

Российский физиологический журнал им. И.М. Сеченова, 2021, T. 107, № 3, стр. 332-351
Пифитрин-альфа тормозит дифференцировку вновь образованных клеток субгранулярной зоны зубчатой извилины у крыс линии Крушинского–Молодкиной при аудиогенном киндлинге
А. А. Куликов 1, *, Е. В. Наслузова 1, Н. А. Дорофеева 1, М. В. Глазова 1, Е. А. Лаврова 1, Е. В. Черниговская 1
1 Институт эволюционной физиологии и биохимии им. И.М. Сеченова, Российской академии наук
Санкт-Петербург, Россия
* E-mail: alexey.kulikov@iephb.ru
Поступила в редакцию 17.12.2020
После доработки 13.01.2021
Принята к публикации 16.01.2021
Аннотация
Одной из задач современной нейробиологии является поиск подходов для предотвращения структурных нарушений мозга, возникающих в результате эпилептиформной активности, в частности, гиппокампа. Известно, что эпилепсия приводит к увеличению пролиферативной активности в нейрогенной нише гиппокампа – в субгранулярном слое зубчатой извилины. В последние годы преобладает представление, что образование новых клеток в большей степени способствуют эпилептогенезу, а не препятствуют развитию нейродегенеративных нарушений, связанных с усилением клеточной гибели. Мы предположили, что проапоптотический белок р53 является одной из возможных терапевтических мишеней при лечении эпилепсии и ее нейродегенеративных последствий. В работе использованы крысы инбредной линии Крушинского-Молодкиной (КМ) с наследственной аудиогенной рефлекторной эпилепсией. При многократных аудиогенных стимуляциях по протоколу киндлинга, являющемуся общепризнанной моделью эпилептогенеза, эпилептиформная активность захватывает лимбическую систему мозга и кору. У крыс линии КМ показано, что при четырехкратных аудиогенных судорожных припадках наблюдается усиление пролиферативной активности, аберрантная миграция вновь образованных клеток в хилус и ускорение их дифференцировки в нейроны. Мы не выявили нарушений в уровне апоптоза и аутофагии на начальной стадии формирования височной эпилепсии. Использование химического ингибитора белка р53 пифитрин-α не привело к изменениям в уровне апоптоза и аутофагии, но вызвало усиление пролиферации и миграции вновь образованных клеток в гранулярный слой зубчатой извилины и в хилус. Однако при инактивации р53 через неделю после судорог число дифференцированных клеток в хилусе снижалось по сравнению с таковым у крыс, которым вводили растворитель блокатора, несмотря на увеличение количества вновь образованных клеток. Эти данные свидетельствует о снижении скорости дифференцировки вновь образованных клеток по нейрональному типу при инактивации белка р53. Уменьшение числа дифференцированных нейронов в хилусе позволяет рассматривать пифитрин-α как потенциальный терапевтический агент для ослабления нейродегенеративных нарушений при эпилепсии.
Нейрогенез у взрослых животных является хорошо сбалансированным процессом, и его любые нарушения могут приводить к серьезным патологиям. Эпилепсия, в отличие от большинства неврологических заболеваний, приводит к увеличению пролиферативной активности в нейрогенной нише гиппокампа – в субгранулярном слое зубчатой извилины [1–6]. Известно, что на ранних стадиях формирования эпилептического статуса наблюдается усиление нейрогенеза в гиппокампе, однако при его длительном течении интенсивность пролиферации может снижаться [5, 7–9]. В последние годы преобладает представление, что образование новых клеток при эпилептическом статусе в большей степени способствует эпилептогенезу, а не препятствуют развитию нейродегенеративных нарушений, связанных с усилением клеточной гибели [10–12]. Это связано с миграцией новообразованных клеток не в гранулярный слой зубчатой извилины, как это происходит в норме, а в хилус, где они образуют эпилептические нейрональные связи. Однако процент новообразованных эктопических нейронов в хилусе относительно невелик. Также незначителен вклад этих нейронов в формирование аберрантных мшистых волокон [13]. Необходимо учитывать, что число нервных связей, образовавшихся за счет возникновения новых гранулярных клеток, зависит также от вызванной судорогами апоптотической гибели как зрелых, так и новобразованных нейронов гиппокампа, значительная часть которых в норме погибает путем апоптоза [14]. Нейрональная гибель, вызванная судорожной активностью, может приводить к когнитивным нарушениям [15], а также вызывать дальнейшее развитие эпилепсии [16, 17].
При эпилептическом статусе также наблюдается активация аутофагии и подавление митохондриального пути запуска апоптоза, что снижает степень дегенеративных процессов в мозге [18]. Активация аутофагии при незначительных повреждающих воздействиях способствует выживаемости клеток. При тяжелых повреждениях активация аутофагии приводит к клеточной гибели [19]. Снижение уровня аутофагии приводит к возникновению спонтанных судорог, что, возможно, связано с нарушением формирования нейрональных связей [20]. Введение ингибитора mTOR и активация аутофагии вызывает снижение спрутинга, нейродегенерации и нейрогенеза [21]. В целом вопрос о значении нейрогенеза, апоптоза и аутофагии в развитии эпилептических состояний остается открытым, так же как и механизмы регуляции этих процессов.
Одной из задач современной нейробиологии является поиск подходов для предотвращения нейродегенеративных последствий судорожной активности. Наиболее традиционный подход – это торможение судорожной активности с использованием антиконвульсантов. Другой подход – попытка воздействовать на сигнальные каскады, регулирующие клеточную гибель [22]. Белок р53 является одной из возможных терапевтических мишеней при лечении эпилепсии и ее нейродегенеративных последствий, так как является одним из ключевых сигнальных белков, активирующих апоптоз при эпилепсии. Судорожная активность, вызванная введением конвульсантов, приводит к значительному увеличению экспрессии р53 в гиппокампе, особенно в поле СА3 [23]. Известно, что фармакологическая или генетическая инактивация белка р53 уменьшает клеточную гибель в гиппокампе при судорогах, вызванных введением каината. При этом продолжительность спонтанных судорог увеличивается [24]. Эти данные указывают не только на важную роль р53 в развитии нейродегенеративных нарушений гиппокампа при эпилепсии, но также и на возможность участия этого белка в формировании эпилептических состояний.
Известно, что белок р53 участвует как в регуляции апоптоза, так и аутофагии [25]. Роль р53 в регуляции аутофагии зависит от его субклеточной локализации. В ядре р53 стимулирует аутофагию, а в цитопламе ингибирует [26]. Несмотря на способность р53 как усиливать, так и подавлять аутофагию, показано, что нокаут р53 приводит к увеличению количества аутофагосом и к последующей клеточной смерти [27], что свидетельствует о его преимущественно ингибирующем действии на аутофагию. Кроме того, р53 участвует в регуляции пролиферации и дифференцировки нейрональных стволовых клеток [28, 29]. Снижение количества р53 ведет к увеличению пролиферативной̆ активности нейронов [30, 31], что может вызывать истощение пула предшественников нейронов. Кроме того, p53 является транскрипционным фактором для ряда генов, отвечающих за нейрогенез [32].
Пифитрин-α является синтетическим ингибитором р53. Предполагают, что пифитрин-α непосредственно связывается с белком р53 [33] и блокирует транслокацию в ядро, но при этом не влияет на синтез белка и его активацию протеинкиназами. При этом уменьшается способность р53 связываться со специфическими сайтами ДНК [34, 35]. Luo и соавт. показали, что блокада р53 пифитрином-α при ишемии коры не только приводит к увеличению числа нейрональных предшественников в субвентрикулярной зоне, но и способствует их выживаемости, миграции в поврежденные участки коры, дифференцировке по нейрональному типу и встраиванию в нейрональные сети [36].
