

Российский физиологический журнал им. И.М. Сеченова, 2021, T. 107, № 6-7, стр. 773-784
Роль гистондеацетилаз I и IIa (HDAC1, HDAC4/5) и сигнального пути MAPK38 в регуляции атрофических процессов при функциональной разгрузке скелетных мышц
Т. Л. Немировская *
Институт медико-биологических проблем РАН
Москва, Россия
* E-mail: nemirovskaya@bk.ru
Поступила в редакцию 02.02.2021
После доработки 10.03.2021
Принята к публикации 11.03.2021
Аннотация
Различные формы функциональной разгрузки мышц можно обнаружить у больных при длительном постельном режиме, при инсультах и спинальных поражениях, во время иммобилизации мышц в травматологии, в условиях невесомости и т.п. В основном при разгрузке страдают постуральные мышцы (например, камбаловидная мышца – m. soleus). В основе перестройки скелетных мышц при функциональной разгрузке лежит их атрофия из-за увеличения протеолиза и падения интенсивности белкового синтеза [1, 2]. Обзор посвящен исследованию роли гистондеацетилаз I и IIa (HDAC1, HDAC4/5), а также сигнального пути MAPK38 в активации транскрипционных факторов FOXO и миогенина, которые участвуют в экспрессии генов Е3 убиквитинлигаз atrogin-1, MuRF-1 при функциональной разгрузке скелетных мышц.
Скелетные мышцы обладают высокой степенью пластичности и могут отвечать на состояние функциональной разгрузки (при иммобилизации мышц в травматологии, при гравитационной разгрузке, у больных, помещенных в условия длительного постельного режима, во время инсультов и спинальных поражений, повреждений нерва и др.) атрофией [3–5]. Постуральная мышца m. soleus подвержена при этом наибольшим изменениям. Не так много работ исследует триггерные механизмы белковой деградации мышц во время ранних сроков (от нескольких часов до 3 дней) их функциональной разгрузки. Показано, что атрофия, вызванная функциональной разгрузкой, обусловлена не только снижением скорости анаболических процессов, но и резким ускорением протеолиза [1, 2, 6, 7], в результате чего уменьшается содержание сократительных и цитоскелетных белков. Распад мышечных белков контролирует 4 протеолитические системы: кальпаиновая, убиквитин-протеасомная, лизосомальная (аутофаги/лизосомы), каспазная. Степень вовлечения каждой из этих систем в развитие атрофии при различных ее моделях (кахексия, сепсис, денервация, иммобилизация, вывешивание) разная. Вклад убиквитин-протеасомной системы в деградацию белков m.soleus особенно велик, т.к. она отвечает за 80–90% круговорота цитоплазматического белка, доступного для протеолиза [8]. Особое внимание этой мышце уделяется потому, что она наиболее задействована в удержании позы (прямостояние) у человека, и даже при стоянии (т.е. при отсутствии движения) ЭМГ от этой мышцы фиксирует максимальную активность. Около 70% от объема мышечного волокна занимают сократительные белки, и эта фракция при атрофии снижается быстрее, чем многие другие [9]. Протеолиз миофибриллярных белков осуществляется преимущественно под действием убиквитин-протеасомной системы [10], в результате чего исследование триггерных механизмов, запускающих экспрессию Е3 лигаз, является актуальной задачей.
В мышце убиквитин-протеасомная система выполняет задачу удаления саркомерных белков при изменении двигательной активности. Специфичность Е3‑лигаз для определенных групп белков обеспечивает высокую избирательность для их деградации. Из известных в настоящее время Е2- и Е3-лигаз синтез только некоторых из них увеличивается при снижении мышечной массы. Это свидетельствует о том, что мышечная атрофия – это активный процесс, который контролируется специфическими сигнальными путями и транскрипционными программами. Только несколько из множества известных убиквитин-лигаз активируется при атрофических процессах, вызванных функциональной разгрузкой мышц [1]. Хорошо известны экспрессирующиеся в большом количестве при атрофии Е3-лигазы атроген-1 (MAFbx) и MuRF1. MuRF-1 участвует в расщеплении миофибриллярных белков [11], в то время как атроген-1 (MAFbx) контролирует как расщепление цитоскелетных белков, так и процессы, связанные с синтезом белка [12]. Мыши, у которых не синтезируются атроген-1 и MuRF1, устойчивы к мышечной атрофии, вызванной денервацией [13]. Не так давно установлено, что достоверное увеличение экспрессии MuRF1 и MAFbx наблюдается уже в первые сутки гравитационной разгрузки мышц, а к третьим суткам достигает своего пика [14]. Именно ранние этапы развития атрофии изучены хуже всего. До последнего времени основное внимание исследователи обращали на возможность фосфорилирования/дефосфорилирования транскрипционного фактора FOXO1/3 для регуляции экспрессии генов атрогина-1/MAFbx и MuRF-1 во время функциональной разгрузки мышц (рис. 1), т.к. именно FOXO1/3 контролирует их экспрессию [15].
