

Российский физиологический журнал им. И.М. Сеченова, 2021, T. 107, № 2, стр. 135-153
Канал TRPC6 в подоцитах почечных гломерул
В. Б. Михайлова 1, *, А. В. Карпушев 1, Ю. С. Юдина 1
1 Национальный медицинский исследовательский центр им. В.А. Алмазова
Санкт-Петербург, Россия
* E-mail: hastar.teerend@yandex.ru
Поступила в редакцию 18.10.2020
После доработки 02.12.2020
Принята к публикации 10.12.2020
Аннотация
Важнейшим компонентом фильтрационного аппарата почечных гломерул являются специализированные клетки висцерального эпителия – подоциты. Физиологическая функция подоцитов критически зависит от надлежащей регуляции внутриклеточного содержания Ca2+; избыточный вход Ca2+ в клетки может привести к нарушению морфологии клеток, подоцитопатии, апоптозу и последующему повреждению гломерулы в целом. Подоцитопатия является одной из основных характеристик протеинурии и фокально-сегментарного гломерулосклероза. Одним из ключевых белков, ответственных за вход Ca2+ в подоцитах, является ионный канал TRPC6. С момента первого обнаружения мутации в гене, кодирующем TRPC6, внимание научного сообщества сосредоточено на исследовании роли этого ионного канала в возникновении и развитии заболеваний почек. Как усиление, так и снижение функциональной активности TRPC6 сопряжено с проявлением тяжелых нефротических синдромов, ведущих к терминальной стадии хронической болезни почек. В обзоре приведены материалы, касающиеся регуляции активности TRPC6 и роли этого канала в патогенезе гломерулярных заболеваний.
Хроническая болезнь почек (ХБП), представляющая собой прогрессирующее снижение функции органов, происходящее на протяжении длительного периода времени, заключается в нарушении фильтрации плазмы крови, регуляции водно-электролитного баланса, а также продукции эритропоэтина. Терминальная стадия ХБП имеет летальный исход, и для ее преодоления требуется применение диализа или пересадка почки. Тем не менее, даже небольшое снижение почечной функции влечет за собой значительное увеличение риска развития патологии и наступления смерти, часто от сердечно-сосудистых нарушений [1, 2].
Большинство случаев ХБП является неизбежным и естественным следствием системных патологий, таких как сахарный диабет, системная красная волчанка, гипертония или СПИД, которые приводят к почечной недостаточности и обычно сопровождаются дисфункцией различных систем органов. Однако некоторые формы ХБП, например первичный фокально-сегментарный гломерулосклероз (ФСГС), поликистоз почек, некоторые формы гломерулонефрита и некоторые обструктивные состояния, препятствующие нормальному оттоку мочи, возникают вследствие патологических процессов, которые первоначально проявляются в самой почке. Причинами таких патофизиологических состояний могут быть аутоиммунные реакции, либо причины следует искать в структурно-функциональных нарушениях, развивающихся в гломерулах, канальцах нефронов, почечном интерстиции или сосудистой сети. Отличительными чертами большинства форм ХБП выступают воспаление и фиброз почек. В свою очередь, снижение функции почек достаточно для того, чтобы вызвать системные нарушения, включая гипертонию, атеросклероз, дислипидемию, анемию, метаболический ацидоз, мышечную атрофию и остеопатию [3].
Известно, что дисфункция гломерулярных подоцитов и последующая их гибель лежат в основе возникновения и развития почечных заболеваний, таких как ФСГС, мембранозная нефропатия, диабетическая нефропатия и волчаночный нефрит [4–6]. Характерные деструктивные изменения в подоцитах заключаются в реорганизации актинового цитоскелета в отростках тела клетки, педикулах или ножках (foot process), что приводит к сглаживанию ножек и гипертрофии подоцитов. Результатом в конечном итоге является отслоение подоцитов от базальной мембраны и их появление в моче как целых клеток (подоцитурия), так и структурных белков (нефрина, подоцина), уменьшение количества подоцитов в гломерулах (подоцитопения) [7, 8]. Изменения числа или структуры подоцитов ведут к деструктуризации важнейшего компонента гломерулярного фильтрационного барьера – щелевой диафрагмы и развитию протеинурии [9]. Одним из молекулярных механизмов, определяющих функцию подоцитов или ее нарушение является Ca2+-зависимая внутриклеточная сигнализация [10]. Ca2+-проницаемые неселективные катионные каналы семейства TRPC (Transient Receptor Potential Canonical), экспрессирующиеся в различных органах, включая сердце, мозг и почку, являются воротами для входа наружного Ca2+ в клетку. Последние полтора десятка лет большое внимание научного сообщества привлечено к роли канала TRPC6 в регуляции почечной функции [11]. Обнаруженные мутации в гене этого канала приводят к развитию наследуемого ФСГС [12–14].
TRPC-каналы принадлежат к суперсемейству катионных каналов TRP, первый одноименный представитель которого был обнаружен у Drosophila melanogaster [11, 15]. У позвоночных подсемейство TRPC включает семь членов, которые могут быть распределены по четырем подгруппам: TRPC1, TRPC2, TRPC4/5 и TRPC3/6/7. В функциональном отношении каналы TRPC являются Ca2+-проницаемыми неселективными катионными каналами, представляют собой тетрамеры, причем разные члены этой группы могут образовывать гетеротетрамеры, хотя точная стехиометрия эндогенно экспрессирующихся каналов TRPC еще недостаточно изучена. Известно, что TRPC6 может коиммунопреципитироваться с TRPC3, но не с TRPC5 в лизатах тканей коркового слоя почки.
В обзоре представлены материалы по регуляции функциональной активности TRPC6 и роли этого канала в патогенезе гломерулярных заболеваний.
КАНАЛЫ TRPC В ПОДОЦИТАХ
Подоциты представляют собой дифференцированные клетки висцерального эпителия, прикрепленные к поверхности базальной мембраны почечных гломерул, где, взаимодействуя друг с другом, образуют наиболее селективный компонент гломерулярного фильтрационного аппарата [16]. Одной из наиболее важных ультраструктурных особенностей подоцитов является наличие щелевой диафрагмы, специализированного соединения между смежными ножками. Щелевая диафрагма образует фильтр тонкой очистки, который позволяет просеивать молекулы в зависимости от их размера и формы [17, 18]. Диафрагма состоит из молекул, обычно экспрессирующихся в областях адгезивных контактов, таких как P-кадгерин, ZO-1 и FAT [19–21], а также тех, которые специфично экспрессируются в подоцитах, включая нефрин [17, 22], нефрин-подобный белок NEPH1 [23, 24] и подоцин [25]. Примечательно, что трансмембранный белок нефрин, являясь одним из компонентов внеклеточного матрикса, одновременно выступает в качестве внутриклеточной сигнальной молекулы, используя для передачи сигналов фосфорилирование тирозиновых остатков в цитоплазматических доменах [19]. Реакция подоцитов на повреждения и стресс в результате воздействия токсинов или повышения внутрикапиллярного давления заключается в сглаживании ножек и потере щелевой диафрагмы [26]. Если действие стрессорных факторов происходит в течение длительного периода времени, то количество подоцитов в пораженных гломерулах в конечном итоге уменьшается, и потеря около 25% подоцитов обычно приводит к гломерулосклерозу и утрате функции всего нефрона [27]. Таким образом, развитие ФСГС происходит в результате дисфункции подоцитов, гибели и отслоения клеток или изменений в ультраструктуре ножек подоцитов и межподоцитарной щелевой диафрагмы [28–30].
Известно, что TRPC1, TRPC3, TRPC4, TRPC5 и TRPC6 экспрессируются в подоцитах [31–34]. Электрофизиологическими и фармакологическими методами было показано, что TPRC3, TRPC5 и TRPC6 ответственны за вход Ca2+ в подоциты [33]. Каналы TRPC6 обнаруживаются в ножках подоцитов в области щелевой диафрагмы [13, 35–38]. Также TRPC6 наблюдаются в главных ножках (major process) и в теле клеток [39]. В разных клеточных компартментах TRPC6 выполняют различные функции и, по-видимому, находятся в различных макромолекулярных комплексах. Например, белок подоцин взаимодействует с TRPC6, расположенным на мембране ножек, но не тела клетки [40]. Коиммунопреципитация TRPC6 с TRPC3 из лизата клеточных линий подоцитов свидетельствует о том, что, по крайней мере, некоторые эндогенно экспрессирующиеся функциональные каналы являются гетеромерными TRPC6/TRPC3, как это обнаружено и в большинстве других тканей. Показано, что такие гетеромеры обладают повышенной механочувствительностью по сравнению с гомомерами любого типа [41].