Целью настоящей работы было исследование нейрогенеза, апоптоза и аутофагии, а также выявление нарушений в механизмах регуляции этих процессов у крыс линии Крушинского–Молодкиной (КМ), подвергнутых кратковременному аудиогенному киндлингу, являющемуся общепризнанной моделью эпилептогенеза [37–39]. Для выяснения роли проапоптозного белка р53 в развитии структурных изменений гиппокампа на модели начальной стадии эпилептогенеза использовали пифитрин-α, химический ингибитор проапоптотического белка р53. Также целью исследования было оценить возможность использования подобных препаратов для снижения степени нейродегенеративных последствий судорожной активности у больных эпилепсией.
МЕТОДЫ ИССЛЕДОВАНИЯ
В экспериментах были использованы крысы инбредной линии КМ с наследственной аудиогенной эпилепсией, эти животные являются моделью рефлекторной аудиогенной эпилепсии человека [40]. Первые аудиогенные стимулы вызывают у этих крыс рефлекторные судороги, сходные по механизму их инициации с характерными для рефлекторной эпилепсии у человека. Аудиогенная судорожная активность при этом возникает в стволовых отделах мозга [41]. Однако при многократных стимуляциях по протоколу киндлинга (экспериментальная модель эпилептогенеза, основанная на усилении судорожного ответа после повторяющейся эпилептогенной стимуляции) эпилептиформная активность захватывает лимбическую систему мозга и кору, о чем свидетельствует изменение характера судорожного припадка и регистрация электрофизиологических показателей коры, гиппокампа и амигдалы [41]. Аудиогенный киндлинг является моделью височной эпилепсии [37, 42]. Были отобраны крысы КМ, отвечающие на звуковой стимул полным и стабильным клонико-тоническим судорожным припадком. Крыс содержали по пять животных в индивидуальных клетках вивария при 12-часовом световом дне и свободном доступе к пище и воде. Все процедуры с использованием животных проводились согласно этическим принципам, изложенным в Европейской конвенции по защите позвоночных животных (№ 123 01/01/1991), используемых для экспериментальных и других научных целей, и были одобрены этической комиссией Института эволюционной физиологии и биохимии им. И.М. Сеченова РАН.
В опытах были использованы 5 групп крыс, схема экспериментов представлена на рис. 1.
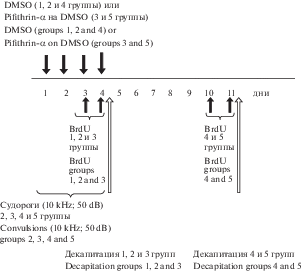
Контролем служили крысы линии КМ, которые не подвергались звуковой стимуляции и получали в течение 4-х дней инъекции DMSO, эти животные были декапитированы через 5 ч после последней инъекции (1-я группа). Также дополнительным контролем служили крысы, которые подвергались звуковой стимуляции (10 kHz, 50 dB) в течение 4-х дней и получали инъекции DMSO за 1 ч до каждого предъявления звуковой стимуляции. Крысы этих экспериментальных групп также были декапитированы через 5 ч (2-я группа) или через 7 дней (4-я группа) после последней инъекции.
Для выяснения роли белка р53 крысы подвергались звуковой стимуляции (10 kHz, 50 dB) в течение 4-х дней и получали инъекции пифитрин-α (pifithrin-α hydrobromide (R&D Systems #1267)), растворенного в DMSO (2 мг/кг массы тела) за 1 ч до каждого предъявления звуковой стимуляции. Крысы этих экспериментальных групп были декапитированы через 5 ч (3-я группа) или через 7 дней (5-я группа) после последней инъекции. Все животные дважды получали инъекции BrdU i.p. (BrdU, Sigma) (100 мг/кг) в последние 2 дня перед декапитацией.
Мозг разделяли на две половины в сагиттальной плоскости. Далее из одной половины иссекали дорсальный гиппокамп для приготовления проб для Вестерн-блот анализа. Вторую половину мозга фиксировали в 4%-ном параформальдегиде на фосфатном буфере для последующего иммуногистохимического анализа.
Иммуногистохимический метод
Чередующиеся серии срезов толщиной 10 мкм изготовляли на криостате. Во всех экспериментальных группах был проведен сравнительный морфометрический анализ зубчатой извилины и хилуса гиппокама крыс исследуемых экспериментальных групп с помощью подсчета числа ядер клеток, меченных DAPI, на единицу площади, что позволило охарактеризовать нарушения клеточного состава этих отделов гиппокампа. Уровень пролиферации и локализация новообразованных клеток были оценены при подсчете иммунопозитивных клеток, выявленных флуоресцентным иммуногистохимическим методом с помощью антител к BrdU (1 : 100, #11170376001, Roche Molecular Biochemicals), или маркера пролиферации белка Ki67 (1 : 300, #AB9260, Millipore). Направленность дифференцировки новообразованных клеток была определена методом двойного иммунофлуоресцентного выявления клеток, прошедших клеточный цикл (выявление BrdU), и коэкспрессии в этих клетках белка-маркера нейронов NeuN (1 : 500, #12943, Cell Signaling Technology) и GFAP (1 : 500, #1117514A, Invitrogen), которые выявляли с помощью вторичных антител, конъюгированных с Alexa Fluor 488 (1 : 1000, #913909, Invitrogen) и Alexa Fluor 568 (1 : 1000, #762708, Invitrogen). Ядра клеток выявляли DAPI (1 : 2000, #D9542, Sigma-Aldrich).
Уровень апоптоза нейронов и их локализация в гиппокампе были определены методом TUNEL с помощью ApopTag® Plus In Situ Apoptosis Fluorescein Detection Kit (Merck Millipore, #S7110) согласно инструкции производителя. В качестве метки в этом наборе использовался дигоксигенин. Ядра клеток выявляли DAPI в концентрации 1 : 2000. Иммуногистохимические реакции проводились одновременно на всех срезах экспериментальных крыс в стандартных условиях. Для оценки специфичности иммунного окрашивания в каждом случае делали отрицательный контроль (выполнение иммуногистохимической реакции без первичных антител), результатом чего было отсутствие иммунного окрашивания. Анализ производили с помощью светового микроскопа Zeiss Axio Imager A1 и на флуоресцентном микроскопе Leica DMI 6000B.