Рис. 1.
Схема регуляции экспрессии Е3-лигаз MAFbx/MuRF1 сигналингом Akt/FOXO3. pAkt фосфорилирует транскрипционный фактор FOXO3. Нефосфорилириванный FOXO3 способен проникать в ядро и активировать экспрессию MAFbx и MuRF1.

Однако не так давно мы показали, что фосфорилирование транскрипционного фактора FOXO3 и транскрипционных факторов сигнального пути NF-κB не всегда предотвращало повышение экспрессии Е3-лигазы MuRF-1 разгруженной мышцы [16, 17]. Так, при введении препаратов, снижающих степень атрофии m. soleus, уровень pFOXO3 был таким же, как у контрольных крыс, а экспрессия MuRF-1 у них оставалась высокой. Ингибирование фосфорилирования транскрипционных факторов сигнального пути NF-kB при функциональной разгрузке крыс в нашем эксперименте также не приводило к снижению экспрессии MuRF-1. Мало того, экспрессия MuRF-1 при ингибировании NF-kB была выше, чем у вывешенных без препарата животных. Эти результаты привели к предположению, что во всех этих случаях в мышце мог активно работать другой механизм, контролирующий работу генов atrogin-1/MAFbx и MuRF-1, вероятно, не связанный с фосфорилированием/дефосфорилированием их транскрипционных факторов. В настоящее время обсуждается участие HDACs (гистондеацетилаз) 1; 4/5, а также p38 MAPK (p38 митогенактивируемой протеинкиназы) в регуляции метаболизма скелетных мышц [18–20].
Обзор посвящен выявлению их роли в регуляции экспрессии Е3-лигаз при функциональной разгрузке мышц.
1. РОЛЬ HDAC В РЕГУЛЯЦИИ ЭКСПРЕССИИ УБИКВИТИНЛИГАЗ ПРИ ФУНКЦИОНАЛЬНОЙ РАЗГРУЗКЕ МЫШЦ
Ферменты HDACs класса IIa рассматриваются как основные транскрипционные репрессоры. Фосфорилирование этих ферментов приводит к их выходу из ядра, что, в свою очередь, сопровождается активацией транскрипционных факторов и увеличению синтеза белка [21]. Известен кальций-зависимый механизм фосфорилирования/дефосфорилирования гистондеацетилаз (HDAC) класса IIа и их ядерно-цитоплазматическое перемещение, позволяющее ядерным HDAC блокировать экспрессию ряда генов [22]. Учитывая эти данные, можно ожидать дефосфорилирование HDACs и накопление их в ядерной фракции m. soleus крыс при функциональной разгрузке, т.к. при разгрузке мышц наблюдается накопление ионов кальция в цитоплазме волокон [23]. Роль HDAC в регуляции экспрессии убиквитинлигаз при функциональной разгрузке мышц изучена слабо, особенно возможность их активизации на ранних этапах разгрузки. Как мы уже упоминали, стимуляция экспрессии Е3-убиквитинлигаз MuRF1 и atrogin-1/MAFbx осуществляется посредством сигнального пути Akt/FOXO3-1 (рис. 1) [24]. В последние годы выявлены и другие механизмы регуляции их экспрессии: через гистондеацетилазу 4, миогенин, HDAC1 [25, 26]. В то же время, работа этих регуляторных механизмов в условиях функциональной разгрузки изучена недостаточно. В дополнение к регулированию транскрипции гена посредством ацетилирования гистонов, каталитическая активность HAT и HDAC регулирует экспрессию гена путем изменения статуса ацетилирования и функции транскрипционных факторов, в частности, и FoxO. В настоящее время имеется ограниченные сведения о конкретных HDAC, которые регулируют статус ацетилирования FoxO в скелетных мышцах при нормальных условиях и тех, которые способствуют снижению ацетилирования и активации FoxO во время катаболических состояний. В последнее время мы провели ряд исследований, в которых изучили возможность работы альтернативного механизма, контролирующего работу генов Е3-лигаз, связанного с деацетилазной активностью различных HDAC. Особое внимание в них было уделено выявлению роли гистондеацетилаз (HDAC1, HDAC4/5) в активации транскрипционных факторов FOXO3 и миогенина при функциональной разгрузке мышц в экспрессии генов atrogin-1 и MuRF-1. Для этого применяли метод селективного ингибирования HDACs (HDAC1, HDAC4 и HDAC5) на ранних этапах функциональной разгрузки мышц.