МУТАЦИИ В TRPC6, АССОЦИИРОВАННЫЕ С ФСГС
Среди каналов семейства TRPC наибольшее внимание исследователей привлекает к себе TRPC6. Критическое значение этого ионного канала для нормальной функции подоцитов подчеркивается рядом работ, сообщающих об идентификации мутаций в гене этого канала, связанных с нефротическими синдромами, в частности с ФСГС (табл. 1). В 2005 г. была впервые идентифицирована мутация в гене канала TRPC6, ассоциированная с ФСГС-аутосомно-доминантного типа наследования [14]. Развитие заболевания у пациентов с обнаруженной мутацией происходило во взрослом возрасте. Терминальная стадия наступала через 4–20 лет после первоначального проявления заболевания, и болезнь не рецидивировала после трансплантации [42]. Биофизические характеристики этой мутации (P112Q) были изучены в гетерологической экспрессионной системе (клетках линии HEK-293), и в результате было показано, что P112Q является усиливающей функцию канала мутацией (gain-of-function). Биохимические данные свидетельствовали о том, что P112Q вызывает увеличение количества TRPC6 на мембране, что указывает на изменения в транспортировке канала к плазматической мембране или интернализации. Позднее были выявлены еще несколько мутаций в TRPC6 у пациентов с ФСГС-аутосомно-доминантного типа наследования, большинство из которых также оказались gain-of-function [12, 13, 43–46]. Анализ клинических характеристик пациентов с этими мутациями формирует представление о роли TRPC6 в гломерулярных заболеваниях и свидетельствует о том, что хроническая гиперактивация TRPC6 и усиленный вход Ca2+ являются причиной развития почечных патологий. Ряд мутаций был обнаружен у пациентов с ранним, в детском возрасте, развитием ФСГС [12, 45]. Кроме того, были идентифицированы мутации, также ассоциированные с ФСГС, но приводящие к снижению функции канала (loss-of-function) [47].
Таблица 1.
Мутации в TRPC6, ассоциированные с ФСГС Table 1. Mutations in TRPC6 associated with FSGS
| Мутация Mutation |
Функциональный тип мутации
по электрофизиологическим данным Functional type of mutation according to electrophysiological data |
Ссылка Reference |
|---|---|---|
| P112Q | Gain-of-function | [14] |
| R895C, E897K | Gain-of-function | [13] |
| N143S, S270T, K874X | Нет отличий от дикого типа No difference from wild-type |
[13] |
| M132T | Gain-of-function | [12, 52] |
| N125S, L395A, G757D, L780P, R895L | Loss-of-function | [47] |
| P15S, A404V | Нет данных No data |
[53, 54] |
| N110H | Gain-of-function | [55] |
| G109S, N125S, L780P | Нет данных No data |
[56] |
| R175Q | Gain-of-function | [46] |
| R360H | Нет отличий от дикого типа No difference from wild-type |
[47, 57] |
| H218L, R895L | Gain-of-function | [45] |
| C121S, D130V, G162R, I111I | Нет данных No data |
[58] |
| Q889K | Gain-of-function | [44] |
Что известно на сегодняшний день о молекулярных механизмах, связывающих мутации в TRPC6 и клинические проявления нефротических синдромов? В одной из работ было показано, что следствием экзогенной экспрессии gain-of-function мутации (R895C) в культивируемых подоцитах является активация киназы, регулируемой внеклеточными сигналами (ERK1/2 – Extracellular-Regulated Kinase) [48]. В другой работе было продемонстрировано, что мутации P112Q, R895C и E897K ведут к конститутивной активации транскрипционных факторов NFAT [49]. Причем, эти эффекты блокировались ингибиторами кальцинейрина (CN), Ca2+/CaM-зависимой протеинкиназы (CaMK) и фосфатидилинозитол-3-киназы (PI3K). В свою очередь, в трансгенных мышах подоцит-специфическая экспрессия активной формы NFATc1 вызывала развитие протеинурии и гломерулосклероза [50]. Мутации в TRPC6 могут реализоваться в нарушении взаимодействия с белками-партнерами, что особенно важно в поддержании надлежащей морфологии ножек подоцитов. Известно, что TRPC6 взаимодействует со структурными белками щелевой диафрагмы, подоцином и нефрином [13, 35]. Взаимодействие с подоцином происходит между цитозольными С-концевыми доменами обоих белков и является функционально значимым для воротного механизма TRPC6 [36]. Взаимодействие между TRPC6 и нефрином регулирует доставку канала на клеточную поверхность подоцита [37, 51]. Нарушение взаимодействия между каналом TRPC6 и нефрином показано для мутации Y284F [51].
Несмотря на то, что TRPC6 стал объектом исследований в первую очередь в связи с наследственными нефротическими синдромами, следует отметить, что наследственные формы ФСГС довольно редки по сравнению с ненаследственными или приобретенными. Актуальным является вопрос о причинах и механизмах нарушения регуляции TRPC6 дикого типа у пациентов с ненаследственными формами гломерулярных заболеваний.
РЕГУЛЯЦИЯ АКТИВНОСТИ КАНАЛА TRPC6 В ПОДОЦИТАХ
Наиболее изученным, каноническим, путем регуляции активности канала TRPC6 в подоцитах является сигнальный каскад, запускаемый стимуляцией рецепторов, сопряженных с G-белками (GPCR – G Protein-Coupled Receptor) (рис. 1). Примером может послужить активация TRPC6 ангиотензином II (Ang II) [59]. Ang II, действуя через ангиотензиновые рецепторы AT1, играет критическую роль в развитии протеинурии и в прогрессировании почечных повреждений при ФСГС [60]. Стимуляция AT1-рецепторов вызывает вход Ca2+ из внеклеточной среды [61]. Было показано, что увеличение экспрессии AT1 в подоцитах трансгенных крыс приводит к развитию протеинурии и ФСГС [62].
Рис. 1.
Схема GPCR-зависимой активации TRPC6. Стимуляция GPCR активирует сигнальный путь, включающий последовательно активацию G-белка GQ, PLC (1), продукцию DAG (2), активацию TRPC6 (3). DAG активирует NADPH-оксидазу NOX2 (4) с образованием комплекса, содержащего каталитическую, gp91phox, и регуляторную, p47phox, субъединицы. Продукция АФК NADPH-оксидазой сопряжена с увеличением количества TRPC6 на клеточной мембране. Подоцин фиксирует каналы TRPC6 в холестерол-обогащенных мембранных доменах. Источник: [39] с модификацией. Fig. 1. Scheme of GPCR-dependent activation of TRPC6. Stimulation of GPCR activates a signaling pathway that includes G-protein GQ, PLC activation (1), production of DAG (2), which acts on the TRPC6 channel (3). DAG activates NOX2 NADPH oxidase (4) with the formation of a complex containing the catalytic, gp91phox, and regulatory, p47phox, subunits. Generation of ROS by NADPH oxidase is associated with an increase in the TRPC6 surface expression. Podocin tethers TRPC6 in cholesterol-rich membrane domains. Source: [39] with modification.

С помощью метода локальной фиксации потенциала в конфигурации whole-cell в подоцитах изолированных и декапсулированных гломерул крысы или в подоцитах иммортализованной клеточной линии было продемонстрировано, что Ang II в наномолярных концентрациях индуцирует катионный ток выходящего выпрямления. Отсутствие тока в подоцитах с нокдауном TRPC6 свидетельствовало об участия канала TRPC6 в генерации Ang II-вызванного катионного тока [59]. Кроме того, эффект Ang II полностью блокировался микромолярными концентрациями La3+ [59], что также подтверждает роль TRPC6, т.к. активность канала TRPC5, экспрессируемого в подоцитах, усиливается La3+ [34].
Сходный с Ang II эффектом на катионный ток канала TRPC6 обладает другой агонист GPCR, ATP. Действующий через P2Y-рецепторы ATP также индуцировал в подоцитах катионный ток выходящего выпрямления [63]. Эффект ATP блокировался сурамином, относительно неселективным ингибитором P2Y-рецепторов. Ответы на другие агонисты P2Y-рецепторов, ADP, UTP или UDP, показывают, что в активации канала TRPC6 играют роль различные подтипы рецепторов, P2Y1, P2Y2 и P2Y6 [63]. Отметим, что ответы как на ATP так и Ang II в подоцитах устранялись введением в цитоплазму клетки гуанозин-5'-O-2-тиодифосфата (GDP-βS), аналога GDP, устойчивого к гидролизу и фосфорилированию, это указывает на то, что Ang II- и ATP-вызванная активация TRPC6 опосредуется G-белками [59, 63]. В обоих случаях эффекты лигандов подавлялись блокированием фосфолипаза С (PLC)-зависимого сигнального пути агентом D609 [59, 63].
В гетерологической экспрессионной системе, клетках линии СНО-К1, был исследован сигнальный каскад активации TRPC6. Было продемонстрировано участие гетеротримерных G-белков, PLC и диацилглицерола (DAG) в передаче сигнала на канал TRPC6 при стимуляции GPCR (рис. 1). Причем было показано, что конечным эффектором является именно DAG, поскольку активация TRPC6 имеет протеинкиназа С (PKC)-независимый характер [64]. Однако более позднее исследование показало, что ингибирование PKC или ее активация соответственно усиливают или подавляют индуцированный агонистом вход Ca2+ в клетках, экспрессирующих экзогенный TRPC6 [65]. Заметим, что инозитолтрифосфат (IP3), образуемый фосфолипазой вместе с DAG, не имел эффектов на TRPC6-опосредуемый ток [64].