Вестерн-блот
Для приготовления проб из мозга иссекали гиппокамп. Образцы гомогенизировали в лизирующем буфере, содержащем ингибиторы протеаз и фосфатаз. Лизаты центрифугировали 15 мин при 12000 g при комнатной температуре, затем отбирали супернатант, после чего к двум частям отобранного супернатанта добавляли 1 часть 3× SDS буфера (0.2 М Tris-HCl, pH 6.7; 6% SDS; 15% глицерина; 0.003% Bromphenolblue и 10% B-меркаптоэтанола) и инкубировали 10 мин при температуре 96°С. Белки в пробах разделяли с помощью электрофореза в полиакриламидном геле по Лэммли (SDS-PAGE). Далее белковые фракции переносили на нитроцеллюлозную мембрану (Amersham Biosciences, Freiburg, Германия). Мембраны инкубировали в 5%-ном обезжиренном молоке или в 3%-ном растворе BSA в течение 40 мин. После этого мембраны инкубировали в растворе первых антител к исследуемым белкам β-Tubulin (1:1000; #2148, Cell Signaling), actin (1 : 1000, #ab3280, Abcam), p53 (1 : 1000; #2524, Cell Signaling), p(Ser15)-p53 (1 : 1000; #12571, Cell Signaling), clCasp3 (1 : 1000; #9661, Cell Signaling); CytC (1 : 1000, Abcam, #ab133504);); LC3B (1 : 1000; #NB100-2220, Novus Biologicals); Beclin-1 (1 : 1000; #PD017, MBL), p62 (1 : 1000; #PM045, MBL); Bcl-2 (1 : 500; #610538, Transduction laboratory), p(Ser70)-Bcl2 (1 : 1000; #05-843R, Milipore) в течение ночи при 4°С. Мембраны промывали TBS-T и помещали в раствор вторых антител на 1 ч: antiRabbit (1 : 10 000, #A5420, Sigma-Aldrich) или antiMouse (1 : 50 000, #A9044, Sigma-Aldrich). Для визуализации результатов использовали SuperSignal™ West Dura Extended Duration Substrate (#34075, ThermoFisher Scientific). Количество белка оценивалось c помощью BioRad ChemiDoc Touch и на фотопленках в программе ImageJ денситометрическим методом. Уровень специфического сигнала был нормирован по оптической плотности тубулина, что необходимо для осуществления загрузочного контроля. Денситометрический анализ проводили с помощью программы ImageJ. Данные были выражены в условных единицах оптической плотности.
Статистический анализ результатов
В исследовании использовалась малая выборка животных, из-за чего все полученные данные обрабатывались статистически с помощью непараметрического теста Краскела–Уолиса и post-hoc критерия Данна с помощью программного обеспечения GraphPad Prism 7 (GraphPad Software, США). При оценке достоверности отличий между группами число животных в каждой группе было одинаковым. В каждой экспериментальной группе крыс было исследовано по 5 животных. Данные представлены в виде среднего значения по каждой группе животных ± стандартная ошибка для среднего значения. Достоверными считались отличия при уровне значимости p < 0.05.
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
С помощью выявления маркера пролиферирующих клеток BrdU показано, что киндлинг приводит к увеличению числа меченых клеток в гранулярном слое зубчатой извилины через 4 ч и через 7 дней после окончания последнего судорожного припадка, а в хилусе только через 7 дней (рис. 2A, B). Увеличения числа меченных BrdU клеток не наблюдалось в субвентрикулярной зоне (рис. 2E). Число вновь образованных клеток, дифференцированных по нейрональному типу в гранулярном слое увеличивается только через 7 дней после завершения судорожной активности, о чем свидетельствует колокализация в них BrdU и NeuN (рис. 3A). Показано, что киндлинг приводит также к усилению миграции вновь образованных нейронов из субгранулярной зоны зубчатой извилины в хилус, причем не только через 7 дней, но и непосредственно сразу после завершения последнего судорожного припадка (рис. 3B, E). То есть процесс усиления пролиферации, дифференцировки и эктопической миграции вновь образованных нейронов, очевидно, запускается в результате первых судорожных припадков и продолжается как минимум неделю после их окончания.
Рис. 2.
Иммунофлуоресцентный анализ пролиферирующих клеток в гранулярном слое зубчатой извилины (A), в хилусе (B) и в субвентрикулярной области (E) интактных крыс линии КМ (группа 1), крыс линии КМ, подвергнутых четырем аудиогенным судорожным припадкам и выведенным из эксперимента через 4 ч (группа 2) и через 7 дней (группа 4) после последних судорог и крыс, получавших инъекции пифитрин-α за 1 ч до предъявления звукового стимула и выведенных из эксперимента через 4 ч (группа 3) и через 7 дней (группа 5) после последних судорог. По оси ординат – число BrdU позитивных клеток. По оси абсцисс – номера экспериментальных групп. Данные представлены в виде средней ± SEM. Скобкой обозначены достоверные различия р < 0.05. На микрофотографиях BrdU иммунопозитивные клетки (красный), NeuN (зеленый) и DAPI (синий) и наложение у контрольных крыс (1-я группа) (C) и крыс, получавших инъекции пифитрин-α за 1 ч до предъявления звукового стимула и выведенных из эксперимента через 7 дней (группа 5) (D). Стрелки указывают на BrdU позитивные клетки. Наконечниками стрелок указаны NeuN позитивные ядра. Fig. 2. Immunofluorescence analysis of proliferating cells in the granular layer of the dentate gyrus (A), in the hilus (B) and in the subventricular region (E). The 1st group – intact KM strain rats, the 2nd and the 4th groups – KM rats with 4 audiogenic seizures were sacrificed 4 h or 7 days following the last seizure, the 3rd groups and the 5th – KM rats with 4 audiogenic seizures that were administered pifitrin-α injection 1 h prior to acoustic stimulus and were sacrificed 4 h or 7 days following the last seizure. The y-axis of this graph represents a number of BrdU positive cells. The x-axis of this graph – number of experimental groups. Data are presented as mean ± SEM. Hooks mark a significant difference р < 0.05. Representative micrographs show BrdU (red), NeuN (green) positive cells and merge image in the group 1 (C) and 5 (D). Cells nuclei are stained by DAPI (blue). Arrows point BrdU positive cells. Arrows heads point NeuN positive cells.
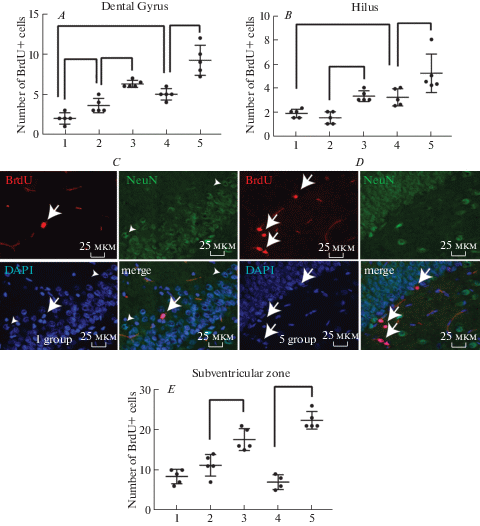
Рис. 3.
Иммунофлуоресцентный анализ дифференцированных по нейрональному (A, B) и глиальному (C, D) пути клеток в гранулярном слое зубчатой извилины (A) и в хилусе (B) интактных крыс линии КМ (группа 1), крыс линии КМ, подвергнутых четырем аудиогенным судорожным припадкам и выведенным из эксперимента через 4 ч (группа 2) и через 7 дней (группа 4) после последних судорог и крыс, получавших инъекции пифитрин-α за 1 ч до предъявления звукового стимула и выведенных из эксперимента через 4 ч (группа 3) и через 7 дней (группа 5) после последних судорог. По оси ординат – число BrdU позитивных клеток. По оси абсцисс – номера экспериментальных групп. Данные представлены в виде средней ± SEM. Скобкой обозначены достоверные различия р < 0.05. На микрофотографиях BrdU иммунопозитивные клетки (красный), NeuN (зеленый) (E) или GFAP (зеленый) и DAPI (синий) (F) и наложение у крыс, получавших инъекции DMSO за 1 ч до предъявления звукового стимула и выведенных из эксперимента через 7 дней (группа 4). Стрелки указывают на BrdU позитивные клетки. Fig. 3. Immunofluorescence analysis of neuronal (A, B) or glial (C, D) differentiation in the granular layer of the dentate gyrus (A) and in the hilus (B). The 1st group – intact KM strain rats, the 2nd and the 4th groups – KM rats with 4 audiogenic seizures were sacrificed 4 h or 7 days following the last seizure, the 3rd groups and the 5th – KM rats with 4 audiogenic seizures that were administered pifitrin-α injection 1 h prior to acoustic stimulus and were sacrificed 4 h or 7 days following the last seizure. The y-axis of this graph represents a number of double labeled BrdU/GFAP or BrdU/NeuN cells. The x-axis of this graph – number of experimental groups. Data are presented as mean ± SEM. Hooks mark a significant difference р < 0.05. Representative micrographs show BrdU (red), NeuN (green) (E) or GFAP (green) (F) positive cells and merge images in the group 4. Cell nuclei are stained by DAPI (blue). Arrows point BrdU positive cells.

