

Российский физиологический журнал им. И.М. Сеченова, 2021, T. 107, № 4-5, стр. 647-660
Взаимодействие механизмов угнетения квантового выделения ацетилхолина при активации ваниллоидных (TRPV1) и пуриновых рецепторов в нервно-мышечном синапсе мыши
А. Ю. Архипов 1, Н. В. Жиляков 1, А. И. Маломуж 1, Д. В. Самигуллин 1, 2, *
1 Казанский институт биохимии и биофизики Казанского научного центра РАН
Казань, Россия
2 Казанский национальный исследовательский технический университет им. А.Н. Туполева
Казань, Россия
* E-mail: samid75@mail.ru
Поступила в редакцию 19.01.2021
После доработки 05.02.2021
Принята к публикации 05.02.2021
Аннотация
Основной целью исследования стало изучение взаимосвязи между сигнальными путями регуляции квантового выделения ацетилхолина (АХ) в периферическом синапсе, которые инициируются активацией ваниллоидных и пуриновых рецепторов. В электрофизиологических экспериментах, проведенных на нервно-мышечном синапсе m. Levator Auris Longus мыши, было установлено, что частота миниатюрных потенциалов концевой пластинки (мПКП) и квантовый состав потенциалов концевой пластинки (ПКП) уменьшаются в присутствии агониста ваниллоидных рецепторов (TRPV1) капсаицина. Данный эффект полностью устранялся соединением SB 366791, специфическим конкурентным антагонистом TRPV1-рецепторов. АТФ, так же, как и капсаицин, снижал частоту мПКП и квантовый состав ПКП. На фоне антагониста TRPV1 угнетающий эффект АТФ на секрецию АХ реализовывался в полном объеме. В то же время на фоне активации TRPV1-каналов капсаицином действие АТФ как на спонтанную, так и на вызванную секрецию АХ отсутствовало. Было сделано предположение, что в основе механизмов действия АТФ и капсаицина может лежать изменение входа Са2+ в нервное окончание. Для проверки этой гипотезы были проведены эксперименты по оценке изменений пресинаптического уровня кальция (Са2+-транзиента) при помощи флуоресцентного кальциевого красителя при стимуляции нерва. Амплитуда Са2+-транзиента не изменялась ни при аппликации АТФ, ни при добавлении капсаицина. Таким образом, в нервно-мышечном синапсе млекопитающих, наряду с пуринергическим путем регуляции АХ, имеет место и механизм модуляции нейросекреции, опосредованный активацией TRPV1-каналов. Запуск этих механизмов приводит к угнетению процессов как спонтанного, так и вызванного выделения квантов АХ из двигательного нервного окончания. Оба пути регуляции не сопровождаются изменением Са2+-транзиента, но имеют общее звено в регуляции квантового выброса медиатора.
ВВЕДЕНИЕ
Синаптическая передача в нервно-мышечном соединении опосредуется высвобождением из нервного окончания молекул ацетилхолина (АХ), которые связываются с рецепторами АХ в постсинаптической области сарколеммы, что приводит к возникновению спонтанных или вызванных потенциалов концевой пластинки [1]. Модуляция синаптической передачи может опосредоваться как собственно молекулами АХ [2], так и синаптически активными молекулами, способными выделяться как из двигательной нервной терминали, так и из глиальных клеток. В качестве таких регуляторов могут выступать молекулы аденозин-5'-трифосфата (АТФ) и его производные, глутамат, N-ацетиласпартилглутамат, вещество P (нейропептид из семейства тахикининов), оксид азота (NO) и др. [3–7].
Одним из наиболее активно изучаемых регуляторов нервно-мышечной трансмиссии является АТФ, который выделяется из синаптических везикул вместе с АХ [8]. Не только АТФ, но и его производные (АДФ, АМФ и аденозин) способны оказывать модуляторное действие на синаптическую передачу, активируя широкий спектр пуриновых рецепторов, среди которых выделяют две большие группы: рецепторы аденозина, или P1, и рецепторы P2, которые активируются АТФ, АДФ, уридин-5'-дифосфатом и уридин-5'-трифосфатом [8, 9]. АТФ и аденозин снижают уровень квантового высвобождения АХ, активируя пресинаптические пуриновые рецепторы [10, 11]. Предположительно, вышеупомянутые эффекты связаны с подавлением входа кальция в нервное окончание [10, 12].
АТФ является важным медиатором химически индуцированной ноцицепции и боли, высвобождаясь из поврежденной и/или воспаленной ткани [13]. АТФ сенситизирует каналы первого типа подсемейства TRPV, называемые ваниллоидными каналами (англ. “transient receptor potential cation channels”, TRPV1), активируемые капсаицином или протонами [14, 15]. Помимо протонов, в качестве эндогенных активаторов могут выступать такие воспалительные агенты, как “эндованиллоиды” (анандамид, N-арахидоноил дофамин и др.) [14, 16]. Получены данные, указывающие на присутствие TRPV1-каналов в периферических нервно-мышечных синапсах [17]. Активация этих каналов приводит к снижению количества выделяемых квантов АХ при стимуляции двигательного нерва [18].