С рассмотренным фармакологическим анализом ответов на Ang II и ATP согласуются данные по активации TRPC6-опосредованных катионных токов в подоцитах мембранно-проницаемым аналогом DAG, 1-олеоил-2-ацетил-sn-глицеролом (OAG) [38, 66]. Вполне вероятно, что активация GPCR дает начало двум отдельным процессам: мобилизации ионных каналов на клеточной поверхности, т.е. встраиванию новых каналов TRPC6, и увеличению вероятности открытия каналов. Было показано, что стимуляция GPCR увеличивает количество TRPC6 на мембране, что может происходить всего через 5 с после начала действия агониста GPCR и при концентрациях агониста ниже, чем требуется для активации TRPC6 [67]. В соответствии с этим механизмом индукции TRPC6-опосредованного тока утверждается, что OAG стимулирует увеличение экспрессии TRPC6 на поверхности подоцитов [66]. Очень похожие эффекты усиления активности TRPC6 наблюдаются в подоцитах и в гетерологической экспрессионной системе при обработке клеток метаболитом арахидоновой кислоты – 20-гидрокси-эйкозатетраеновой кислотой (20-HETE – 20-HydroxyEicosaTetraEnoic acid), которая может высвобождаться из прегломерулярных микрососудов и воздействовать на другие типы клеток гломерул через паракринные сигнальные механизмы [68, 69].
В настоящее время большое количество экспериментальных данных указывает на сигнальную роль активных форм кислорода (АФК) в модуляции каналов TRPC6 в подоцитах (рис. 1). Обработка подоцитов H2O2 значительно увеличивает количество каналов TRPC6 на мембране и усиливает катионные токи [70]. Следует заметить, что Ang II- и ATP-вызванная активация TRPC6 в подоцитах блокируется гасителями АФК или ингибиторами продукции АФК, т.е. ингибиторами NADPH-оксидазы NOX2 [59, 63]. Кроме того, показано, что ATP и Ang II, так же как и 20-HETE, увеличивают выработку АФК в подоцитах [68]. Также наблюдается увеличение продукции АФК под действием OAG, а эффект последнего на TRPC6 также блокируется ингибиторами NOX2 [38]. Реципрокная коиммунопреципитация каталитической субъединицы NOX2 gp91phox и канала TRPC6 свидетельствует об образовании комплекса NOX2–TRPC6 в культивируемых подоцитах. Причем, образованию таких комплексов препятствует нокдаун гена структурного белка щелевой диафрагмы подоцина, что позволяет предположить, что последний играет роль соединительного мостика между NOX2 и TRPC6 (рис. 1) [38]. Вполне вероятно, что такие комплексы образуются в богатых стеролами мембранных доменах, организованных подоцином, образующим олигомеры более высокого порядка [25, 71]. Взаимодействие с подоцином также может связывать TRPC6 с цитоскелетом через CD2-ассоциированный белок (CD2AP) [25]. Комплекс NOX2–TRPC6 может служить для создания относительно высоких концентраций АФК в непосредственной близости от ионного канала без значительных изменений в концентрации АФК в глубине цитоплазмы. Известно, что DAG стимулирует активность NADPH-оксидазы, способствуя сборке активных мультимерных комплексов NOX2, содержащих каталитическую, gp91phox, и регуляторную, p47phox, субъединицы [72–75], что в свою очередь приводит к увеличению количества TRPC6 на мембране [38]. Важно отметить, что эффект увеличения количества TRPC6 блокируется агентами, элиминирующими АФК, ингибирующими NOX2, включая ингибитор малого G-белка Rac1, и дезорганизацией липидных рафтов с помощью нокдауна гена подоцина [38]. Редокс-регуляция, по-видимому, характерна для многих членов суперсемейства TRP [76]. Молекулярные и биофизические механизмы, посредством которых АФК увеличивают количество TRPC6 на мембране, на сегодняшний день не известны. Однако в ряде работ показано, что канал TRPA1 (Transient Receptor Potential Ankyrin) демонстрирует сходную чувствительность к АФК [77], которые, по-видимому, активируют TRPA1 за счет окисления остатков цистеина внутри анкирин-подобных повторов в цитозольном N-конце [78, 79]. TRPC6 имеет сходные анкирин-подобные повторы в своем N-конце, хотя их количество значительно меньше, чем у TRPA1 [80]. Высказываются также предположения о возможном участии активных форм азота в регуляции активности TRPC6, например, в NO-зависимом S-нитрозилировании TRPC6, как способе стабилизации ионного канала в открытом состоянии [70].
В подоцитах существует еще один путь активации каналов TRPC6. Это активация механическими стимулами, которая, по-видимому, принципиально отличается от процессов, происходящих во время передачи сигналов от GPCR [36, 81, 82]. Катионный ток в подоцитах возникает в ответ на растяжение клеточной мембраны [36]. Растяжение мембраны, вызванное кратковременным помещением клеток в гипоосмотический внешний раствор, который вызывает набухание клеток, используется как наиболее простой для воспроизводимости результатов экспериментальный механический стимул, хотя не является физиологически адекватным для подоцитов. Очевидно, что набухание клеток в гипоосмотической среде приводит к быстрым структурным перестройкам цитоскелета, которые, в свою очередь, изменяют внутреннее натяжение мембраны. Нет сомнений, что катионные токи, вызванные растяжением мембраны в подоцитах, опосредуются каналами TRPC6, поскольку они устраняются нокдауном TRPC6 и селективным ингибитором TRPC6 SAR-7334 [36, 81]. Также эти токи блокируются микромолярными концентрациями La3+, токсином паука GsMTx4, который подавляет механически вызванную активацию широкого диапазона каналов, в том числе, TRPC1 и TRPC6 [36]. Сигнальные пути механической активации TRPC6 в подоцитах, по-видимому, отличаются от таковых при GPCR-зависимой активации. Механическая активация TRPC6 сохраняется в присутствии гасителей АФК, а также при введении в клетки GDP-βS, при обработке ингибиторами PLC или фосфолипазы А (PLA) [36]. Напротив, активация каналов TRPC6 с помощью OAG или 20-HETE блокируется гасителями АФК [38, 68]. Следовательно, причиной механической активации TRPC6 в подоцитах не может быть вызванная растяжением активация GPCR, фосфолипаз или NADPH-оксидаз, хотя есть доказательства того, что это может происходить в клетках других типов [83]. Кроме того, показано существование, по крайней мере, одной мутации в TRPC6 (N143S), ассоциированной с ФСГС, которая устраняет механическую активацию ионного канала, в то же время позволяя активировать его с помощью OAG или воздействия на GPCR [84]. Некоторыми исследователями выдвигается гипотеза, что каналы TRPC6 являются непосредственно механочувствительными [70, 85]. Гипотеза подтверждается регистрацией токов TRPC6-каналов в изолированных фрагментах мембран (конфигурация inside-out) трансфицированных СНО клеток в ответ на отрицательное давление [85]. Однако механочувствительность TRPC6 может зависеть от взаимодействия с другими структурными белками или от свойств локальных участков мембраны, которые будут варьировать в клетках различных тканей и органов и даже в разных компартментах одной и той же клетки. Механика клеточной поверхности зависит от взаимодействия с нижележащим цитоскелетом, который может влиять на базовое натяжение или искривление плазматической мембраны. По этим причинам каналы TRPC6 в ножках подоцитов и в теле клетки могут иметь различный характер механочувствительности и активации [70]. Функционирование каналов TRPC6 как механосенсоров коррелирует с распределением экспрессии TRPC6 в тканях, которые подвергаются гидростатическим силам. Как механосенсор TRPC6 является основным кандидатом на роль преобразователя внешних механических сил в активацию внутриклеточных сигнальных каскадов в подоцитах.