2. ФУНКЦИЯ И МЕХАНИЗМ КОНТРОЛЯ HDAC1 ЭКСПРЕССИИ Е3-ЛИГАЗ ПРИ ФУНКЦИОНАЛЬНОЙ РАЗГРУЗКЕ МЫШЦ
В работе Beharry и соавт. показано, что FoxO3 активирует HDAC1 [25]. Авторы ингибировали работу различных гистондеацетилаз в культуре клеток и обнаружили, что HDAC1 является ключевым регулятором FOXO3 и запускает мышечную программу атрофии. Это связано с деацетилазной активностью HDAC1, т.к. для активации экспрессии MuRF-1 и atrogin-1/MAFbx требуется деацетилирование FOXO3a [27]. Активность FoxO3a репрессируется в базальных условиях посредством обратимого ацетилирования лизина ацетилтрансферазой (HAT), которая начинает работать во время катаболических состояний [27]. Снижение активности HAT в скелетной мышце индуцировало транскрипционную активность FoxO3, в то время как увеличение активности HAT предотвращало ядерную локализацию FoxO3, убирая ее транскрипционную активность по отношению к генам-мишеням.
Где и каким образом в клетке могут происходить эти процессы ацетилирования/деацетилирования FOXO?
Мы предположили, что ядерно-цитоплазматический транспорт HDAC и HAT может контролировать способность к блокированию активности промоторов генов, кодирующих Е3-убиквитинлигазы in vivo при функциональной разгрузке мышц. Для проверки гипотезы мы провели эксперимент с ингибированием HDAC1 при трехдневной функциональной разгрузке у крыс (модель вывешивания). Ингибирование HDAC1 при разгрузке специфическим ингибитором снизило степень атрофии m.soleus и экспрессию в ней atrogin-1/MAFbx (но не MuRF1) по-сравнению с вывешенными без ингибитора животными [28]. Снижение экспрессии атрогена-1 не было связано с различиями в фосфорилировании Akt и FOXO3 между вывешенными группами. Очевидно, что в данном случае работает другой механизм, регулирующий экспрессию атрогена-1 (не связанный с фосфорилированной/ дефосфорилированной формой FOXO3а). Мы обнаружили также, что у вывешенных животных в ядерной фракции m. soleus уровень HDAC1 существенно повышен, а уровень гистонацетилтрансферазы P300 (HAT) существенно понижен (p < 0.05) относительно уровня контроля. Ингибирование HDAC1 предотвращало эти изменения. Ранее Senf и соавт. отмечали, что снижение активности P300 связано с увеличением экспрессии MAFbx/Atrogin-1 при атрофии, вызванной денервацией [29]. Мы обнаружили также резкое увеличение содержания гистона Ac-H3 в m. soleus только в группе с ингибированием HDAC1 при вывешивании крыс [28]. Связь ДНК с гистонами приводит к плотной упаковке хроматина, которая ограничивает связывание белков, участвующих в транскрипции генов, с ДНК. Итак, мы показали, что экспрессия Е3-лигазы atrogin-1/MAFbx связана с активностью HDAC1 следующим образом. При функциональной разгрузке m.soleus HDAC1 дефосфорилируется и поступает в ядро, HAC P300 в ядре перестает ацетилировать лизин на FOXO3a, что позволяет ему запустить экспрессию atrogin-1, так как гистон H3 в это время перестает плотно связывать место контакта FOXO3a с ДНК. Схема регуляции HDACI экспрессии atrogin-1 приведена на рис. 2.
Рис. 2.