Таким образом, механизм снижения выделения АХ из нервного окончания может запускаться активацией как пуриновых рецепторов, так и TRPV1-каналов. В связи с этим встал вопрос о наличии или отсутствии взаимосвязи между сигнальными путями регуляции выделения АХ, которые инициируются пуриновыми и ваниллоидными рецепторами. Также встает вопрос о том, могут ли в механизмах модуляторного действия пуринов и агонистов TRPV1-рецепторов на квантовую секрецию АХ в периферических нервных окончаниях теплокровных быть задействованы цепочки, связанные с изменением пресинаптического уровня кальция. Поиск ответов на эти вопросы и стал целью настоящего исследования.
МЕТОДЫ ИССЛЕДОВАНИЯ
Экспериментальные процедуры выполняли в соответствии с инструкциями по использованию лабораторных животных Казанского федерального университета и Казанского медицинского университета в соответствии с руководством NIH по уходу и использованию лабораторных животных. Протокол эксперимента соответствовал требованиям Директивы Совета европейских сообществ 86/609/EEC.
Исследования были проведены на изолированных нервно-мышечных препаратах мыши (линия BALB/c, m. Levator Auris Longus). Нервно-мышечные препараты помещали в ванночку объемом 3 мл, дно которой было покрыто смолой Sylgard (Dow Corning, США). Препараты растягивали на 15% от их исходного размера в покое при помощи микроигл из нержавеющей стали. Через ванночку протекал раствор со скоростью 3 мл/мин. следующего состава (в мМ): 137 NaCl; 5 KCl; 1 MgCl2; 2 CaCl2; 1 Na2HPO4; 15 NaHCO3; 11 глюкоза. Раствор аэрировали карбогеном (95% O2, 5% CO2), pH раствора составлял 7.2–7.4, температуру в ванночке поддерживали встроенными элементами Пельтье на уровне 20.0 ± 0.3°C.
Нерв раздражали прямоугольными стимулами длительностью 0.25 мс супрамаксимальной амплитуды с частотой 0.5 Гц при помощи всасывающего электрода. Во всех экспериментальных сериях сокращения нервно-мышечного препарата блокировали путем добавления в омывающий раствор μ-конотоксина GIIIB 2 мкМ (Peptide Institute Inc., Япония).
В ходе экспериментов методами внутриклеточного отведения потенциала осуществлялась регистрация вызванных и спонтанных (миниатюрных) потенциалов концевой пластинки (ПКП и мПКП). Электроды для измерения потенциалов изготавливались на приборе P-97 Micropipette Puller (Sutter Instrument, США). Сопротивление электродов составляло порядка 30 МОм. Заполнялись электроды 3-х молярным раствором KCl. Регистрация и анализ постсинаптических сигналов осуществлялись с помощью установки для электрофизиологических исследований, включающей в себя: микроскоп BX 51 (Olympus, Япония) с водно-иммерсионным объективом (увеличение 60×, апертура 1.00), двухканальный усилитель биопотенциалов AxoClamp 900A (Axon Instruments, США), стимулятор 2100 (A-M Systems, США), аналого-цифровой преобразователь Digidata 1440A (Axon Instruments, США) с частотой дискретизации 100 кГц, микроманипуляторы MPC-200 (Sutter Instruments, США) и компьютер. Для регистрации мембранного потенциала и непрерывной записи событий во время эксперимента к усилителю также был подключен медленный аналого-цифровой преобразователь MiniDigi 1B (Axon Instruments, США) с частотой дискретизации 1 кГц.
Обработка и регистрация данных производилась в программном пакете pCLAMP 10.4. Величина мембранного потенциала, при котором начинали регистрацию, варьировала в диапазоне 65–75 мВ. Результаты экспериментов, в которых мембранный потенциал покоя изменялся в течение опыта более, чем на 10 мВ не анализировались. В каждом эксперименте в одном синаптическом контакте регистрировали 30–50 вызванных ПКП, после чего, в течение 2-х минут записывали мПКП в контроле и после действия фармакологических агентов. Квантовый состав находили путем деления усредненных амплитуд вызванных ПКП на среднюю амплитуду мПКП. Для коррекции квантового состава на нелинейность суммации использовали поправку Мартина [19].
Для регистрации относительного изменения уровня Са2+ в пресинаптической клетке (Са2+-транзиента) использовалась фотометрическая система на базе микроскопа Olympus BX 51, оснащенная высокоскоростной чувствительной камерой Red Shirt Imaging Neuro CCD-smq camera (Red Shirt Imaging, США). Регистрация Са2+‑транзиента осуществлялась с использованием специфического Са2+-чувствительного флуоресцентного красителя Oregon Green 488 Bapta 1 hexapotassium salt (Molecular Probes, США). Загрузку кальциевого красителя (1 мМ) в двигательные моторные окончания осуществляли через культю нерва [20].
Для записи и обработки данных в экспериментах по измерению Са2+-транзиента использовались программы Turbo-SM и ImageJ. Данные представлены в виде отношения: (ΔF/F0 – 1) × 100%, где ΔF – интенсивность флуоресценции во время стимуляции, F0 – интенсивность флуоресценции в состоянии покоя.