Подоцин, как отмечалось выше, играет важную роль в образовании комплекса NOX2–TRPC6 [38]. Кроме того, было показано, что экспрессия экзогенного подоцина значительно усиливает активацию TRPC6, вызванную OAG и стимуляцией GPCR [35]. На основании результатов работы с MEC-2, ортологом подоцина, ответственным за механотрансдукцию в сенсорных нейронах C. elegans, было высказано предположение, что подоцин также играет роль в механоактивации TRPC6 [35]. Однако оказалось, что функция подоцина имеет более сложный характер, чем у MEC-2. В то время как GPCR- или OAG-зависимая активация TRPC6 уменьшается или исчезает после нокдауна гена подоцина, механоактивация TRPC6 значительно усиливается [36]. Т.е., одна из функций подоцина может заключаться в подавлении механической активации TRPC6 в подоцитах, что может быть особенно важно в ножках клеток, испытывающих давление со стороны гломерулярных капилляров под действием сил циркулирующей в них жидкости [30, 86, 87]. Как и другие члены семейства стоматин-подобных белков (STOML – Stomatin-like protein), подоцин имеет холестерол-связывающий прохибитин-домен (PHB), также известный как стоматин/прохибитин/флотиллин/HflKC домен [88]. Возможно, что подоцин фиксирует каналы TRPC6 в стерол-обогащенных мембранных доменах, которые могут быть относительно устойчивыми к определенным типам конформационных изменений, вызванных механическими раздражителями. К этому следует добавить, что вызванная растяжением активация TRPC6 в подоцитах усиливается после разрушения F-актина цитохалазином-D [36]. Поскольку подоцин, обладая CD2AP-связывающим мотивом, взаимодействует с цитоскелетом, можно предположить, что последний так или иначе вовлечен в подавление активации TRPC6 растяжением [25]. Таким образом, независимо от того, каков биохимический или биофизический механизм действия подоцина, очевидно, что этот структурный элемент фильтрационного аппарата щелевой диафрагмы модулирует доминантный способ активации TRPC6, т.е. подоцин способствует DAG- и АФК-зависимой и, следовательно, GPCR-зависимой активации TRPC6, но снижает чувствительность ионного канала к механическому воздействию, которое в подоцитах не связано с GPCR. Предположительно, механическая активация TRPC6 может рассматриваться как патофизиологический процесс, происходящий в клетках в силу снижения экспрессии подоцина. Этот структурный белок весьма специфичен, и его экспрессия, кроме гломерулярных подоцитов, была обнаружена только в семенниках [89]. Поэтому подоцин-зависимый тип модуляции TRPC6 является уникальным механизмом в ножках подоцитов. Однако возможно, что и другие белки семейства STOML, имеющие, как подоцин, PHB-домен, также играют сходную роль в модуляции активности каналов семейства TRPC в других тканях [90]. Выше было отмечено, что у C. elegans ортолог подоцина, MEC-2, необходим для механической активации катионных каналов из семейства ENaC/Deg, к которому принадлежит эпителиальный натриевый канал (ENaC – Epithelial Sodium Channel) дистального отдела нефрона [35, 91]. Показано, что стоматин в составе комплекса более высокого порядка ингибирует протон-управляемые токи, опосредованные кислото-чувствительными ионными каналами (ASIC – Acid-Sensing Ion Channel) у мышей [92, 93]. Особенно интересно, что STOML-3 усиливает вызванную смещением мембран активацию механочувствительного канала PIEZO1 в нейронах спинальных ганглиев, причем, с порогом активации по смещению мембраны порядка 10 нм [94]. Таким образом, белки с PHB-доменом регулируют механочувствительность клеток, а также доминантный способ активации мультимодальных каналов. Предполагается, что, по крайней мере, часть этих эффектов происходит посредством регуляции натяжения клеточной мембраны в покое [95]. Преобладание того или иного способа активации ионного канала с участием белка с PHB-доменом, вероятно, зависит от особенностей структуры цитоскелета, мембраны и типа канала.
Фосфорилирование белка также играет важную роль в модуляции активности каналов TRPC6. В подоцитах TRPC6 был обнаружен в комплексе, содержащим тирозинкиназу семейства Src, которая фосфорилирует TRPC6 по нескольким аминокислотным остаткам и тем самым увеличивает количество канала на мембране [51, 82]. Среди таких сайтов особенно важным является Y284, поскольку именно фосфорилирование этого аминокислотного остатка необходимо для доставки TRPC6 на клеточную поверхность. Также фосфорилирование Y284 регулирует взаимодействие с другими компонентами щелевой диафрагмы, например нефрином [51]. Кроме того, активация Src в подоцитах происходит в ответ на генерацию АФК [82].
Учитывая, что мутации в TRPC6 связаны с наследственными формами ФСГС, нарушение надлежащей регуляции ионного канала дикого типа при наличии патогенных факторов может быть связано с развитием приобретенных форм ФСГС. Актуальным является вопрос о причинах и механизмах нарушения регуляции TRPC6 у пациентов с ненаследственными формами гломерулярных заболеваний. Например, у пациентов с различными формами ХБП показано увеличение экспрессии TRPC6 дикого типа в гломерулах [96]. Инкубация иммортализованных подоцитов мышей с образцами сыворотки и плазмы, взятыми у пациентов с первичным ФСГС, заболевание которых рецидивировало после трансплантации, вызывала увеличение количества TRPC6 на поверхности клеток и сопровождалось одновременной потерей подоцина [81]. Однако нокдаун подоцина сам по себе не изменял количество TRPC6 на мембране подоцитов. Поэтому вероятно, что эффекты на TRPC6 и подоцин представляют собой независимые друг от друга сигнальные пути. Кроме того, наблюдалось заметное усиление активации каналов TRPC6 за счет растяжения мембраны.
TRPC6 И Ca2+-СИГНАЛИЗАЦИЯ
Поскольку TRPC6 опосредует вход Ca2+ в клетку, канал управляет Ca2+-зависимыми сигнальными путями. Принципиально, любой Ca2+- или CaM-зависимый белок, локализованный в пределах доступного диффузионного радиуса, окружающего канал TRPC6, может представлять собой начало нисходящего TRPC6-зависимого сигнального пути. На сегодняшний день известно, что TRPC6-опосредованный вход Ca2+ активирует серин/треонин фосфатазу CN (рис. 2) [50]. CN широко экспрессируется клетками многих тканей, становится каталитически активным при взаимодействии с Ca2+ или Ca2+-CaM [97, 98] и участвует во множестве сигнальных каскадов. Возможный диапазон сигнальных путей, запускаемых активацией TRPC6, можно оценить по количеству CN-дефосфорилируемых белков, включая компоненты цитоскелета, митоген-активируемые протеинкиназы, белки, вовлеченные в клеточный цикл и апоптоз, ионные каналы и транскрипционные факторы [99, 100]. Хорошо изучен сигнальный путь, приводящий к активации семейства транскрипционных факторов, известных как ядерные факторы активированных Т-клеток (NFAT – Nuclear Factor of Activated T-cells) [50]. Белки семейства NFAT, NFATc1-4 и NFAT5 были первоначально описаны как часть транскрипционного регуляторного комплекса, индуцируемого сигналами в активированных Т-клетках, которые приводят к PLC-зависимому увеличению внутриклеточного Ca2+ и активации CaM и CN [101]. Активированный CN дефосфорилирует цитозольный NFAT, вызывая его транслокацию в ядро (рис. 2). Оказавшись внутри ядра, NFAT снова фосфорилируется ядерными киназами, после чего он образует комплексы с другими транскрипционными регуляторными белками [102]. Этот сигнальный путь затрагивается при иммуносупрессии ингибиторами CN, циклоспорином и такролимусом, что подало идею использования этих агентов при нефротических синдромах, таких как первичный ФСГС. В свою очередь, сами каналы TRPC6 также регулируются на уровне транскрипции CN–NFAT-зависимым путем, что порождает положительную обратную связь, которая может вызвать конститутивную активацию TRPC6 (рис. 2) [49, 103,]. Было показано, что именно этот механизм обуславливает Ang II-вызванное увеличение экспрессии TRPC6 в почечных гломерулах [103]. В другой работе была продемонстрирована роль Ang II/TRPC6/NFAT-пути в повреждении подоцитов при развитии диабетической нефропатии. При этом такролимус вызывал снижение экспрессии как TRPC6, так и NFAT и уменьшал степень повреждения подоцитов [104].
Рис. 2.
Схема сигнального каскада активации NFAT. Вход Ca2+ через канал TRPC6 активирует кальцинейрин CN (1). Активированный CN дефосфорилирует цитозольный NFAT (2), вызывая его транслокацию в ядро (3). NFAT стимулирует транскрипцию различных генов, включая TRPC6 (4), что увеличивает количество TRPC6 на клеточной мембране (5). Источник: [11] с модификациями. Fig. 2. Scheme of the signaling pathway of activation of NFAT. Ca2+ entry through the TRPC6 channel activates calcineurin CN (1). Activated CN dephosphorylates cytosolic NFAT (2), causing its translocation to the nucleus (3). NFAT stimulates the transcription of various genes including TRPC6 (4), which further increases the TRPC6 surface expression (5). Source: [11] with modification.

TRPC6-зависимые сигнальные пути в ножках подоцитов, вероятно, отличаются от таковых в теле клетки. По-видимому, так же, как и в теле клетки, в ножках вход Ca2+ через каналы TRPC6 активирует CN. Активированный CN дефосфорилирует синаптоподин, что обеспечивает его протеолиз катепсином L (рис. 3) [105]. Синаптоподин является регуляторным белком цитоскелета, ответственным за поддержание нормальной морфологии ножек подоцитов [105, 106]. Следовательно, длительная активация TRPC6 и, соответственно, активация CN будет способствовать дестабилизации ножек. В сообщениях о терапии нефротического синдрома предполагается, что именно с воздействием на указанный сигнальный путь связаны защитные эффекты циклоспорина или такролимуса [107, 108]. Т.е. действие этих агентов обусловлено не иммуносупрессией, а, вероятнее всего, стабилизацией ножек и предотвращением отслоения подоцитов [105]. Кроме того, в экспериментах на культивируемых подоцитах было показано, что синаптоподин имеет тенденцию лимитировать количество TRPC6 на клеточной поверхности [109]. Следовательно, как и в случае динамики транскрипции в теле подоцита, длительная активация TRPC6 в ножках может способствовать росту количества каналов на мембране вследствие положительной обратной связи. Вход Ca2+ через каналы TRPC6 активирует CN, что обеспечивает протеолиз синаптоподина, это, в свою очередь, дестабилизирует цитоскелет, нарушает морфологию ножек подоцитов и увеличивает число TRPC6 на клеточной мембране.
Рис. 3.
