreception in health and disease. Respir Physiol Neurobiol 184(2): 158–164. https://doi.org/10.1016/j.resp.2012.03.019
Li HF, Yu J (2009) Airway chemosensitive receptors in vagus nerve perform neuro-immune interaction for lung-brain communication. Adv Exp Med Biol 648: 421–426. https://doi.org/10.1007/978-90-481-2259-2_48
Valdés V, Mosqueira M, Rey S (2003) Inhibitory effects of NO on carotid body: contribution of neural and endothelial nitric oxide synthase isoforms. Am J Physiol Lung Cell Mol Physiol 284(1): 57–68.
Дополнительные материалы отсутствуют.
Инструменты
Российский физиологический журнал им. И.М. Сеченова