Эксперименты осуществляли по следующему протоколу: записывали контрольные сигналы, после чего апплицировали вещество с помощью системы общей перфузии в течение 20 мин и регистрировали сигналы под действием вещества. Применяли следующие вещества: капсаицин (1 мкМ, 10 мкМ), SB 366791 (5 мкМ) и АТФ (100 мкМ). В экспериментах с капсаицином и SB 366791 вещества предварительно растворяли в DMSO. В экспериментах, проведенных с этими агентами, в контрольный раствор добавляли такую же концентрацию DMSO, которая присутствовала в растворе с апплицируемым веществом. В конечном итоге концентрация DMSO в растворе не превышала 0.01%.
Экспериментальные данные представлены как среднее значение ± ошибка среднего. В электрофизиологических экспериментах n – количество синапсов, соответствующее количеству нервно-мышечных препаратов. В экспериментах по регистрации кальциевого транзиента n – количество синапсов в нервно-мышечных препаратах от 4–6 животных. Для статистической обработки результатов использовали двухсторонний критерий Стьюдента. Проверку выборки на нормальное распределение проводили с помощью критерия Андерсона–Дарлинга. В экспериментах с тремя группами данных применяли поправку Бонферрони на мно́жественную проверку гипотез. Различия считали статистически значимыми при p < 0.05.
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
На начальном этапе исследования оценивали влияние активации ваниллоидных и пуриновых рецепторов на процесс спонтанной квантовой секреции АХ. Частота мПКП в контроле в синапсах m. LAL составила 1.2 ± 0.1 имп/с (n = 15). На рис. 1 представлены фрагменты нативных записей миниатюрных потенциалов концевой пластинки и гистограммы распределения амплитуд мПКП в контроле и под действием капсаицина. Капсаицин в концентрациях 1 и 10 мкМ приводил к снижению частоты на 13.3 ± 0.7% (p < 0.05, n = 7) и 13.2 ± 4.6% (p < 0.05, n = 4) соответственно (рис. 1, 2). Так как не наблюдалось выраженного дозозависимого эффекта капсаицина на частоту (и, как показано ниже, на квантовый состав ПКП) в дальнейшем использовали агонист в концентрации 1 мкМ.
Рис. 1.
Миниатюрные потенциалы концевой пластинки (mEPPs), зарегистрированные в нервно-мышечных синапсах m. LAL в контроле (Control), под действием капсаицина (CAP) и АТФ (ATP). Представлены данные в рамках двух репрезентативных экспериментов: верхняя панель эксперимент с капсаицином, нижняя с АТФ. Слева – фрагменты нативных записей мПКП; в центре – гистограммы распределения амплитуд мПКП; справа – гистограммы распределения межимпульсных интервалов мПКП. Кривые на гистограммах – функции плотности вероятности для распределений Гаусса и Пуассона (черные – контроль, серые – после аппликации вещества).
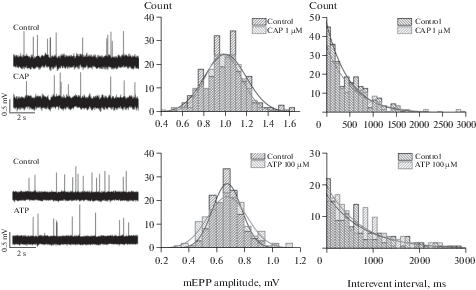
Рис. 2.
Влияние агониста TRPV1-каналов капсаицина (CAP; 1 и 10 мкМ), блокатора TRPV1-каналов SB 366791 (SB; 5 мкМ) и АТФ (ATP; 100 мкМ) на частоту мПКП (Frequency mEPP), выраженную в процентах от контрольного значения. В экспериментах с SB 366791 и АТФ капсаицин использовали в концентрации 1 мкМ. Данные представлены как среднее ± стандартная ошибка среднего. n = 4–8, *p < 0.05 по сравнению с контролем.
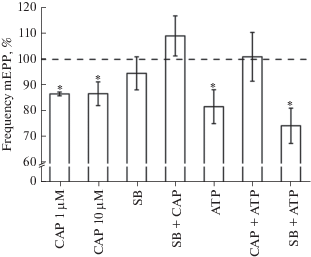
Конкурентный антагонист TRPV1-каналов SB 366791 в концентрации 5 мкМ не оказывал влияния на спонтанную секрецию АХ, и частота мПКП составляла 94.6 ± 6.3% относительно контроля (p > 0.05, n = 8; рис. 2), в то же время на фоне SB 366791 эффект капсаицина полностью отсутствовал (109.0 ± 7.7%; p > 0.05, n = 8; рис. 2). Следовательно, снижение уровня спонтанного выделения АХ под действием капсаицина опосредовано активацией TRPV1-каналов.
Ранее неоднократно было показано, что АТФ угнетающе влияет на процесс спонтанной нейросекреции АХ [10, 21, 22]. В наших экспериментах АТФ в концентрации 100 мкМ приводил к снижению частоты мПКП на 18.2 ± 6.5% (p < 0.05, n = 6; рис. 1, 2).