Схема Ca2+-зависимой активации кальцинейрина и кальпаина. Вход Ca2+ через канал TRPC6 активирует кальцинейрин CN (1) и кальпаин-1 (2). Активированный CN дефосфорилирует синаптоподин (3), что обеспечивает его протеолиз катепсином L (4). Кальпаин-1 осуществляет протеолиз талина-1 (5). Потеря синаптоподина и талина дестабилизирует цитоскелет подоцитов. Источник: [11] с модификациями. Fig. 3. Scheme of Ca2+-dependent activation of calcineurin and calpain. Ca2+ entry through the TRPC6 channel activates calcineurin CN (1) and calpain-1 (2). Activated CN dephosphorylates synaptopodin (3), which ensures its proteolysis by cathepsin L (4). Calpain-1 carries out the proteolysis of talin-1 (5). Loss of synaptopodin and talin destabilizes the cytoskeleton of podocytes. Source: [11] with modification.
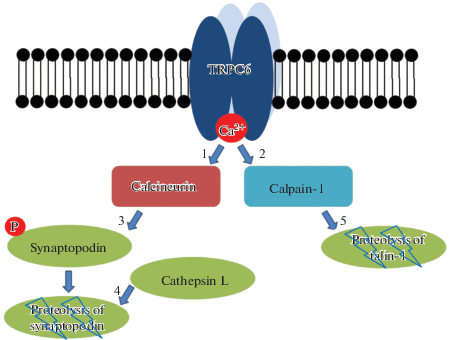
CN является лишь одной из широкого спектра Ca2+-зависимых сигнальных молекул в подоцитах. Недавнее исследование показало, что TRPC6-зависимый вход Ca2+ активирует кальпаин-1 и приводит к структурной дестабилизации и повреждению подоцитов через потерю белка талин-1 и активацию CN (рис. 3) [110]. Кальпаин принадлежит к семейству нелизосомных цистеиновых протеаз, которые активируются ионами Ca2+; его гиперактивность тесно связана с повреждением почек [111]. Талины являются одним из важнейших белков адгезии, соединяющие интегрины с актиновым цитоскелетом [112]. Есть две изоформы талина у позвоночных: талин-1 и талин-2. Талин-1 широко экспрессируется в цитоплазме и может расщепляться кальпаином [113]. Кроме того, TRPC6 может активировать кальпаин через ERK1/2 Ca2+-независимым образом, тем самым регулируя перестройку цитоскелета, адгезию и подвижность подоцитов [114].
ЗАКЛЮЧЕНИЕ
В настоящее время существует острая потребность в эффективных методах лечения многих форм ХБП. Основу современной терапии составляют подавление ренин-ангиотензиновой системы, иммуносупрессия, лечение дислипидемии и других метаболических и системных нарушений, возникающих в результате почечной дисфункции [115]. Во многих случаях эти подходы в конечном итоге терпят неудачу. Например, часто первичный ФСГС не поддается лечению глюкокортикоидами, прогрессирует до терминальной стадии ХБП [116, 117] и рецидивирует у 40% пациентов, которые получают трансплантат почки [118]. Следовательно, существует сильная мотивация для выявления новых терапевтических мишеней для лечения ХБП [119]. Одной из потенциальных и, возможно, очень перспективных мишеней являются каналы TRPC и, в частности, TRPC6, поскольку анализ экспериментальных и клинических данных свидетельствует, что этот ионный канал играет решающую роль в повреждении подоцитов при заболеваниях почек. Поскольку каналы TRPC имеют достаточно широкое распространение в организме, вполне возможно, что острое фармакологическое ингибирование TRPC6 может привести к негативным побочным эффектам. Например, отмечено, что резкое торможение этих каналов приводит к дефициту когнитивных функций [120–122]. В этом случае существенный вклад в поиск и разработку лекарственных средств вносит изучение механизмов регуляции каналов TRPC в почечных клетках и исследование TRPC-зависимых сигнальных каскадов.
Список литературы
Weaver D.J., Mitsnefes M. Cardiovascular disease in children and adolescents with chronic kidney disease. Semin. Nephrol. 38(6): 559–569. 2018.
Taal M.W. Chronic kidney disease 10 years on: what have we learned? Curr. Opin. Nephrol. Hypertens. 21(6): 607–611. 2012.
Kiefer M.M., Ryan M.J. Primary care of the patient with chronic kidney disease. Med. Clin. North. Am. 99(5): 935–952. 2015.
Somlo S., Mundel P. Getting a foothold in nephrotic syndrome. Nat. Genet. 24(4): 333–335. 2000.
Kerjaschki D. Caught flat-footed: podocyte damage and the molecular bases of focal glomerulosclerosis. J. Clin. Invest. 108(11): 1583–1587. 2001.
Pena-Polanco J.E., Fried L.F. Established and emerging strategies in the treatment of chronic kidney disease. Semin. Nephrol. 36(4): 331–342. 2016.
Tryggvason K., Wartiovaara J. Molecular basis of glomerular permselectivity. Curr. Opin. Nephrol. Hypertens. 10(4): 543–549. 2001.
Kaplan J.M., Kim S.H., North K.N., Rennke H., Correia L.A., Tong H.Q., Mathis B.J., Rodríguez-Pérez J.C., Allen P.G., Beggs A.H., Pollak M.R. Mutations in ACTN4, encoding α-actinin-4, cause familial focal segmental glomerulosclerosis. Nat. Genet. 24(3): 251–256. 2000.
Fissell W.H., Miner J.H. What is the glomerular ultrafiltration barrier? J. Am. Soc. Nephrol. 29(9): 2262–2264. 2018.
Kerjaschki D. Polycation-induced dislocation of slit diaphragms and formation of cell junctions in rat kidney glomeruli: the effects of low temperature, divalent cations, colchicine, and cytochalasin B. Lab. Invest. 39(5): 430–440. 1978.
Hall G., Wang L., Spurney R.F. TRPC Channels in Proteinuric Kidney Diseases. Cells. 9(1): e44. 2019.
Heeringa S.F., Möller C.C., Du J., Yue L., Hinkes B., Chernin G., Vlangos C.N., Hoyer P.F., Reiser J., Hildebrandt F. A novel TRPC6 mutation that causes childhood FSGS. PLoS One. 4(11): e7771. 2009.
Reiser J., Polu K.R., Moller C.C., Herbert S., Villegas I., Vila‑Casado C., McGee M., Sugimoto H., Brown D., Kalluri R., Mundel P., Smith P.L., Clapham D.E., Pollak M.R. TRPC6 is a glomerular slit diaphragm‑associated channel required for normal renal function. Nat. Genet. 37(7): 739–744. 2005.
Winn M.P., Conlon P.J., Lynn K.L., Farrington M.K., Creazzo T., Hawkins A.F., Daskalakis N., Kwan S.Y., Ebersviller S., Burchette J.L., Pericak‑Vance M.A., Howell D.N., Vance J.M., Rosenberg P.B. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science. 308(5729): 1801–1804. 2005.
Montell C., Rubin G.M. Molecular characterization of the Drosophila trp locus: a putative integral membrane protein required for phototransduction. Neuron. 2(4): 1313–1323. 1989.
Pavenstädt H., Kriz W., Kretzler M. Cell biology of the glomerular podocyte. Physiol. Rev. 83(1): 253–307. 2003.
Wartiovaara J., Öfverstedt L.-G., Khoshnoodi J., Zhang J.J., Mäkelä E., Sandin S., Ruotsalainen V., Cheng R.H., Jalanko H., Skoglund U., Tryggvason K. Nephrin strands contribute to a porous slit diaphragm scaffold as revealed by electron tomography. J. Clin. Invest. 114(10): 1475–1483. 2004.
Haraldsson B., Nyström J., Deen W.M. Properties of the glomerular barrier and mechanisms of proteinuria. Physiol. Rev. 88(2): 451–487. 2008.
Martin C.E., Jones N. Nephrin signaling in the podocyte: An updated view of signal regulation at the slit diaphragm and beyond. Front. Endocrinol. (Lausanne). 9: 302. 2018.
Reiser J., Kriz W., Kretzler M., Mundel P. The glomerular slit diaphragm is a modified adherens junction. J. Am. Soc. Nephrol. 11(1): 1–8. 2000.
Schnabel E., Anderson J.M., Farquhar M.G. The tight junction protein ZO-1 is concentrated along slit diaphragms of the glomerular epithelium. J. Cell. Biol. 111(3): 1255–1263. 1990.
Ruotsalainen V., Ljungberg P., Wartiovaara J., Lenkkeri U., Kestilä M., Jalanko H., Holmberg C., Tryggvason K. Nephrin is specifically located at the slit diaphragm of glomerular podocytes. Proc. Natl. Acad. Sci. USA. 96(14): 7962–7967. 1999.
Sellin L., Huber T.B., Gerke P., Quack I., Pavenstädt H., Walz G. NEPH1 defines a novel family of podocin interacting proteins. FASEB J. 17(1): 115–117. 2003.
Grahammer F., Wigge C., Schell C., Kretz O., Patrakka J., Schneider S., Klose M., Kind J., Arnold S.J., Habermann A., Bräuniger R., Rinschen M.M., Völker L., Bregenzer A., Rubbenstroth D., Boerries M., Kerjaschki D., Miner J.H., Walz G., Benzing T., Fornoni A., Frangakis A.S., Huber T.B. A flexible, multilayered protein scaffold maintains the slit in between glomerular podocytes. J. CIin. Insight. 1(9): e86177. 2016.