Схема предполагаемой регуляции экспрессии Е3-лигаз с помощью HDAC1. К увеличению экспрессии Е3-убиквитинлигаз может вести накопление HDAC в ядре. В нормальном состоянии фосфорилированный и ацетилированный FOXO3 а также фосфорилированная HDAC1 не поступают в ядро. При дефосфорилировании они проникают в ядро, HDAC1 деацетилирует FOXO3. HAT P300 (гистонацетилтрансфераза P300) в ядре перестает ацетилировать лизин на FOXO3a, что позволяет ему запустить экспрессию atrogin-1, так как гистон H3 в это время перестает плотно связывать место контакта FOXO3a с ДНК.
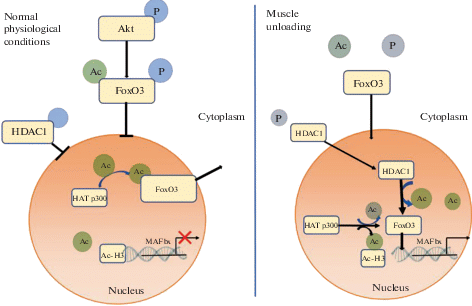
Итак, HDAC1 контролирует экспрессию Е3-лигазы atrogin-1/MAFbx при функциональной разгрузке мышц. Вероятно, контроль экспрессии atrogin-1 (наряду с фосфорилированием/дефосфорилированием сайта FOXO3a) осуществляется с помощью его ацетилирования/деацетилирования.
3. ФУНКЦИЯ И МЕХАНИЗМ КОНТРОЛЯ HDAC4/5 ЭКСПРЕССИИ Е3-ЛИГАЗ ПРИ ФУНКЦИОНАЛЬНОЙ РАЗГРУЗКЕ МЫШЦ
Ферменты класса IIa HDAC (например, HDAC4/5) также участвуют в реакциях со специфическими транскрипционными факторами и оказывают влияние на регуляцию генов в скелетных мышцах. Не так много работ, в которых бы исследовали их роль в экспрессии Е3-лигаз при функциональной разгрузке мышц. Четыре гистондеацетилазы класса IIa (HDAC4,5,7 и 9) являются тканеспецифичными и принимают участие в регуляции метаболизма скелетных и сердечной мышц [20, 21]. Из них HDAC4,5 известны как возможные регуляторы экспрессии генов MuRF-1 и atrogin-1. HDAC класса II (HDAC4 и HDAC5) способствуют нейрогенной атрофии через их транскрипционную репрессию Dach2, которая обычно действует для подавления миогенин-зависимой индукции генов, связанных с атрофией MuRF-1 и atrogin-1/MAFbx [30]. В настоящее время не так много сведений о регуляции его работы в мышце при разгрузке. Миогенин – мышечно специфичный транскрипционный фактор, вовлеченный в координацию развития скелетной мышцы, миогенез и ее восстановление. Однако имеется сообщение о том, что удаление миогенина у взрослой мыши также снижает экспрессию MuRF-1 и atrogin-1 в мышце при денервации и способствует увеличению ее устойчивости к атрофии [30]. На рис. 3 представлена схема работы миогенина при денервации мышцы [31]. Авторы предположили, что на ранней стадии процесс атрофии может запускаться HDAC4/5. Она способна активировать работу гена транскрипционного фактора миогенина, который поступает в ядро и приводит к экспрессии MuRF-1. На более поздних сроках разгрузки мышц включается сигнальный путь mTORC1, который активирует дальнейшую экспрессию E3-лигаз, запуская каскад Akt-FoxO (рис. 3).
Рис. 3.
Временная регуляция сигнальных каскадов при разгрузке мышц. Иллюстрация гипотезы о том, что атрофия мышц является результатом активации ряда сигнальных каскадов, одни из которых активируются на ранних этапах, другие на более поздних этапах разгрузки. На ранней стадии процесс атрофии может запускаться HDAC4/5. Она способна активировать экспрессию гена транскрипционного фактора миогенина, который поступает в ядро и приводит к экспрессии MuRF-1. На более поздних сроках разгрузки мышц включается сигнальный путь mTORC1, который активирует дальнейшую экспрессию E3-лигаз, запуская каскад Akt-FoxO (модифицированный рисунок H . Tang и соавт. [31]).