Поскольку АТФ способен модулировать работу TRPV1-каналов [14], то необходимо было оценить возможность реализации угнетающего действия нуклеозидтрифосфата посредством активации ваниллоидных рецепторов. На фоне антагониста TRPV1-каналов SB 366791 в концентрации 5 мкМ угнетающий эффект АТФ реализовывался в полном объеме, и частота мПКП снижалась на 25.5 ± 6.8% (p < 0.05, n = 7; рис. 2). В то же время на фоне активации TRPV1-каналов капсаицином действие АТФ на спонтанную квантовую секрецию устранялось, и частота мПКП соответствовала уровню, зарегистрированному в присутствии только капсаицина (101.0 ± 9.4%; p > 0.05, n = 8; рис. 2). Во всех экспериментах с добавлением АТФ наблюдалось увеличение времени спада мПКП, которое в среднем составляло 3.0 ± 0.2 мс (n = 7) в контроле. При аппликации только нуклеозидтрифосфата время спада увеличивалось на 28.3 ± 11.0% (p < 0.05, n = 6); в экспериментах, где АТФ добавляли на фоне антагониста TRPV1-каналов, данный параметр увеличивался на 57.7 ± 16.3% (p < 0.05, n = 7). В экспериментальной серии, когда апплицировали АТФ после капсаицина, время спада возрастало на 65.0 ± 7.9% (p < 0.5, n = 8). Во всех других сериях (капсаицин 1 мкМ; капсаицин 10 мкМ; SB 366791; капсаицин 1 мкМ на фоне SB 366791) изменений во времени спада мПКП не наблюдалось (p > 0.05 во всех случаях).
Поскольку при активации TRPV1-рецепторов угнетающее влияние АТФ на частоту мПКП полностью отсутствует, то есть основания полагать наличие определенной взаимосвязи между сигнальными путями регуляции спонтанного выделения АХ, которые инициируются пуриновыми и ваниллоидными рецепторами. Возможность наличия такой взаимосвязи регуляторных механизмов была проверена далее в экспериментах с регистрацией вызванной квантовой секреции АХ.
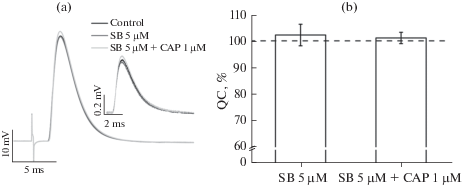
Среднее значение квантового состава в контроле для препарата m. LAL составило 34.4 ± 2.5% (n =25). Под действием капсаицина (1 мкМ) квантовый состав ПКП снижался на 16.0 ± 3.6% (p < 0.05, n = 7; рис. 3). Повышение концентрации капсаицина до 10 мкМ не приводило к увеличению угнетающего эффекта, квантовый состав снижался на 15.4 ± 5.7% (p < 0.05, n = 4; рис. 3). В присутствии антагониста TRPV1-каналов SB 366791 (5 мкМ) квантовый состав ПКП не изменялся (102.2 ± 4.1%; p > 0.05, n = 8; рис. 4), однако ингибирующий эффект капсаицина устранялся полностью (101.1 ± 2.1%; p > 0.05, n = 8; рис. 4).
Рис. 3.
Угнетающее влияние капсаицина (CAP) в концентрациях 1 мкМ (n = 7) и 10 мкМ (n = 4) на вызванное стимуляцией нерва выделение АХ. Панель (a) – нативные записи вызванных и миниатюрных потенциалов концевой пластинки (усредненные по 50 сигналам) в отдельно взятом эксперименте в контроле и после аппликации капсаицина. Панель (b) – относительное изменение величины квантового состава потенциалов концевой пластинки (QC), выраженное в процентах от контрольного значения (по всем экспериментам в серии). Данные представлены как среднее ± стандартная ошибка среднего. *p < 0.05 по сравнению с контролем.
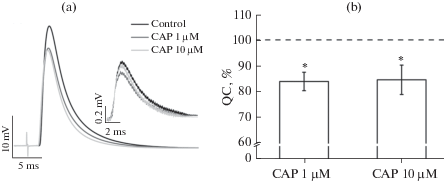
Рис. 4.
Отсутствие влияния блокатора TRPV1-каналов SB 366791 (SB; 5 мкМ, n = 8), а также эффекта капсаицина (CAP; 1мкМ, n = 8) в присутствии этого блокатора на вызванное стимуляцией нерва выделение АХ. Панель (a) – нативные записи вызванных и мПКП (усредненные по 50 сигналам) в отдельно взятом эксперименте в контроле и после аппликации фармакологических агентов. Панель (b) – относительное изменение величины квантового состава ПКП (QC), выраженное в процентах от контрольного значения (по всем экспериментам в серии). Данные представлены как среднее ± стандартная ошибка среднего.
Далее, как и в экспериментах со спонтанной квантовой секрецией, оценивали возможность взаимосвязи между пуринергическим и ваниллоидным сигнальными путями регуляции вызванного освобождения АХ.
АТФ в концентрации 100 мкМ снижал квантовый состав ПКП на 14.9 ± 3.3% (p < 0.05, n = 6; рис. 5). Данный угнетающий эффект АТФ на фоне антагониста TRPV1-каналов SB 366791 в концентрации 5 мкМ сохранялся, и квантовый состав снижался на 22.5 ± 2.0% (p < 0.05, n = 7; рис. 5). Далее было установлено, что АТФ на фоне активации TRPV1-каналов капсаицином не способен оказывать своего угнетающего влияния на вызванную квантовую секрецию АХ (квантовый состав составил 99.8 ± 4.8%; p > 0.05, n = 7; рис. 5).