Schwarz K., Simons M., Reiser J., Saleem M.A., Faul C., Kriz W., Shaw A.S., Holzman L.B., Mundel P. Podocin, a raft-associated component of the glomerular slit diaphragm, interacts with CD2AP and nephrin. J. Clin. Invest. 108(11): 1621–1629. 2001.
Kriz W., Shirato I., Nagata M., LeHir M., Lemley K.V. The podocyte’s response to stress: the enigma of foot process effacement. Am. J. Physiol. Renal Physiol. 304(4): 333–347. 2013.
Kim Y.H., Goyal M., Kurnit D., Wharram B., Wiggins J., Holzman L., Kershaw D., Wiggins R. Podocyte depletion and glomerulosclerosis have a direct relationship in the PAN-treated rat. Kidney Int. 60(3): 957–968. 2001.
Kriz W., Elger M., Hosser H., Hähnel B., Provoost A., Kränzlin B., Gretz N. How does podocyte damage result in tubular damage? Kidney Blood Press. Res. 22(1–2): 26–36. 1999.
Kriz W., Lemley K.V. A potential role for mechanical forces in the detachment of podocytes and the progression of CKD. J. Am. Soc. Nephrol. 26(2): 258–269. 2015.
Kriz W., Lemley K.V. Potential relevance of shear stress for slit diaphragm and podocyte function. Kidney Int. 91(6): 1283–1286. 2017.
Goel M., Sinkins W.G., Zuo C.D., Estacion M., Schilling W.P. Identification and localization of TRPC channels in the rat kidney. Am. J. Physiol. Renal Physiol. 290(5): 1241–1252. 2006.
Ilatovskaya D.V., Levchenko V., Ryan R.P., Cowley A.W. Jr., Staruschenko A. NSAIDs acutely inhibit TRPC channels in freshly isolated rat glomeruli. Biochem. Biophys. Res. Commun. 408(2): 242–247. 2011.
Ilatovskaya D.V., Staruschenko A. TRPC6 channel as an emerging determinant of the podocyte injury susceptibility in kidney diseases. Am. J. Physiol. Renal Physiol. 309(5): 393-397. 2015.
Tian D., Jacobo S.M., Billing D., Rozkalne A., Gage S.D., Anagnostou T., Pavenstadt H., Hsu H.H., Schlondorff J., Ramos A., Greka A. Antagonistic regulation of actin dynamics and cell motility by TRPC5 and TRPC6 channels. Sci. Signal. 3(145): ra77. 2010.
Huber T.B., Schermer B., Müller R.U., Höhne M., Bartram M., Calixto A., Hagmann H., Reinhardt C., Koos F., Kunzelmann K., Shirokova E., Krautwurst D., Harteneck C., Simons M., Pavenstädt H., Kerjaschki D., Thiele C., Walz G., Chalfie M., Benzing T. Podocin and MEC-2 bind cholesterol to regulate the activity of associated ion channels. Proc. Natl. Acad. Sci. USA. 103(46): 17079–17086. 2006.
Anderson M., Kim E.Y., Hagmann H., Benzing T., Dryer S.E. Opposing effects of podocin on the gating of podocyte TRPC6 channels evoked by membrane stretch or diacylglycerol. Am. J. Physiol. Cell Physiol. 305(3): 276–289. 2013.
Kim E.Y., Choi K.J., Dryer S.E. Nephrin binds to the COOH terminus of a largeconductance Ca2+-activated K+ channel isoform and regulates its expression on the cell surface. Am. J. Physiol. Renal Physiol. 295(1): 235–246. 2008.
Kim E.Y., Anderson M., Wilson C., Hagmann H., Benzing T., Dryer S.E. NOX2 interacts with podocyte TRPC6 channels and contributes to their activation by diacylglycerol: essential role of podocin in formation of this complex. Am. J. Physiol. Cell Physiol. 305(9): 960–971. 2013.
Dryer S.E., Reiser J. TRPC6 channels and their binding partners in podocytes: role in glomerular filtration and pathophysiology. Am. J. Physiol. Renal Physiol. 299(4): 689–701. 2010.
Roselli S., Gribouval O., Boute N., Sich M., Benessy F., Attié T., Gubler M.C., Antignac C. Podocin localizes in the kidney to the slit diaphragm area. Am. J. Pathol. 60(1): 131–139. 2002.
Quick K., Zhao J., Eijkelkamp N., Linley J.E., Rugiero F., Cox J.J., Raouf R., Gringhuis M., Sexton J.E., Abramowitz J., Taylor R., Forge A., Ashmore J., Kirkwood N., Kros C.J., Richardson G.P., Freichel M., Flockerzi V., Birnbaumer L., Wood J.N. TRPC3 and TRPC6 are essential for normal mechanotransduction in subsets of sensory neurons and cochlear hair cells. Open Biol. 2(5): 120068. 2012.
Winn M.P., Conlon P.J., Lynn K.L., Howell D.N., Slotterbeck B.D., Smith A.H., Graham F.L., Bembe M., Quarles L.D., Pericak-Vance M.A., Vance J.M. Linkage of a gene causing familial focal segmental glomerulosclerosis to chromosome 11 and further evidence of genetic heterogeneity. Genomics. 58(2): 113–120. 1999.
Santín S., Ars E., Rossetti S., Salido E., Silva I., García-Maset R., Giménez I., Ruíz P., Mendizábal S., Luciano Nieto J., Peña A., Camacho J.A., Fraga G., Cobo M.A., Bernis C., Ortiz A., de Pablos A.L., Sánchez-Moreno A., Pintos G., Mirapeix E., Fernández-Llama P., Ballarín J., Torra R., Zamora I., López-Hellin J., Madrid A., Ventura C., Vilalta R., Espinosa L., García C., Melgosa M., Navarro M., Giménez A., Cots J.V., Alexandra S., Caramelo C., Egido J., San José M.D., de la Cerda F., Sala P., Raspall F., Vila A., Daza A.M., Vázquez M., Ecija J.L., Espinosa M., Justa M.L., Poveda R., Aparicio C., Rosell J., Muley R., Montenegro J., González D., Hidalgo E., de Frutos D.B., Trillo E., Gracia S., de los Ríos F.J. TRPC6 mutational analysis in a large cohort of patients with focal segmental glomerulosclerosis. Nephrol. Dial. Transplant. 24(10): 3089–3096. 2009.
Zhu B., Chen N., Wang Z.H., Pan X.X., Ren H., Zhang W., Wang W.M. Identification and functional analysis of a novel TRPC6 mutation associated with late onset familial focal segmental glomerulosclerosis in Chinese patients. Mutat. Res. 664(1–2): 84–90. 2009.
Gigante M., Caridi G., Montemurno E., Soccio M., d’Apolito M., Cerullo G., Aucella F., Schirinzi A., Emma F., Massella L., Messina G., De Palo T., Ranieri E., Ghiggeri G.M., Gesualdo L. TRPC6 mutations in children with steroid-resistant nephrotic syndrome and atypical phenotype. Clin. J. Am. Soc. Nephrol. 6(7): 1626–1634. 2011.
Hofstra J.M., Lainez S., van Kuijk W.H., Schoots J., Baltissen M.P., Hoefsloot L.H., Knoers N.V., Berden J.H., Bindels R.J., van der Vlag J., Hoenderop J.G., Wetzels J.F., Nijenhuis T. New TRPC6 gain-of-function mutation in a non-consanguineous Dutch family with late-onset focal segmental glomerulosclerosis. Nephrol. Dial. Transplant. 28(7): 1830–1838. 2013.
Riehle M., Büscher A.K., Gohlke B.O., Kaßmann M., Kolatsi-Joannou M., Bräsen J.H., Nagel M., Becker J.U., Winyard P., Hoyer P.F., Preissner R., Krautwurst D., Gollasch M., Weber S., Harteneck C. TRPC6 G757D loss-of-function mutation associates with FSGS. J. Am. Soc. Nephrol. 27(9): 2771–2783. 2016.
Chiluiza D., Krishna S., Schumacher V.A., Schlondorff J. Gain-of-function mutations in transient receptor potential C6 (TRPC6) activate extracellular signal-regulated kinases 1/2 (ERK1/2). J. Biol. Chem. 288(25): 18407–18420. 2013.
Schlöndorff J., Del Camino D., Carrasquillo R., Lacey V., Pollak M.R. TRPC6 mutations associated with focal segmental glomerulosclerosis cause constitutive activation of NFAT-dependent transcription. Am. J. Physiol. Cell Physiol. 296(3): 558-569. 2009.
Wang Y., Jarad G., Tripathi P., Pan M., Cunningham J., Martin D.R., Liapis H., Miner J.H., Chen F. Activation of NFAT signaling in podocytes causes glomerulosclerosis. J. Am. Soc. Nephrol. 21(10): 1657-1666. 2010.
Kanda S., Harita Y., Shibagaki Y., Sekine T., Igarashi T., Inoue T., Hattori S. Tyrosine phosphorylation-dependent activation of TRPC6 regulated by PLC-γ1 and nephrin: effect of mutations associated with focal segmental glomerulosclerosis. Mol. Biol. Cell. 22(11): 1824–1835. 2011.