Вероятно, противоречия в результатах об активировании экспрессии Е3 лигаз в разных работах связаны с тем, что эти события изучались в некоторых случаях на ранних, а в некоторых – на поздних сроках разгрузки мышц, когда уже запускаются другие сигнальные механизмы, регулирующие экспрессию генов Е3-убиквитинлигаз. В недавней работе S. Dupré-Aucouturier и соавт. [32] обнаружено снижение степени атрофии m. soleus при ингибировании активности HDAC4/5 во время 14-дневного вывешивания крыс с введением трихостатина. Трихостатин – неспецифический ингибитор HDAC4/5, который влияет также и на некоторые другие сигнальные пути клетки. Авторы удивились тому факту, что экспрессия MuRF-1 у этих крыс была снижена без какого-либо влияния на него транскрипционного фактора Foxo3. Они отметили, что механизм действия препарата им остался неясным. Содержание миогенина в этой работе исследовано не было.
Мы предположили, что ингибирование HDAC4/5 в ней могло предотвратить увеличение содержания миогенина в мышце, что сделало невозможным активацию экспрессии Е3-лигаз. Мы проверили эту гипотезу, применив ингибитор HDAC4 и 5, и обнаружили, что при 3-дневной разгрузке m. soleus HDAC4/5 регулирует экспрессию миогенина и мРНК MAFbx/Atrogin-1. Ингибирование одной только HDAC4 специфическим ингибитором (Tq-, Tasquinimod) показало, что при разгрузке она не контролирует экспрессию всех этих белков [33].
Итак, большее значение в регуляции экспрессии миогенина и MAFbx при разгрузке имеет HDAC5, но не HDAC4 [33]. Можно отметить также, что при 3-дневной разгрузке m. soleus HDAC5 регулирует экспрессию atrogin-1, но не MuRF-1. Вполне вероятно, что на более позднем этапе разгрузки к регуляции HDAC5 присоединяется механизм mTORC1, который активирует дальнейшую экспрессию E3-лигаз, запуская каскад Akt-FoxO (рис. 3).
С помощью какого механизма HDAC5 может влиять на экспрессию Е3 лигаз?
P. Du Bois с соавт. предположили, что PKD1 (протеинкиназа Д) создает в HDAC5 мотивы распознавания фосфо-14-3-3, которые рекрутируют белки шаперона 14-3-3, что в конечном итоге приводит к экспорту HDAC5 из ядра [34]. Это предполагаемый механизм транспорта HDAC5 из ядра. Когда HDAC5 находится в ядре (как в нашем эксперименте с ингибированием HDAC5 [33]), экспрессия MAFbx блокируется. Происходит это при участии транскрипционного фактора миогенина.
Итак, при функциональной разгрузке мышц меняется ядерно-цитоплазматический обмен HDAC1 и HDAC5, что позволяет им регулировать экспрессию atrogin-1 (но не MuRF1): HDAC1 импортируется в ядро, а HDAC5 экспортируется из ядра. Контроль экспрессии MAFbx осуществляется при участии миогенина, а также с помощью ацетилирования/деацетилирования сайта FOXO3 (наряду с его фосфорилированием/ дефосфорилированием).
4. РОЛЬ P38 MAPK В ЭКСПРЕССИИ MURF-1 И ATROGIN-1/MAFBX ПРИ ФУНКЦИОНАЛЬНОЙ РАЗГРУЗКЕ МЫШЦ
p38 MAPK – (митоген-активированная протеинкиназа) принадлежит к MAP-киназному пути. В клетке известны три основные MAP-киназы, которые активируются фосфорилированием: это p38 MAPK, ERK (extracellular signal-related kinase) и JNK (c-Jun NH2-terminal kinase) (рис. 4).
Рис. 4.
Сигнальные пути, регулируемые митогенактивируемыми протеинкиназами. p38 MAPK (p38- митогенактивируемая протеинкиназа), ERK (extracellular signal-related kinase) и JNK (c-Jun NH2-terminal kinase). Модифицированная схема из [35].