Рис. 5.
Угнетающее влияние АТФ (ATP; 100 мкМ) на вызванное стимуляцией нерва выделение АХ; отсутствие эффекта АТФ после предварительной аппликации агониста TRPV1-каналов капсаицина (CAP; 1 мкМ) и проявление угнетающего действия АТФ после блокады TRPV1-каналов SB 366791 (SB; 5 мкМ). Панели а, b и c – нативные записи вызванных и мПКП (усредненные по 50 сигналам) в отдельно взятом эксперименте в контроле и после аппликации фармакологических агентов. Панель d –относительное изменение величины квантового состава ПКП (QC), выраженное в процентах от контрольного значения (по всем экспериментам в серии). Данные представлены как среднее ± стандартная ошибка среднего. n = 6–7, *p < 0.05 по сравнению с контролем.

Рис. 6.
Отсутствие влияния агониста TRPV1-каналов капсаицина (CAP; 1 мкМ, n = 9), блокатора данных каналов SB 366791 (SB; 5 мкМ, n = 18) и АТФ (ATP; 100 мкМ, n = 12) на Ca2+-транзиент в нервном окончании при стимуляции двигательного нерва. Панели а, b и c – изменения интенсивности флуоресценции (∆F/F0) в контроле и после аппликации фармакологических агентов (представлены нативные записи, усредненные по 8 сигналам и зарегистрированные в отдельно взятых экспериментах). Панель d – средние значения и ошибки, выраженные в процентах от контрольного значения амплитуды Са2+-транзиента (по всем экспериментам в серии). Данные представлены как среднее ± стандартная ошибка среднего.

Как и в экспериментах с регистрацией мПКП, у вызванных ПКП также наблюдалось увеличение времени спада сигнала во всех случаях (p < 0.05), когда апплицировали АТФ (только АТФ, АТФ на фоне капсаицина, АТФ на фоне SB 366791). Эти данные предполагают наличие постсинаптического действия нуклеозидтрифосфата, не связанного напрямую с механизмом действия АТФ на процесс нейросекреции АХ.
Поскольку количество квантов АХ, выделившихся в ответ на стимуляцию двигательного нерва, сильно зависит от уровня входящего в нервную терминаль Са2+ [23], то было сделано предположение, что в основе механизмов действия АТФ и капсаицина может лежать изменение входа Са2+. Проверка этого предположения весьма интересна, так как TRPV1-рецепторы представляют собой потенциал независимые кальциевые каналы [24].
При регистрации Са2+-транзиента в нервной терминали, амплитуда которого коррелирует с входом Са2+ в нервное окончание и изменением квантового освобождения медиатора [25], были получены следующие результаты.
Аппликация как агониста (капсаицин, 1 мкМ), так и блокатора (SB 366791, 5 мкМ) TRPV1-рецепторов никак не повлияла на амплитуду Са2+-транзиента (106.7 ± 1.8%; p > 0.05, n = 9 и 101.4 ± 1.3%; p > 0.05, n = 18 соответственно; рис. 6 ). При исследовании механизма действия АТФ также не было обнаружено значимого изменения в Са2+-транзиенте, амплитуда которого составила 97.6 ± 2.5% (p > 0.05, n = 12; рис. 6 ).
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
В настоящем исследовании впервые был обнаружен феномен угнетающего влияния капсаицина на процесс спонтанной квантовой секреции АХ из двигательных нервных окончаний и доказано участие TRPV1-рецепторов в этом регуляторном процессе. Впервые в нервно-мышечном синапсе описан эффект применения соединения SB 366791, специфического конкурентного антагониста TRPV1, который устраняет эффект капсаицина в более низкой концентрации, чем используемый в ряде работ капсазепин. Впервые показано, что предварительная активация TRPV1-каналов полностью препятствует развитию угнетающего действия АТФ как на спонтанную, так и на вызванную секрецию АХ. Впервые показано, что при инициации исследуемых регуляторных механизмов значимых изменений пресинаптического уровня кальция (Са2+-транзиента) при стимуляции нерва не происходит.
Феномен пуринергической модуляции выделения АХ из двигательной нервной терминали позвоночных установлен достаточно давно [8], относительно хорошо исследован [10, 21, 22], и продолжает изучаться по настоящий момент [26]. Полученные в нашей работе данные об угнетающем влиянии АТФ на процессы спонтанной и вызванной квантовой секреции в нервно-мышечном синапсе m. LAL полностью согласуются с данными литературы [21, 22]. Более того, нами получены доказательства наличия и постсинаптического действия АТФ, заключающееся в увеличении времени спада сигналов (как мПКП, так и ПКП). Ранее было доказано, что в основе данного механизма действия АТФ на временные параметры постсинаптических сигналов лежит активация экстрасинаптических P2Y1-рецепторов и ингибирование хлорных каналов мембраны мышечного волокна [27].