Buscher A.K., Kranz B., Buscher R., Hildebrandt F., Dworniczak B., Pennekamp P., Kuwertz-Bröking E., Wingen A.M., John U., Kemper M., Monnens L., Hoyer P.F., Weber S., Konrad M. Immunosuppression and renal outcome in congenital and pediatric steroid-resistant nephrotic syndrome. Clin. J. Am. Soc. Nephrol. 5(11): 2075–2084. 2010.
Obeidova L., Reiterova J., Lnenicka P., Stekrova J., Safrankova H., Kohoutova M., Tesař V. TRPC6 gene variants in Czech adult patients with focal segmental glomerulosclerosis and minimal change disease. Folia. Biol. 58(4): 173–176. 2012.
Mir S., Yavascan O., Berdeli A., Sozeri B. TRPC6 gene variants in Turkish children with steroid-resistant nephrotic syndrome. Nephrol. Dial. Transplant. 27(1): 205–209. 2012.
Barua M., Brown E.J., Charoonratana V.T., Genovese G., Sun H., Pollak M.R. Mutations in the INF2 gene account for a significant proportion of familial but not sporadic focal and segmental glomerulosclerosis. Kidney Int. 83(2): 316–322. 2013.
Santin S., Ars E., Rossett S., Salido E., Silva I., Garcia-Maset R. Giménez I., Ruíz P., Mendizábal S., Luciano Nieto J., Peña A., Camacho J.A., Fraga G., Cobo M.A., Bernis C., Ortiz A., de Pablos A.L., Sánchez-Moreno A., Pintos G., Mirapeix E., Fernández-Llama P., Ballarín J., Torra R., Zamora I., López-Hellin J., Madrid A., Ventura C., Vilalta R., Espinosa L., García C., Melgosa M., Navarro M., Giménez A., Cots J.V., Alexandra S., Caramelo C., Egido J., San José M.D., de la Cerda F., Sala P., Raspall F., Vila A., Daza A.M., Vázquez M., Ecija J.L., Espinosa M., Justa M.L., Poveda R., Aparicio C., Rosell J., Muley R., Montenegro J., González D., Hidalgo E., de Frutos D.B., Trillo E., Gracia S., de los Ríos F.J. TRPC6 mutational analysis in a large cohort of patients with focal segmental glomerulosclerosis. Nephrol. Dial. Transplant. 24(10): 3089–3096. 2009.
Buscher A.K., Konrad M., Nagel M., Witzke O., Kribben A., Hoyer P.F., Weber S. Mutations in podocyte genes are a rare cause of primary FSGS associated with ESRD in adult patients. Clin. Nephrol. 78(1): 47–53. 2012.
Gheissari A., Meamar R., Kheirollahi M., Rouigari M., Dehbashi M., Dehghani L., Abedini A. TRPC6 mutational analysis in Iranian children with focal segmental glomerulosclerosis. Iran J. Kidney Dis. 12(6): 341–349. 2018.
Anderson M., Roshanravan H., Khine J., Dryer S.E. Angiotensin II activation of TRPC6 channels in rat podocytes requires generation of reactive oxygen species. J. Cell. Physiol. 229(4): 434–442. 2014.
Taal M.W., Brenner B.M. Renoprotective benefits of RAS inhibition: from ACEI to angiotensin II antagonists. Kidney Int. 57(5):1803-1817. 2000.
Balla T., Varnai P., Tian Y., Smith R.D. Signaling events activated by angiotensin II receptors: what goes before and after the calcium signals. Endocr. Res. 24(3-4): 335-344. 1998.
Hoffmann S., Podlich D., Hähnel B., Kriz W., Gretz N. Angiotensin II type 1 receptor overexpression in podocytes induces glomerulosclerosis in transgenic rats. J. Am. Soc. Nephrol. 15(6):1475–1487. 2004.
Roshanravan H., Dryer S.E. ATP acting through P2Y receptors causes activation of podocyte TRPC6 channels: role of podocin and reactive oxygen species. Am. J. Physiol. Renal Physiol. 306(9): 1088–1097. 2014.
Hofmann T., Obukhov A.G., Schaefer M., Harteneck C., Gudermann T., Schultz G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature. 397(6716): 259–263. 1999.
Bousquet S.M., Monet M., Boulay G. Protein kinase C-dependent phosphorylation of transient receptor potential canonical 6 (TRPC6) on serine 448 causes channel inhibition. J. Biol. Chem. 285(52): 40534–40543. 2010.
Kim E.Y., Anderson M., Dryer S.E. Insulin increases surface expression of TRPC6 channels in podocytes: role of NADPH oxidases and reactive oxygen species. Am. J. Physiol. Renal Physiol. 302(3): 298–307. 2012.
Cayouette S., Lussier M.P., Mathieu E.L., Bousquet S.M., Boulay G. Exocytotic insertion of TRPC6 channel into the plasma membrane upon Gq protein-coupled receptor activation. J. Biol. Chem. 279(8): 7241–7246. 2004.
Roshanravan H., Kim E.Y., Dryer S.E. 20-Hydroxyeicosatetraenoic acid (20-HETE) modulates canonical transient receptor potential-6 (TRPC6) channels in podocytes. Front. Physiol. 7: 351. 2016.
Basora N., Boulay G., Bilodeau L., Rousseau E., Payet M.D. 20-hydroxyeicosatetraenoic acid (20-HETE) activates mouse TRPC6 channels expressed in HEK293 cells. J. Biol. Chem. 278(34): 31709–31716. 2003.
Dryer S.E., Roshanravan H., Kim E.Y. TRPC channels: Regulation, dysregulation and contributions to chronic kidney disease. Biochim. Biophys. Acta. Mol. Basis. Dis. 1865(6): 1041–1066. 2019.
Huber T.B., Simons M., Hartleben B., Sernetz L., Schmidts M., Gundlach E., Saleem M.A., Walz G., Benzing T. Molecular basis of the functional podocin-nephrin complex: mutations in the NPHS2 gene disrupt nephrin targeting to lipid raft microdomains. Hum. Mol. Genet. 12(24): 3397–3405. 2003.
Burnham D.N., Uhlinger D.J., Lambeth J.D. Diradylglycerol synergizes with an anionic amphiphile to activate superoxide generation and phosphorylation of p47phox in a cell-free system from human neutrophils. J. Biol. Chem. 265(29): 17550–17559. 1990.
Park J.W., Babior B.M. The translocation of respiratory burst oxidase components from cytosol to plasma membrane is regulated by guanine nucleotides and diacylglycerol. J. Biol. Chem. 267(28): 19901–19906. 1992.
Qualliotine-Mann D., Agwu D.E., Ellenburg M.D., McCall C.E., McPhail L.C. Phosphatidic acid and diacylglycerol synergize in a cell-free system for activation of NADPH oxidase from human neutrophils. J. Biol. Chem. 268(32): 23843–23849. 1993.
Tyagi S.R., Neckelmann N., Uhlinger D.J., Burnham D.N., Lambeth J.D. Cell-free translocation of recombinant p47-phox, a component of the neutrophil NADPH oxidase: effects of guanosine 5'-O-(3-thiotriphosphate), diacylglycerol, and an anionic amphiphile. Biochemistry. 31(10): 2765–2774. 1992.
Ogawa N., Kurokawa T., Mori Y. Sensing of redox status by TRP channels. Cell. Calcium. 60(2): 115–122. 2016.
Kozai D., Ogawa N., Mori Y. Redox regulation of transient receptor potential channels. Antioxid. Redox Signal. 21(6): 971–986. 2014.
Samanta A., Kiselar J., Pumroy R.A., Han S., Moiseenkova-Bell V.Y. Structural insights into the molecular mechanism of mouse TRPA1 activation and inhibition. J. Gen. Physiol. 150(5): 751–762. 2018.
Pires P.W., Earley S. Redox regulation of transient receptor potential channels in the endothelium. Microcirculation. 2017 Apr; 24(3):https://doi.org/10.1111/micc.12329. 10.1111/micc.12329
Bouron A., Chauvet S., Dryer S.E., Rosado J.A. Second messenger-operated calcium entry through TRPC6. Adv. Exp. Med. Biol. 898: 201–249. 2016.
Kim E.Y., Roshanravan H., Dryer S.E. Changes in podocyte TRPC channels evoked by plasma and sera from patients with recurrent FSGS and by putative glomerular permeability factors. Biochim. Biophys. Acta. Mol. basis Dis. 1863(9): 2342–2354. 2017.
Kim E.Y., Hassanzadeh Khayyat N., Dryer S.E. Mechanisms underlying modulation of podocyte TRPC6 channels by suPAR: role of NADPH oxidases and Src family tyrosine kinases. Biochim. Biophys. Acta. Mol. Basis Dis. 1864(10): 3527–3536. 2018.
Patel A., Sharif-Naeini R., Folgering J.R., Bichet D., Duprat F., Honoré E. Canonical TRP channels and mechanotransduction: from physiology to disease states. Pflugers Arch. 460(3): 571–581. 2010.
Wilson C., Dryer S.E. A mutation in TRPC6 channels abolishes their activation by hypoosmotic stretch but does not affect activation by diacylglycerol or G protein signaling cascades. Am. J. Physiol. Renal Physiol. 306(9): 1018–1025. 2014.