Методом двойного иммуногистохимического мечения на BrdU и GFAP было показано, что под действием киндлинга только незначительный процент вновь образованных клеток в гранулярном слое зубчатой извилины гиппокампа и в хилусе крыс линии КМ коэкспрессирует GFAP (рис. 3C, D). Следовательно, краткосрочный киндлинг не влияет на образование глиальных клеток в гиппокампе крыс линии КМ.
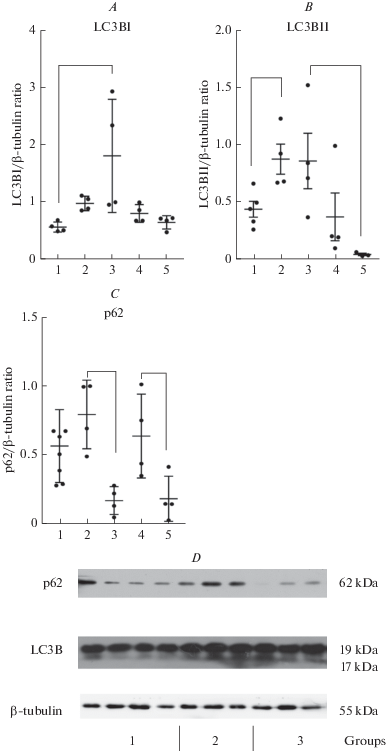
Несмотря на усиление пролиферации в результате киндлинга число клеток, выявленных с помощью подсчета количества ядер, меченных DAPI, на единицу площади гранулярного слоя зубчатой извилины и хилуса не изменялось по сравнению с контролем (рис. 4A, B). С помощью метода TUNEL (Terminal deoxynucleotidyl transferase (TdT) dUTP Nick- End Labeling) был проведен анализ уровня апоптотической гибели клеток, который также не показал различий между контрольными крысами и животными, подвергавшимися звуковой стимуляции (рис. 4C, D). Также с помощью Вестерн-блот анализа не было выявлено значимых различий в уровне экспрессии и активности сигнальных белков апоптоза (рис. 5). Через 4 ч после завершения последнего судорожного припадка было выявлено увеличение содержания белка-маркера аутофагии LC3B1 и LC3BII, что позволяет предположить некоторую активацию этого процесса, очевидно, направленную на защиту клеток гиппокампа в условиях гиперактивации (рис. 6A, B, D). Через 7 дней после завершения судорожных припадков различий в уровне экспрессии белков- регуляторов аутофагии уже не было обнаружено.
Рис. 4.
Анализ числа ядер клеток, окрашенных ядерным красителем DAPI (A, B) и клеток, выявленных методом TUNEL с помощью дигоксигенина (C, D) в гранулярном слое зубчатой извилины (A, C) и в хилусе (B, D) интактных крыс линии КМ (группа 1), крыс линии КМ, подвергнутых четырем аудиогенным судорожным припадкам и выведенным из эксперимента через 4 ч (группа 2) и через 7 дней (группа 4) после последних судорог и крыс, получавших инъекции пифитрин-α за 1 ч до предъявления звукового стимула и выведенных из эксперимента через 4 ч (группа 3) и через 7 дней (группа 5) после последних судорог. По оси ординат — число DAPI и дигоксигенин-позитивных клеток. По оси абсцисс – номера экспериментальных групп. Данные представлены в виде средней ± SEM. Достоверных изменений нет, р > 0.05. Fig. 4. Analysis of DAPI-stained nuclei (A, B) and the number of TUNEL-positive apoptotic cells (C, D) in the granular layer of the dentate gyrus (A, C) and in the hilus (B, D). The 1st group – intact KM strain rats, the 2nd and the 4th groups – KM rats with 4 audiogenic seizures were sacrificed 4 hours or 7 days following the last seizure, the 3rd groups and the 5th - KM rats with 4 audiogenic seizures that were administered pifitrin -α injection 1 hour prior to acoustic stimulus and were sacrificed 4 hours or 7 days following the last seizure. The y-axis of this graph represents a number of DAPI-stained nuclei and a number TUNEL positive cells. The x-axis of this graph – number of experimental groups. Data are presented as mean ± SEM. Difference are not significant, р > 0.05.
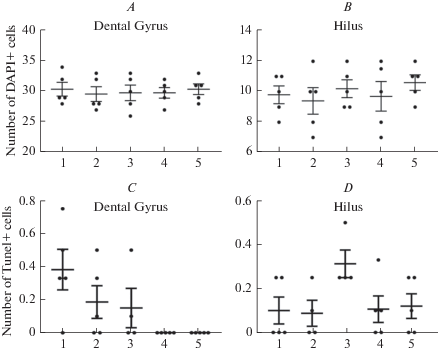
Рис. 5.
Содержание про- и антиапоптотических белков в гиппокампе крыс линии КМ, подвергнутых аудиогенной стимуляции и при химическом ингибировании белка р53, определенное с помощью Вестерн-блота. Показано достоверное снижение содержания р53 при введении пифитрин-α через 4 ч после последнего судорожного припадка (A, E). Содержание расщепленной каспазы 9 (C) и 3 (D), а также антиапоптотического белка Bcl2 (B) во всех изученных группах крыс не изменялось. По оси абсцисс на графике расположены номера экспериментальных групп. По оси ординат— оптическая плотность, измеренная в условных единицах. Данные представлены в виде средней ± SEM. Скобкой обозначены достоверные различия р < 0.05. Fig. 5. Western blot analysis of pro- and antiapoptotic protein expression in the hippocampi of KM rats that were administered pifitrin-α injection and were exposed to acoustic stimuli. Expression of p53 decreased after pifitrin-α administration in the group 3 (were sacrificed 4 h after the last seizure) (A, E). Expression of cleaved caspase 9 (C), 3 (D) and antiapoptotic protein Bcl2 (B) did not change. The y-axis of this graph represents relative optical density. The x-axis of this graph – number of experimental groups. Data are presented as mean ± SEM. Hooks mark a significant difference р < 0.05.

Рис. 6.
Содержание белков (определенное с помощью Вестерн-блота) – регуляторов аутофагии в гиппокампе крыс линии КМ, подвергнутых аудиогенной стимуляции и при химическом ингибировании белка р53. Содержание LC3BI (A, D) и LC3BII (B, D) увеличивалось через 4 ч после последних судорог, а количество LC3BII через 7 дней снижалось (B). Показано достоверное снижение содержания р62 при введении пифитрин-α через 4 ч и через 7 дней после последнего судорожного припадка (C, D). По оси абсцисс – номера экспериментальных групп. По оси ординат — оптическая плотность, измеренная в условных единицах. Данные представлены в виде средней ± SEM. Скобкой обозначены достоверные различия р < 0.05. Fig. 6. Western-blot analysis of autophagy regulator proteins in the hippocampus of KM rats that were administered pifitrin-α injection and were exposed to acoustic stimuli. Expression of LC3BI (A, D) and LC3BII (B, D) increased after 4 h from the last seizure but after 7 days expression of LC3BII decreased (B). We showed that p62 decreased after after pifitrin-α administration the 3rd and 5th groups (C, D). The y-axis of this graph represents relative optical density. The x-axis of this graph – number of experimental groups. Data are presented as mean ± SEM. Hooks marks a significant difference р < 0.05.