Возможность участия ваниллоидных рецепторов в регуляции нервно-мышечной нейротрансмиссии у млекопитающих была обнаружена сравнительно недавно и все еще остается слабо изученным феноменом [17, 18]. Так, на препарате диафрагмы мыши было впервые показано угнетающее влияние капсаицина на процесс вызванной нейросекреции АХ, однако какого-либо эффекта агониста ваниллоидных рецепторов на процесс спонтанного квантового выделения медиатора обнаружено не было [18]. В наших же экспериментах получены результаты, подтверждающие наличие данного механизма, угнетающего вызванное высвобождение квантов АХ из нервного окончания. В то же время впервые обнаружено, что капсаицин способен снижать и уровень спонтанного выделения АХ, причем данный эффект агониста полностью устранялся селективным конкурентным антагонистом TRPV1-каналов SB 366791.
Обнаружение факта снижения спонтанной секреции АХ при активации TRPV1-каналов в препарате m. LAL, в отличие от препарата диафрагмы, свидетельствует о наличии некоторых функциональных особенностей синаптических контактов в разных препаратах. Вероятно, эти особенности и накладывают свой отпечаток на то, что выраженность эффектов АТФ на процессы как спонтанной, так и вызванной секреции АХ несколько ниже, чем в синапсах диафрагмы [21, 22].
Схожесть во влиянии АТФ и капсаицина (угнетение обеими фармакологическими агентами процессов как спонтанной, так и вызванной секреции АХ) может свидетельствовать о возможном наличии общего звена в путях пуринергической и TRPV1-опосредованной регуляции процессов нейросекреции АХ. В настоящем исследовании получены данные, демонстрирующие, что эффект АТФ как на спонтанную, так и на вызванную секрецию АХ отсутствует при предварительной аппликации агониста TRPV1-рецепторов, но сохраняется при блокаде последних селективным антагонистом. Эти результаты позволяют нам заключить: в механизмах, запускаемых активацией как TRPV1-рецепторов, так и пуринорецепторов, имеет место наличие одного общего звена, которое отвечает за снижение количества выделяемых квантов АХ.
В качестве такой мишени могли бы выступить молекулы, тем или иным образом влияющие на метаболизм кальция в нервном окончании. Это могут быть как потенциал-чувствительные кальциевые каналы [12, 28], так и внутриклеточные кальциевые депо [29]. Методика регистрации Са2+-транзиента в нервном окончании как раз и позволяет нам фиксировать изменения интегрального внутриклеточного кальция. Ранее на нейронах крысы было установлено, что активация Р2Y-рецепторов приводит к уменьшению входа ионов кальция через потенциал-зависимые кальциевые каналы N типа [30]. О снижении входа кальция в нервное окончание под действием АТФ свидетельствуют и данные, полученные на нервно-мышечном синапсе лягушки [10, 12]. В свою очередь, TRPV1-каналы проявляют довольно большую селективность к Са2+ и участвуют в регуляции содержания Са2+ в цитозоле клетки [24, 31]. Кроме того, было высказано предположение, что кальций, входящий через TRPV1-каналы в двигательном нервном окончании, способен изменять уровень мембранного PIP2 и приводить, таким образом, к уменьшению уровня выделения АХ [18]. В наших же экспериментах какого-либо изменения интегрального кальция обнаружено не было ни при аппликации АТФ, ни при добавлении капсаицина. Исходя из эти данных, можно предположить, что эффекты активации TRPV1-рецепторов обусловлены увеличением базового уровня кальция, а не кальция, входящего при стимуляции двигательного нерва. Входящий через TRPV1-каналы кальций, в свою очередь, может приводить к активации кальций-зависимых белков, таких как кальмодулин и кальцинейрин, которые, как было установлено ранее, участвуют в механизмах снижения уровня выделения АХ [21, 32, 33]. В то же время подавляющий нейротрансмиссию эффект АТФ можно объяснить активацией метаботропных P2Y-рецепторов, которые, по-видимому, не вносят значительного вклада в изменение входа Са2+ через потенциал-чувствительные кальциевые каналы и выделение Са2+ из депо. При этом имеет место активация той же конечной мишени, регулирующей процесс экзоцитоза синаптических везикул, что и при активации TRPV1-каналов. И в данном случае, в качестве такой мишени может выступать кальмодулин, поскольку показано, что при ингибировании этого белка опосредованный активацией P2Y-рецепторов угнетающий эффект АТФ на выделение АХ полностью отсутствует [20].
Возможное физиологическое значение механизмов регуляции, запускаемых при активации пуриновых и ваниллоидных рецепторов, наиболее вероятно заключается в модуляции процесса нейросекреции АХ по принципу обратной отрицательной связи. Поскольку при квантовом выделении АХ в синаптической щели увеличивается концентрация как АТФ [8], так и протонов [34], то увеличивается и вероятность активации как пуринорецепторов, так и TRPV-каналов. Как следствие этого – снижение уровня выделения последующих порций медиатора. Кроме того, недавно было установлено, что TRPV1-рецепторы участвуют в реализации симпатической регуляции выброса АХ в периферических нервных окончаниях мыши [35].
Таким образом, в нервно-мышечном синапсе млекопитающих, наряду с ранее установленным пуринергическим путем регуляции АХ, имеет место и механизм модуляции нейросекреции, опосредованный активацией TRPV1-каналов. Запуск этих механизмов приводит к угнетению процессов как спонтанного, так и вызванного выделения квантов АХ из двигательного нервного окончания. При этом продемонстрировано, что оба этих механизма регуляции не сопровождаются изменением Ca2+-транзиента в нервном окончании и имеют общее звено. На роль такого звена может претендовать белок кальмодулин, однако это еще необходимо проверить экспериментально в будущем.