Spassova M.A., Hewavitharana T., Xu W., Soboloff J., Gill D.L. A common mechanism underlies stretch activation and receptor activation of TRPC6 channels. Proc. Natl. Acad. Sci. USA. 103(44): 16586–16591. 2006.
Brenner B.M., Troy J.L., Daugharty T.M. The dynamics of glomerular ultrafiltration in the rat. J. Clin. Invest. 50(8): 1776–1780. 1971.
Endlich N., Endlich K. The challenge and response of podocytes to glomerular hypertension. Semin. Nephrol. 32(4): 327–341. 2012.
Browman D.T., Hoegg M.B., Robbins S.M. The SPFH domain-containing proteins: more than lipid raft markers. Trends Cell. Biol. 17(8): 394–402. 2007.
Relle M., Cash H., Brochhausen C., Strand D., Menke J., Galle P.R., Schwarting A. New perspectives on the renal slit diaphragm protein podocin. Mod. Pathol. 24(8): 1101–1110. 2011.
Moshourab R.A., Wetzel C., Martinez-Salgado C., Lewin G.R. Stomatin-domain protein interactions with acid-sensing ion channels modulate nociceptor mechanosensitivity. J. Physiol. 591(22): 5555–5574. 2013.
O'Hagan R., Chalfie M., Goodman M.B. The MEC-4 DEG/ENaC channel of Caenorhabditis elegans touch receptor neurons transduces mechanical signals. Nat. Neurosci. 8(1): 43–50. 2005.
Price M.P., Thompson R.J., Eshcol J.O., Wemmie J.A., Benson C.J. Stomatin modulates gating of acid-sensing ion channels. J. Biol. Chem. 279(51): 53886–53891. 2004.
Brand J., Smith E.S., Schwefel D., Lapatsina L., Poole K., Omerbašić D., Kozlenkov A., Behlke J., Lewin G.R., Daumke O. A stomatin dimer modulates the activity of acid-sensing ion channels. EMBO J. 31(17): 3635–3646. 2012.
Poole K., Herget R., Lapatsina L., Ngo H.D., Lewin G.R. Tuning Piezo ion channels to detect molecular-scale movements relevant for fine touch. Nat. Commun. 5: 3520. 2014.
Lewis A.H., Grandl J. Mechanical sensitivity of Piezo1 ion channels can be tuned by cellular membrane tension. Elife. 4: e12088. 2015.
Möller C.C., Wei C., Altintas M.M., Li J., Greka A., Ohse T., Pippin J.W., Rastaldi M.P., Wawersik S., Schiavi S., Henger A., Kretzler M., Shankland S. J., Reiser J. Induction of TRPC6 channel in acquired forms of proteinuric kidney disease. J. Am. Soc. Nephrol. 18(1): 29–36. 2007.
Rusnak F., Mertz P. Calcineurin: form and function. Physiol. Rev. 80(4): 1483–1521. 2000.
Klee C.B., Crouch T.H., Krinks M.H. Calcineurin: a calcium- and calmodulinbinding protein of the nervous system. Proc. Natl. Acad. Sci. USA. 76(12): 6270–6273. 1979.
Li H., Rao A., Hogan P.G. Interaction of calcineurin with substrates and targeting proteins. Trends Cell. Biol. 21(2): 91–103. 2011.
Song R., Li J., Zhang J., Wang L., Tong L., Wang P., Yang H., Wei Q., Cai H., Luo J. Peptides derived from transcription factor EB bind to calcineurin at a similar region as the NFAT-type motif. Biochimie. 142: 158–167. 2017.
Macian F. NFAT proteins: key regulators of T-cell development and function. Nat. Rev. Immunol. 5(6): 472–484. 2005.
Müller M.R., Rao A. NFAT, immunity and cancer: a transcription factor comes of age. Nat. Rev. Immunol. 10(9): 645–656. 2010.
Nijenhuis T., Sloan A.J., Hoenderop J.G., Flesche J., van Goor H., Kistler A.D., Bakker M., Bindels R.J., de Boer R.A., Möller C.C., Hamming I., Navis G., Wetzels J.F., Berden J.H., Reiser J., Faul C., van der Vlag J. Angiotensin II contributes to podocyte injury by increasing TRPC6 expression via an NFAT-mediated positive feedback signaling pathway. Am. J. Pathol. 179(4): 1719–1732. 2011.
Ma R., Liu L., Jiang W., Yu Y., Song H. FK506 ameliorates podocyte injury in type 2 diabetic nephropathy by down-regulating TRPC6 and NFAT expression. Int. J. Clin. Exp. Pathol. 8(11): 14063–14074. 2015.
Faul C., Donnelly M., Merscher-Gomez S., Chang Y.H., Franz S., Delfgaauw J., Chang J.M., Choi H.Y., Campbell K.N., Kim K., Reiser J., Mundel P. The actin cytoskeleton of kidney podocytes is a direct target of the antiproteinuric effect of cyclosporine A. Nat. Med. 14(9): 931–938. 2008.
Asanuma K., Yanagida-Asanuma E., Faul C., Tomino Y., Kim K., Mundel P. Synaptopodin orchestrates actin organization and cell motility via regulation of RhoA signaling. Nat. Cell. Biol. 8(5): 485–491. 2006.
Gellermann J., Stefanidis C.J., Mitsioni A., Querfeld U. Successful treatment of steroid-resistant nephrotic syndrome associated with WT1 mutations. Pediatr. Nephrol. 25(7): 1285–1289. 2010.
Malina M., Cinek O., Janda J., Seeman T. Partial remission with cyclosporine A in a patient with nephrotic syndrome due to NPHS2 mutation. Pediatr. Nephrol. 24(10): 2051–2053. 2009.
Yu H., Kistler A., Faridi M.H., Meyer J.O., Tryniszewska B., Mehta D., Yue L., Dryer S.E., Reiser J. Synaptopodin limits TRPC6 podocyte surface expression and attenuates proteinuria. J. Am. Soc. Nephrol. 27(11): 3308–3319. 2016.
Verheijden K.A.T., Sonneveld R., Bakker-van Bebber M., Wetzels J.F.M., van der Vlag J., Nijenhuis T. The calcium-dependent protease calpain-1 links TRPC6 activity to podocyte injury. J. Am. Soc. Nephrol. 29(8): 2099–2109. 2018.
Peltier J., Bellocq A., Perez J., Doublier S., Dubois Y.C., Haymann J.P., Camussi G., Baud L. Calpain activation and secretion promote glomerular injury in experimental glomerulonephritis: evidence from calpastatin-transgenic mice. J. Am. Soc. Nephrol. 17(12): 3415–3423. 2006.
Gough R.E., Goult B.T. The tale of two talins - two isoforms to fine-tune integrin signaling. FEBS Letters. 592(12): 2108–2125. 2018.
Franco S.J., Rodgers M.A., Perrin B.J., Han J., Bennin D.A., Critchley D.R., Huttenlocher A. Calpain-mediated proteolysis of talin regulates adhesion dynamics. Nature. Cell. Biol. 6(10): 977–983. 2004.
Farmer L.K., Rollason R., Whitcomb D.J., Ni L., Goodliff A., Lay A.C., Birnbaumer L., Heesom K.J., Xu S.Z., Saleem M.A., Welsh G.I. TRPC6 binds to and activates calpain, independent of its channel activity, and regulates podocyte cytoskeleton, cell adhesion, and motility. J. Am. Soc. Nephrol. 30(10): 1910–1924. 2019.
Turner J.M., Bauer C., Abramowitz M.K., Melamed M.L., Hostetter T.H. Treatment of chronic kidney disease. Kidney Int. 81(4): 351–362. 2012.
Hogan J., Radhakrishnan J. The treatment of idiopathic focal segmental glomerulosclerosis in adults. Adv. Chronic Kidney Dis. 21(5): 434–441. 2014.
Rosenberg A.Z., Kopp J.B. Focal segmental glomerulosclerosis. Clin. J. Am. Soc. Nephrol. 12(3): 502–517. 2017.
Leca N. Focal segmental glomerulosclerosis recurrence in the renal allograft. Adv. Chronic Kidney Dis. 21(5): 448–452. 2014.
Breyer M.D., Susztak K. The next generation of therapeutics for chronic kidney disease. Nat. Rev. Drug Discov. 15(8): 568–588. 2016.
Qin X., Liu Y., Zhu M., Yang Z. The possible relationship between expressions of TRPC3/5 channels and cognitive changes in rat model of chronic unpredictable stress. Behav. Brain Res. 290: 180–186. 2015.
Liu Y., Liu C., Qin X., Zhu M., Yang Z. The change of spatial cognition ability in depression rat model and the possible association with down-regulated protein expression of TRPC6. Behav. Brain Res. 294: 186–193. 2015.
Bröker-Lai J., Kollewe A., Schindeldecker B., Pohle J., Nguyen Chi V., Mathar I., Guzman R., Schwarz Y., Lai A., Weißgerber P., Schwegler H., Dietrich A., Both M., Sprengel R., Draguhn Köhr G., Fakler B., Flockerzi V., Bruns D., Freichel M. Heteromeric channels formed by TRPC1, TRPC4 and TRPC5 define hippocampal synaptic transmission and working memory. EMBO J. 36(18): 2770–2789. 2017.
Дополнительные материалы отсутствуют.
Инструменты
Российский физиологический журнал им. И.М. Сеченова