Введение химического ингибитора проапоптозного белка р53 пифитрин-α привело к значительному увеличению числа пролиферирующих клеток в гранулярном слое зубчатой извилины и в хилусе как через 4 ч, так и через 7 дней после окончания последнего судорожного припадка (рис. 3A–D). Также было показано увеличение количества вновь образованных клеток в субвентрикулярной зоне (рис. 3E). Однако число клеток дифференцированных по нейрональному типу при введении пифитрин-α не отличалось от характерного для контрольных животных во всех изученных отделах гиппокампа и в субвентрикулярной зоне – в отличие от показанного увеличения у животных под воздействием киндлинга (рис. 3C, E). Более того, число дифференцированных клеток в хилусе через неделю после судорог снижалось под действием пифитрин-α по сравнению с таковым у крыс, которым вводили растворитель блокатора. Также не было выявлено влияния пифитрин-α на дифференцировку вновь образованных клеток по глиальному типу (рис. 3C, D, F).
Общее число клеток в гранулярном слое зубчатой извилины и в хилусе крыс, которым на фоне многократных судорожных припадков вводили пифитрин-α, также не отличалось от контрольного уровня через как 4 ч, так и через 7 дней после окончания последнего судорожного припадка (рис. 4A, B). Уровень апоптотической гибели клеток под действием пифитрин-α не изменялся (рис. 4C, D). Однако через 4 ч после судорог было показано снижение экспрессии проапоптозного белка р53 (рис. 5A, E). При этом показано отсутствие нарушений в содержании активной расщепленной каспазы 9 и эффекторной каспазы 3, что свидетельствует об отсутствии активации митохондриального пути запуска апоптоза (рис. 5C–E). Через 7 дней после судорог нарушений в экспрессии и активности про- и антиапоптотических белков уже не наблюдалось (рис. 5A–D).
Под действием ингибитора р53 нарушался баланс белков-регуляторов аутофагии. Через 4 ч после последних судорог наблюдалось значительное снижение содержания белка р62 при неизменяющейся экспрессии и активности других белков – беклин, LC3B1 и LC3BII (рис. 6A, B, D). Через 7 сут после судорог также наблюдалось снижение содержания белка р62, но при этом также уменьшалось количество LC3BII. Полученные данные не позволяют сделать заключение об усилении или снижении уровня аутофагии, но свидетельствуют о влиянии пифитрин-α на экспрессию белков, отвечающих за регуляцию аутофагии.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Интенсивность пролиферации в субгранулярной зоне гиппокама зависит от силы и длительности судорог [9]. Мы показали, что уже четыре судорожных припадка вызывают значительное усиление пролиферативной активности в гиппокампе, при этом в субвентрикулярной зоне усиления нейрогенеза не происходит. Это указывает на то, что гиппокамп более чувствителен к судорожной активности. Повышение пролиферативной активности наблюдается уже непосредственно после завершения последнего четвертого судорожного припадка. Однако после латентного периода в течение 7 дней число пролиферирующих клеток в гранулярном слое еще возрастает. Это указывает на пролонгированное действие даже достаточно краткосрочной судорожной активности на нейрогенез в гиппокампе, что было показано и на других моделях эпилепсии [43–45]. В нормальных условиях вновь образованные нейроны встраиваются в уже существующую нейрональную сеть гранулярного слоя зубчатой извилины [46]. Однако известно, что судорожные припадки приводят к возникновению аберрантного нейрогенеза, происходящего в результате миграции вновь образованных гранулярных клеток в хилус зубчатой извилины, а не в гранулярный клеточный слой слой [11, 47–49]. Эти эктопические клетки встраиваются в нейрональную сеть зубчатой извилины и получают возбуждающую иннервацию от других гранулярных клеток. Показано, что коллатерали их аксонов образуют проекции в область CA3, где образуют синаптические контакты c мшистыми волокнами гранулярных клеток и ГАМК-ергическими интернейронами, иннервирующими пирамидные клетки СА3 [49, 50]. Таким образом, формируются циклические связи, приводящие к гипервозбуждению гиппокампа и усилению эпилептиформной активности. Кроме того, показано, что эктопические гранулярные клетки хилуса характеризуются постоянно повышенной активностью по сравнению с гранулярными клетками зубчатой извилины [51–53], что также приводит к усилению эпилептиформной активности. Мы показали, что уже на ранней стадии эпилептогенеза у крыс линии КМ происходит усиление миграции вновь образованных клеток не только в гранулярный слой зубчатой извилины, но и в большей степени в хилус, где они дифференцируются в нейроны. Причем, повышенное число вновь образованных нейронов в хилусе наблюдается как непосредственно после завершения последнего четвертого судорожного припадка, так и после латентного периода в течение 7 дней. Надо отметить, что в хилусе число дифференцированных нейронов относительно всех вновь образованных клеток выше, чем в гранулярном слое (77 и 76% в хилусе и 50% в гранулярном слое), то есть судорожная активность приводит к усилению процесса дифференцировки клеток по нейрональному пути в хилусе. При этом краткосрочный киндлинг не повлиял на образование глиальных клеток в гиппокампе крыс линии КМ. Таким образом, на начальной стадии эпилептогенеза у крыс линии КМ уже наблюдается усиление аберрантного нейрогенеза в гиппокампе.
Показано, что в норме значительная часть вновь образованных клеток гиппокампа погибает путем апоптоза [54]. Эпилептиформная активность приводит к усилению апоптотической гибели [55, 56]. Причем, апоптотической гибели при эпилепсии подвержены не только вновь образованные, но и зрелые нейроны, сформировавшиеся при развитии гиппокампа [57, 58]. Продолжительность жизни новообразованных клеток и уровень клеточной гибели, как и интенсивность пролиферации, зависят от степени формирования эпилептиформной активности [9, 59].
В фармакологических моделях височной эпилепсии при формировании эпилептичекого статуса наблюдается значительная апоптотическая гибель клеток. Под действием каиновой кислоты методом TUNEL показана локализация гибнущих клеток в хилусе и поле гиппокампа СА3, причем, максимальное число клеток гибнущих путем апоптоза было обнаружено через неделю после эпилептичекого статуса [55, 60]. В пилокарпиновой модели в гиппокампе наибольшее количество апоптотических клеток выявлялось на 3-й день после эпилептичекого статуса [61]. Однако у крыс линии WAR после 14–16-дневного аудигенного киндлинга нейродегенерации в гиппокампе обнаружено не было. Существенная потеря клеток была выявлена только в амигдале [42].
В наших экспериментах аудиогенно-чувствительные крысы линии КМ были подвергнуты четырем судорожным припадкам, что можно рассматривать как начальную стадию формирования эпилептического статуса. Мы не выявили значительных нарушений в уровне апоптотической гибели клеток в гиппокампе этих крыс. Также не было обнаружено нарушений в содержании и активности белков, отвечающих за запуск и подавление апоптоза и аутофагии. В том числе не изменялся и уровень экспрессии и фосфорилирования белка р53 по Ser15, отвечающего за запуск апоптоза [24, 62, 63] и регулирующего также и аутофагию [25–27, 64].