Список литературы
Fagerlund M.J., Eriksson L.I. (2009) Current concepts in neuromuscular transmission. Br. J. A-naesth. 103: 108–114. https://doi.org/10.1093/bja/aep150
Petrov K.A., Nikolsky E.E., Masson P. (2018) Autoregulation of acetylcholine release and micro-pharmacodynamic mechanisms at neuromuscular junction: Selective acetylcholinesterase inhibitors for therapy of myasthenic syndromes. Front. Pharmacol. 9: 1–8. https://doi.org/10.3389/fphar.2018.00766
Kilbinger H. (1996) Modulation of acetylcholine release by nitric oxide. Prog. Brain. Res. 109: 219–224. https://doi.org/10.1016/s0079-6123(08)62105-6
Datar P., Srivastava S., Coutinho E., Govil G. (2005) Substance P: Structure, Function, and Therapeutics. Curr. Top. Med. Chem. 4: 75–103. https://doi.org/10.2174/1568026043451636
Pinard A., Lévesque S., Vallée J., Robitaille R. (2003) Glutamatergic modulation of synaptic plasticity at a PNS vertebrate cholinergic synapse. Eur. J. Neurosci. 18: 3241–3250. https://doi.org/10.1111/j.1460-9568.2003.03028.x
Malomouzh A.I., Nikolsky E.E., Lieberman E.M., Sherman J.A., Lubischer J.L., Grossfeld R.M., Urazaev A.K. (2005) Effect of N-acetylaspartylglutamate (NAAG) on non-quantal and spontaneous quantal release of acetylcholine at the neuromuscular synapse of rat. J. Neurochem. 94: 257–267. https://doi.org/10.1111/j.1471-4159.2005.03194.x
Ribeiro J.A., Cunha R.A., Correia-de-Sa P., Sebastiao A.M. (1996) Purinergic regulation of acetylcholine release. Prog. Brain Res. 109: 231–241. https://doi.org/10.1016/s0079-6123(08)62107-x
Burnstock G. (2007) Physiology and pathophysiology of purinergic neurotransmission. Physiol. Rev. 87: 659–797. https://doi.org/10.1152/physrev.00043.2006
Surprenant A., North R.A. (2009) Signaling at purinergic P2X receptors. Annu. Rev. Physiol. 71: 333–359. https://doi.org/10.1146/annurev.physiol.70.113006.100630
Grishin S., Shakirzyanova A., Giniatullin A., Afzalov R., Giniatullin R. (2005) Mechanisms of ATP action on motor nerve terminals at the frog neuromuscular junction. Eur. J. Neurosci. 21: 1271–1279. https://doi.org/10.1111/j.1460-9568.2005.03976.x
Ginsborg B.L., Hirst G.D.S. (1972) The effect of adenosine on the release of the transmitter from the phrenic nerve of the rat. J. Physiol. 224: 629–645. https://doi.org/10.1113/jphysiol.1972.sp009916
Khaziev E.F., Samigullin D.V., Tsentsevitsky A.N., Bukharaeva E.A., Nikolsky E.E. (2018) ATP Reduces the Entry of Calcium Ions into the Nerve Ending by Blocking L-type Calcium Channels. Acta Naturae. 10: 93–96. https://doi.org/10.32607/20758251-2018-10-2-93-96
Tominaga M., Wada M., Masu M. (2001) Potentiation of capsaicin receptor activity by metabotropic ATP receptors as a possible mechanism for ATP-evoked pain and hyperalgesia. Proc. Natl. Acad. Sci. USA. 98: 6951–695. https://doi.org/10.1073/pnas.111025298
Morales-Lázaro Sara L., Simon Sidney A., Rosenbaum T. (2013) The role of endogenous molecules in modulating pain through TRPV1. J. Physiol. 591: 3109–3121. https://doi.org/10.1113/jphysiol.201
Lishko P.V., Procko E., Jin X., Phelps C.B., Gaudet R. (2007) The Ankyrin Repeats of TRPV1 Bind Multiple Ligands and Modulate Channel Sensitivity. Neuron. 54: 905–918. https://doi.org/10.1016/j.neuron.2007.05.027
Fernández-Carvajal A., Fernández-Ballester G., Devesa I., González-Ros J.M., Ferrer-Montiel (2011) New strategies to develop novel pain therapies: Addressing thermoreceptors from different points of view. Pharmaceuticals. 5: 16–48. https://doi.org/10.3390/ph5010016
Thyagarajan B., Krivitskaya N, Potian J.G., Hognason K., Garcia C.C., McArdle J.J. (2009) Capsaicin protects mouse neuromuscular junctions from the neuroparalytic effects of botulinum neurotoxin. J. Pharmacol. Exp. Ther. 331: 361–371. https://doi.org/10.1124/jpet.109.156901
Thyagarajan B., Potian J.G., Baskaran P., McArdle J.J. (2014) Capsaicin modulates acetylcholine release at the myoneural junction. Eur. J. Pharmacol. 744: 211–219. https://doi.org/10.1016/j.ejphar.2014.09.044
Martin A.R. (1976) The effect of membrane capacitance on non-linear summation of synaptic potentials. J. Theor. Biol. 59: 179–187. https://doi.org/10.1016/S0022-5193(76)80031-8