Известно, что р53 участвует не только в регуляции клеточной гибели, но и отвечает за пролиферацию и дифференцировку нейрональных стволовых клеток [28–30, 65]. Белок р53 является транскрипционным фактором для ряда генов, отвечающих за дифференцировку клеток по нейрональному типу [30, 66]. В опытах in vitro было показано значительное снижение уровня дифференцировки нейронов при ингибировании p53 пифитрином-α или siRNA p53 [67]. В экспериментах на первичной культуре клеток эмбриональной коры [68] и на клетках линии РС12 было выявлено усиление роста и регенерации аксонов [69, 70] при повышенной экспрессии р53. Однако в опытах in vivo было показано, что блокада р53 пифитрином-α при ишемии коры не только приводит к увеличению числа нейрональных предшественников в субвентрикулярной зоне, но и способствует их выживаемости, миграции в поврежденные участки коры, дифференцировке по нейрональному типу и встраиванию в нейрональные сети [36].
Эти противоречивые данные являлись предпосылкой к нашему исследованию роли ингибирования р53 в формировании нарушений в структуре гиппокампа на модели формирования височной эпилепсии. Мы показали, что ингибирование р53 привело к значительному увеличению числа пролиферирующих клеток в гранулярном слое зубчатой извилины и в хилусе как непосредственно, так и через 7 дней после окончания четырехкратных судорожных припадков. Это подтверждает данные литературы об активирующем влиянии снижения уровня р53 на пролиферацию [29]. Аберрантный нейрогенез, описанный при эпилептиформных состояниях различной этиологии, по современным представлениям является одним из факторов, приводящих к усилению болезни [10–12]. Уже при краткосрочном киндлинге мы показали увеличение числа клеток, дифференцированных по нейрональному типу у крыс, которые не получали инъекций пифитрин-α, что может являться одним из факторов развития заболевания. Однако под действием пифитрин-α через неделю после судорог, несмотря на увеличение количества вновь образованных клеток в хилусе, число дифференцированных клеток снижалось по сравнению с таковым у крыс, которым вводили растворитель блокатора. Эти данные свидетельствует о подавлении дифференцировки вновь образованных клеток по нейрональному типу при инактивации белка р53.
Можно предположить, что увеличение выявляемых пролиферирующих клеток связано со снижением апоптоза вновь образованных клеток при ингибировании одного из основных путей запуска апоптотической гибели. Однако количество клеток под влиянием пифитрин-α не изменялось, и не было выявлено нарушений в уровне апоптоза, оцененного с помощью TUNEL. Анализ уровня экспрессии и активности сигнальных белков апоптоза, отвечающих за его инициацию и подавление, не выявил значимых отличий от характерных для контрольных животных и крыс, подвергавшихся судорожным припадкам. Исключение составлял только проапоптозный белок р53, содержание которого в гиппокампе значительно снизилось под действием пифитрин-α.
Пониженное содержания в гиппокампе р53 также может быть одной из причин усиления аутофагии, так как известно, что, несмотря на способность р53 как усиливать, так и подавлять аутофагию, нокаут р53 приводит к увеличению количества аутофагосом и к последующей клеточной смерти [27], это свидетельствует о преимущественно его ингибирующем действии на аутофагию за счет подавления синтеза белка LC3В. Однако мы не выявили достоверного увеличения активной формы LC3В при химическом ингибировании р53. Напротив, наблюдалось снижение содержания LC3ВII – активной формы этого белка, и при этом уменьшалось содержание белка р62. р62 связывается с мембраной аутофагосом, и снижение его количества свидетельствует о деградации аутофагосом и об активации аутофагии. Однако об уровне аутофагии нельзя судить на основании изменений в экспрессии одного белка [71], а значимых отличий по другим белкам-регуляторам аутофагии под действием ингибитора р53 нами обнаружено не было.
Таким образом, на модели формирования височной эпилепсии у крыс линии КМ уже после четырех аудиогенных судорожных припадков наблюдается усиление пролиферативной активности, аберрантная миграция вновь образованных клеток в хилус и ускорение их дифференцировки в нейроны. При этом мы не выявили нарушений в уровне апоптоза и аутофагии. Блокада активности проапоптозного белка р53 при введении его химического ингибитора пифитрин-α также не повлияла на апоптоз и аутофагию. Однако введение пифитрин-α привело к усилению пролиферации и миграции вновь образованных клеток в гранулярный слой зубчатой извилины и в хилус, при этом действие ингибитора негативно отразилось на скорости и характере дифференцировки вновь образованных клеток. Снижение числа дифференцированных нейронов в хилусе позволяет рассматривать пифитрин-α как потенциальный терапевтический подход для уменьшения нейродегенеративных нарушений при эпилепсии, так как его использование может уменьшить уровень гибели зрелых нейронов и при этом предотвратить усиление аберрантного нейрогенеза.
Список литературы
Blümcke I., Schewe J.-C., Normann S., Brüstle O., Schramm J., Elger C.E., Wiestler O.D. Increase of nestin-immunoreactive neural precursor cells in the dentate gyrus of pediatric patients with early-onset temporal lobe epilepsy. Hippocampus. 11(3): 311–321. 2001.
Crespel A., Rigau V., Coubes P., Rousset M.C., De Bock F., Okano H., Baldy-Moulinier M., Bockaert J., Lerner-Natoli M. Increased number of neural progenitors in human temporal lobe epilepsy. Neurobiol. Disease. 19(3): 436–450. 2005.
Jessberger S., Parent J.M. Epilepsy and Adult Neurogenesis. Cold Spring Harb. Perspect. Biol. 7(12): a020677. 2015.
Kokaia M. Seizure-induced neurogenesis in the adult brain. Eur. J. Neurosci. 33(6): 1133–1138. 2011.
Scharfman H.E., Gray W.P. Relevance of seizure-induced neurogenesis in animal models of epilepsy to the etiology of temporal lobe epilepsy. Epilepsia. 48. Suppl 2: 33–41. 2007.
Thom M., Martinian L. Fau-Williams G., Williams G. Fau- toeber K., Stoeber K. Fau-Sisodiya S.M., Sisodiya S.M. Cell proliferation and granule cell dispersion in human hippocampal sclerosis. (0022-3069 (Print)). 2005.
Romcy-Pereira R.N., Garcia-Cairasco N. Hippocampal cell proliferation and epileptogenesis after audiogenic kindling are not accompanied by mossy fiber sprouting or fluoro-jade staining. Neuroscience. 119(2): 533–546. 2003.
Bengzon J., Kokaia Z., Elmer E., Nanobashvili A., Kokaia M., Lindvall O. Apoptosis and proliferation of dentate gyrus neurons after single and intermittent limbic seizures. Proc. Natl. Acad. Sci. USA. 94(19): 10432–10437. 1997.
Mohapel P., Ekdahl C.T., Lindvall O. Status epilepticus severity influences the long-term outcome of neurogenesis in the adult dentate gyrus. Neurobiol. Disease. 15(2): 196–205. 2004.
Cho K.O., Lybrand Z.R., Ito N., Brulet R., Tafacory F., Zhang L., Good L., Ure K., Kernie S.G., Birnbaum S.G., Scharfman H.E., Eisch A.J., Hsieh J. Aberrant hippocampal neurogenesis contributes to epilepsy and associated cognitive decline. Nat. Commun. 6: 6606. 2015.