Samigullin D.V., Khaziev E.F., Zhilyakov N.V., Sudakov I.A., Bukharaeva E.A, Nikolsky E.E. (2017) Calcium Transient Registration in Response to Single Stimulation and During Train of Pulses in Mouse Neuromuscular Junction. Bionanoscience. 7: 162–166. https://doi.org/10.1007/s12668-016-0318-6
De Lorenzo S., Veggetti M., Muchnik S., Losavio A. (2006) Presynaptic inhibition of spontaneous acetylcholine release mediated by P2Y receptors at the mouse neuromuscular junction. Neuroscience. 142: 71–85. 2006.https://doi.org/10.1016/j.neuroscience.2006.05.062
Galkin A.V., Giniatullin R.A., Mukhtarov M.R., Švandová I., Grishin S.N., Vyskočil F. (2001) ATP but not adenosine inhibits nonquantal acetylcholine release at the mouse neuromuscular junction. Eur. J. Neurosci. 13: 2047–2053. 2001.https://doi.org/10.1046/j.0953-816X.2001.01582.x
Augustine G.J. (2001) How does calcium trigger neurotransmitter release? Curr. Opin. Neurobiol. 11: 320–326. https://doi.org/10.1016/S0959-4388(00)00214-2
Caterina M.J., Schumacher M.A., Tominaga M, Rosen T.A., Levine J.D, Julius D. (1997) The capsaicin receptor: A heat-activated ion channel in the pain pathway. Nature. 389: 816–824. https://doi.org/10.1038/39807
Samigullin D.V., Zhilyakov N.V., Khaziev E.F., Bukharaeva E.A., Nikolsky E.E. (2018) Calcium Transient and Quantal Release in Mouse Neuromuscular Junction Under Extracellular Calcium Concentration Change. Bionanoscience. 8: 984–987. https://doi.org/10.1007/s12668-018-0558-8
Ziganshin A.U., Khairullin A.E., Hoyle C.H.V., Grishin S.N. (2020) Modulatory roles of ATP and adenosine in cholinergic neuromuscular transmission. Int. J. Mol. Sci. 21: 1–15. https://doi.org/10.3390/ijms21176423
Voss A.A. (2009) Extracellular ATP inhibits chloride channels in mature mammalian skeletal muscle by activating P2Y1 receptors. J. Physiol. 587: 5739–5752. https://doi.org/10.1113/jphysiol.2009.179275
Gaydukov A.E., Melnikova S.N., Balezina O.P. (2009) Facilitation of acetylcholine secretion in mouse motor synapses caused by calcium release from depots upon activation of L-type calcium channels. Bull. Exp. Biol. Med. 148: 163–166. https://doi.org/10.1007/s10517-009-0678-9
Khuzakhmetova V.F., Samigullin D.V., Bukharaeva E.A. (2014) The role of presynaptic ryanodine receptors in regulation of the kinetics of the acetylcholine quantal release in the mouse neuromuscular junction. Biochem. Suppl. Ser. A Membr. Cell Biol. 8: 144–152. https://doi.org/10.1134/S199074781305005X
Filippov A.K., Brown D.A., Barnard E.A. (2000) The P2Y1 receptor closes the N-type Ca2+ channel in neurones, with both adenosine triphosphates and diphosphates as potent agonists. Br. J. Pharmacol. 129: 1063–1066. https://doi.org/10.1038/sj.bjp.0703185
Marshall I.C.B., Owen D.E., Cripps T.V., Davis J.B., McNulty S., Smart D. (2003) Activation of vanilloid receptor 1 by resiniferatoxin mobilizes calcium from inositol 1,4,5-trisphosphate-sensitive stores. Br. J. Pharmacol. 138: 172–176. https://doi.org/10.1038/sj.bjp.0705003
Tarasova E.O., Gaydukov A.E., Balezina O.P. (2018) Calcineurin and Its Role in Synaptic Transmission. Biochem. 83: 674–689. https://doi.org/10.1134/S0006297918060056
Ando K., Kudo Y., Aoyagi K., Ishikawa R., Igarashi M., Takahashi M. (2013) Calmodulin-dependent regulation of neurotransmitter release differs in subsets of neuronal cells. Brain Res. 1535: 1–13. https://doi.org/10.1016/j.brainres.2013.08.018
Zhang Z., Nguyen K.T, Barrett E.F., David G. (2010) Vesicular ATPase Inserted into the Plasma Membrane of Motor Terminals by Exocytosis Alkalinizes Cytosolic pH and Facilitates Endocytosis. Neuron. 68: 1097–1108. https://doi.org/10.1016/j.neuron.2010.11.035
Rodrigues A.Z.C., Wang Z.M., Messi M.L., Delbono O. (2019) Sympathomimetics regulate neuromuscular junction transmission through TRPV1, P/Q- and N-type Ca 2+ channels. Mol. Cell. Neurosci. 95: 59–70. https://doi.org/10.1016/j.mcn.2019.01.007
Дополнительные материалы отсутствуют.
Инструменты
Российский физиологический журнал им. И.М. Сеченова