Danzer S.C. Contributions of Adult-Generated Granule Cells to Hippocampal Pathology in Temporal Lobe Epilepsy: A Neuronal Bestiary. Brain Plast. 3(2): 169–181. 2018.
Santos V.R., Melo I.S., Pacheco A.L.D., Castro O.W. Life and death in the hippocampus: What’s bad? Epilepsy Behav. 106595. 2019.
Liu J.X., Hu M., Zhu K., Chen X.L., Zhang J.S., Yuan B., Liu Y. Residual neurogenesis in chronically epileptic hippocampus of mice. Epilepsy Res. 127: 40–49. 2016.
Mohapel P., Ekdahl C.T., Lindvall O. Status epilepticus severity influences the long-term outcome of neurogenesis in the adult dentate gyrus. Neurobiol. Dis. 15(2): 196–205. 2004.
Kotloski R., Lynch M., Lauersdorf S., Sutula T. Repeated brief seizures induce progressive hippocampal neuron loss and memory deficits. Prog. Brain Res. 135: 95–110. 2002.
Walker M. Neuroprotection in epilepsy. Epilepsia. 48. Suppl 8: 66–68. 2007.
Zhang X., Cui S.S., Wallace A.E., Hannesson D.K., Schmued L.C., Saucier D.M., Honer W.G., Corcoran M.E. Relations between brain pathology and temporal lobe epilepsy. J Neurosci. 22(14): 6052–6061. 2002.
Wang B.H., Hou Q., Lu Y.Q., Jia M.M., Qiu T., Wang X.H., Zhang Z.X., Jiang Y. Ketogenic diet attenuates neuronal injury via autophagy and mitochondrial pathways in pentylenetetrazol-kindled seizures. Brain Res. 1678: 106–115. 2018.
Rodriguez-Vargas J.M., Ruiz-Magana M.J., Ruiz-Ruiz C., Majuelos-Melguizo J., Peralta-Leal A., Rodriguez M.I., Munoz-Gamez J.A., De Almodovar M.R., Siles E., Rivas A.L., Jaattela M., Oliver F.J. ROS-induced DNA damage and PARP-1 are required for optimal induction of starvation-induced autophagy. Cell Res. 22(7): 1181–1198. 2012.
Mcmahon J., Huang X., Yang J., Komatsu M., Yue Z., Qian J., Zhu X., Huang Y. Impaired autophagy in neurons after disinhibition of mammalian target of rapamycin and its contribution to epileptogenesis. J. Neurosci. 32(45): 15704–15714. 2012.
Giorgi F.S., Biagioni F., Lenzi P., Frati A., Fornai F. The role of autophagy in epileptogenesis and in epilepsy-induced neuronal alterations. J. Neural Transm. (Vienna). 122(6): 849–862. 2015.
Meldrum B.S. Implications for neuroprotective treatments. Prog. Brain Res. 135: 487–495. 2002.
Ding D.-X., Tian F.-F., Guo J.-L., Li K.a.I., He J.-X., Song M.-Y., Li L.I., Huang X.I.A. Dynamic expression patterns of ATF3 and p53 in the hippocampus of a pentylenetetrazole-induced kindling model. Mol. Med. Reports. 10(2): 645–651. 2014.
Engel T., Tanaka K., Jimenez-Mateos E.M., Caballero-Caballero A., Prehn J.H., Henshall D.C. Loss of p53 results in protracted electrographic seizures and development of an aggravated epileptic phenotype following status epilepticus. Cell. Death. Dis. 1: e79. 2010.
White E. Autophagy and p53. Cold Spring Harb. Perspect. Med. 6(4): a026120. 2016.
Tasdemir E., Chiara M. M., Morselli E., Criollo A., D’amelio M., Djavaheri-Mergny M., Cecconi F., Tavernarakis N., Kroemer G. A dual role of p53 in the control of autophagy. Autophagy. 4(6): 810–814. 2008.
Scherz-Shouval R., Weidberg H., Gonen C., Wilder S., Elazar Z., Oren M. p53-dependent regulation of autophagy protein LC3 supports cancer cell survival under prolonged starvation. Proc. Natl. Acad. Sci. USA. 107(43): 18511–18516. 2010.
Meletis K., Wirta V., Hede S.-M., Nistér M., Lundeberg J., Frisén J. p53 suppresses the self-renewal of adult neural stem cells. Development. 133(2): 363–369. 2006.
Grigor’eva Y.S.,Glazova M. The role of p53 in the proliferation and differentiation of neural stem cells in organotypic cultures of mouse hippocampus. J. Neurosci. Behav. Physiol. 45(6): 627–636. 2015.
Glazova M. The role of p53 protein in the regulation of neuronal differentiation. J. Neurosci. Behav. Physiol. 46(9): 984–991. 2016.
Luo Y., Kuo C.C., Shen H., Chou J., Greig N.H., Hoffer B.J., Wang Y. Delayed treatment with a p53 inhibitor enhances recovery in stroke brain. Ann. Neurol. 65(5): 520–530. 2009.
Zhou D., Zhang Z., He L.M., Du J., Zhang F., Sun C.K., Zhou Y., Wang X.W., Lin G., Song K.M., Wu L.G., Yang Q. Conversion of fibroblasts to neural cells by p53 depletion. Cell Rep. 9(6): 2034–2042. 2014.
Culmsee C., Zhu X., Yu Q.S., Chan S.L., Camandola S., Guo Z., Greig N.H., Mattson M.P. A synthetic inhibitor of p53 protects neurons against death induced by ischemic and excitotoxic insults, and amyloid beta-peptide. J. Neurochem. 77(1): 220–228. 2001.
Pietrancosta N., Moumen A., Dono R., Lingor P., Planchamp V., Lamballe F., Bähr M., Kraus J.-L., Maina F. Imino-tetrahydro-benzothiazole Derivatives as p53 Inhibitors: Discovery of a Highly Potent in Vivo Inhibitor and Its Action Mechanism. J. Med. Chem. 49(12): 3645–3652. 2006.
Sohn D., Graupner V., Neise D., Essmann F., Schulze-Osthoff K., Janicke R.U. Pifithrin-[alpha] protects against DNA damage-induced apoptosis downstream of mitochondria independent of p53. Cell. Death Differ. 16(6): 869–878. 2009.
Luo Y., Kuo C.-C., Shen H., Chou J., Greig N.H., Hoffer B.J., Wang Y. Delayed treatment with a p53 inhibitor enhances recovery in stroke brain. Ann. Neurol. 65(5): 520–530. 2009.
Garcia-Cairasco N., Umeoka E.H., De Oliveira J.C. The Wistar Audiogenic Rat (WAR) strain and its contributions to epileptology and related comorbidities: history and perspectives. J. Epilepsy Behav. 71: 250–273. 2017.
Vinogradova L.V. Audiogenic kindling in Wistar and WAG/Rij rats: Kindling-prone and kindling-resistant subpopulations. J. Epilepsia. 49(10): 1665–1674. 2008.
Vinogradova L.V. Audiogenic kindling and secondary subcortico-cortical epileptogenesis: Behavioral correlates and electrographic features. J. Epilepsy Behav. 71: 142–153. 2017.
Poletaeva I., Surina N., Kostina Z., Perepelkina O., Fedotova I. The Krushinsky-Molodkina rat strain: The study of audiogenic epilepsy for 65 years. Epilepsy & Behavior. 71: 130–141. 2017.
Faingold C.L. Neuronal networks in the genetically epilepsy-prone rat. Adv. Neurol. 79: 311–321. 1999.
Galvis-Alonso O.Y., Cortes De Oliveira J.A., Garcia-Cairasco N. Limbic epileptogenicity, cell loss and axonal reorganization induced by audiogenic and amygdala kindling in wistar audiogenic rats (WAR strain). Neuroscience. 125(3): 787–802. 2004.
Parent J.M., Yu T.W., Leibowitz R.T., Geschwind D.H., Sloviter R.S., Lowenstein D.H. Dentate Granule Cell Neurogenesis Is Increased by Seizures and Contributes to Aberrant Network Reorganization in the Adult Rat Hippocampus. J. Neurosci. 17(10): 3727–3738. 1997.
Gray W.P., Sundstrom L.E. Kainic acid increases the proliferation of granule cell progenitors in the dentate gyrus of the adult rat. Brain Res. 790(1–2): 52–59. 1998.
Jessberger S., Römer B., Babu H., Kempermann G. Seizures induce proliferation and dispersion of doublecortin-positive hippocampal progenitor cells. Exp. Neurol. 196(2): 342–351. 2005.
Kempermann G. Adult Neurogenesis, in Neuroscience in the 21st Century: From Basic to Clinical. New York. Springer. 161–178. 2013.
Danzer S.C. Depression, stress, epilepsy and adult neurogenesis. Exp. Neurol. 233(1): 22–32. 2012.
Parent J.M. Adult neurogenesis in the intact and epileptic dentate gyrus. In: Progress in Brain Research. Elsevier. 529–817. 2007.
Scharfman H.E., Pierce J.P. New insights into the role of hilar ectopic granule cells in the dentate gyrus based on quantitative anatomic analysis and three-dimensional reconstruction. Epilepsia. 53. Suppl. 1: 109–115. 2012.
Buckmaster P.S., Zhang G.F., Yamawaki R. Axon sprouting in a model of temporal lobe epilepsy creates a predominantly excitatory feedback circuit. J. Neurosci. 22(15): 6650–6658. 2002.
Dashtipour K., Tran P.H., Okazaki M.M., Nadler J.V., Ribak C.E. Ultrastructural features and synaptic connections of hilar ectopic granule cells in the rat dentate gyrus are different from those of granule cells in the granule cell layer. Brain Res. 890(2): 261–271. 2001.
Murphy B.L., Pun R.Y., Yin H., Faulkner C.R., Loepke A.W., Danzer S.C. Heterogeneous integration of adult-generated granule cells into the epileptic brain. J. Neurosci. 31(1): 105–117. 2011.
Scharfman H.E., Goodman J.H., Sollas A.L. Granule-Like Neurons at the Hilar/CA3 Border after Status Epilepticus and Their Synchrony with Area CA3 Pyramidal Cells: Functional Implications of Seizure-Induced Neurogenesis. J. Neurosci. 20(16): 6144–6158. 2000.
Ryu J.R., Hong C.J., Kim J.Y., Kim E.K., Sun W., Yu S.W. Control of adult neurogenesis by programmed cell death in the mammalian brain. Mol. Brain. 9: 43. 2016.
Liang S., Zhang L., Yu X., Zhang S., Zhang G., Ding P. Neuroprotective Effect of Electric Conduction Treatment on Hippocampus Cell Apoptosis in KA Induced Acute Temporal Lobe Epileptic Rats. Brain Stimul. 9(6): 933–939. 2016.
Henshall D.C., Simon R.P. Epilepsy and apoptosis pathways. J. Cerebr. Blood Flow Metab. 25(12): 1557–1572. 2005.
Zhu H., Dahlstrom A., Hansson H.A. Characterization of cell proliferation in the adult dentate under normal conditions and after kainate induced seizures using ribonucleotide reductase and BrdU. Brain Res. 1036(1–2): 7–17. 2005.
Ekdahl C.T., Zhu C., Bonde S., Bahr B.A., Blomgren K., Lindvall O. Death mechanisms in status epilepticus-generated neurons and effects of additional seizures on their survival. Neurobiol. Dis. 14(3): 513–523. 2003.
Cardoso A., Lukoyanova E.A., Madeira M.D., Lukoyanov N.V. Seizure-induced structural and functional changes in the rat hippocampal formation: comparison between brief seizures and status epilepticus. Behav. Brain Res. 225(2): 538–546. 2011.
Pollard H., Charriaut-Marlangue C., Cantagrel S., Represa A., Robain O., Moreau J., Ben-Ari Y. Kainate-induced apoptotic cell death in hippocampal neurons. Neuroscience. 63(1): 7–18. 1994.
Roux P.P., Colicos M.A., Barker P.A., Kennedy T.E. p75 neurotrophin receptor expression is induced in apoptotic neurons after seizure. J. Neurosci. 19(16): 6887–6896. 1999.
Ding D.X., Tian F.F., Guo J.L., Li K., He J.X., Song M.Y., Li L., Huang X. Dynamic expression patterns of ATF3 and p53 in the hippocampus of a pentylenetetrazole-induced kindling model. Mol. Med. Rep. 10(2): 645–651. 2014.
Fujikawa D.G. Prolonged seizures and cellular injury: understanding the connection. Epilepsy Behav. 7. Suppl. 3: S3–S11. 2005.
Nikoletopoulou V., Markaki M., Palikaras K., Tavernarakis N. Crosstalk between apoptosis, necrosis and autophagy. Biochim. Biophys. Acta. 1833(12): 3448–3459. 2013.
Van Lookeren Campagne M., Gill R. Tumor-suppressor p53 is expressed in proliferating and newly formed neurons of the embryonic and postnatal rat brain: comparison with expression of the cell cycle regulators p21Waf1/Cip1, p27Kip1, p57Kip2, p16Ink4a, cyclin G1, and the proto-oncogene Bax. J. Comp. Neurol. 397(2): 181–198. 1998.
Zhou D., Zhang Z., He L.-M., Du J., Zhang F., Sun C.-K., Zhou Y., Wang X.-W., Lin G., Song K.-M., Wu L.-G., Yang Q. Conversion of Fibroblasts to Neural Cells by p53 Depletion. Cell Reports. 9(6): 2034–2042. 2014.
Eom H.S., Park H.R., Jo S.K., Kim Y.S., Moon C., Jung U. Ionizing radiation induces neuronal differentiation of Neuro-2a cells via PI3-kinase and p53-dependent pathways. Int. J. Radiat. Biol. 91(7): 585–595. 2015.
Di Giovanni S., Knights C.D., Rao M., Yakovlev A., Beers J., Catania J., Avantaggiati M.L., Faden A.I. The tumor suppressor protein p53 is required for neurite outgrowth and axon regeneration. The EMBO J. 25(17): 4084–4096. 2006.
Hughes A.L., Gollapudi L., Sladek T.L., Neet K.E. Mediation of nerve growth factor-driven cell cycle arrest in PC12 cells by p53. Simultaneous differentiation and proliferation subsequent to p53 functional inactivation. J. Biol. Chem. 275(48): 37829–37837. 2000.
Montano X. P53 associates with trk tyrosine kinase. Oncogene. 15(3): 245–256. 1997.
Klionsky D.J., Abdelmohsen K., Abe A., Abedin M.J. Guidelines for the use and interpretation of assays for monitoring autophagy. J. Autophagy. 12(1): 1–222. 2016.
Дополнительные материалы отсутствуют.
Инструменты
Российский физиологический журнал им. И.М. Сеченова