В целом известно, что p38 MAP-киназный сигнальный путь запускает процессы транскрипции и регулирует клеточную подвижность, и этот сигнальный путь участвует в регуляции мышечной массы. Его поведение при функциональной разгрузке мышц исследовали мало. Активацию p38 MAPK наряду с атрофией мышц при вывешивании крыс отмечали T. L. Hilder и соавт., E. Dupont и соавт. [36, 37]. Последние отмечали, что увеличение активности MAPK в клетке ведет к фосфорилированию цитоплазматических и/или ядерных белков, а МАP-киназный путь частично ответственен за регуляцию процессов пролиферации и дифференциации модулированием генной экспрессии. Эти авторы полагали, что увеличение фосфорилирования p38 MAPK после 14-дневного вывешивания крыс может быть связано с изменением миозинового фенотипа и регулированием экспрессии тяжелых цепей миозина. Мы подтвердили эту гипотезу, показав, что ингибирование p38 MAPK при разгрузке m. soleus препятствует увеличению экспрессии быстрых изоформ тяжелых цепей миозина из-за активации сигнального каскада кальцинейрин/NFAT [38]. Но этим функция p38 MAPK не исчерпывается. Y. Liu и соавт. в 2005 г. обнаружили, что ингибирование р38 в культуре миотуб С2С12 с помощью SB203580 или куркумина вело к снижению убиквитин-конъюгатной активности [22]. Таким образом, p38 MAPK способна регулировать активность убиквитин-протеасомного сигнального пути, однако эта ее функция in vivo при разгрузке мышц не изучалась. Ранее мы обнаружили, что пик экспрессии Е3-лигаз наблюдается к 3-му дню вывешивания крыс [14, 39]. В то же время содержание p38 MAPK во всех предыдущих работах исследовали на более поздних этапах развития атрофии и не ассоциировали с регуляцией работы убиквитин-протеасомной системы. Исключение составляет одна работа, в которой обнаружили повышение уровня фосфорилированной p38 MAPK в m. soleus крыс через 6 ч после иммобилизации конечности [40]. Экспрессию Е3-лигаз в этой работе не тестировали.
Мы предположили, что p38MAPK может принимать участие в регуляции экспрессии MuRF-1 и атрогена-1, и, соответственно, атрофических процессов в мышце при ее функциональной разгрузке. Для проверки гипотезы мы ингибировали фосфорилирование p38MAPK специфическим селективным ингибитором VX 745 в m.soleus крыс линии Вистар в течение 3-суточного вывешивания крыс. Мы обнаружили, что ингибирование p38 MAP-киназы предотвращает атрофию m. soleus крыс, вызванную разгрузкой. Происходило это за счет снижения экспрессии кальпаина-1, Е3-лигазы MuRF1 (но не atrogin-1/MAFbx), а также снижения убиквитинирования белков [39]. Механизм влияния p38 MAP-киназы на экспрессию Е3-лигазы MuRF1 может быть следующим. Как упоминалось выше, индуцированная разгрузкой экспрессия MuRF1 и MAFbx контролируется транскрипционным фактором FoxO [15]. Уровень фосфорилирования FOXO3 был снижен только в m. soleus крыс, вывешенных без ингибирования p38 MAP-киназы, а там, где вводили ингибитор, он не отличался от группы контроля. Т.к. только нефосфорилированная форма FOXO3 способна проникать в ядро и активировать экспрессию MuRF1, можно предположить, что ингибирование p38 MAP-киназы при вывешивании крыс препятствовало снижению уровня pFOXO3 и экспрессии MuRF1 в этой группе. Наши данные коррелируют с предыдущими исследованиями, показывающими, что активность FoxO3 может регулироваться с помощью передачи сигналов киназы p38α MAP. Например, транскрипционная активность FoxO3 снижалась в мышцах мышей с мышечно-специфическим удалением киназы p38α MAP [41]. Передача сигналов киназы p38 MAP также регулирует ядерную транслокацию и транскрипционную активность FoxO3 в миобластах [42]. На маркеры процессов трансляции и элонгации белков ингибирование p38 MAP-киназы при вывешивании никакого дополнительного влияния в нашем эксперименте не оказывало [39]. Мы показали также, что в эту регуляцию вовлечены IL-6 и PGC-1α.
Наше исследование впервые показало, что p38 MAP-киназа принимает участие в регуляции экспрессии MuRF1 (но не MAFbx), при функциональной разгрузке m.soleus.
Механизм, активирующий работу гена MuRF-1 может быть связан с фосфорилированием/дефосфорилированием транскрипционного фактора FOXO3.
Итак, 1. на раннем этапе функциональной разгрузки (3 дня) HDAC1, HDAC4/5 контролируют экспрессию Е3-лигазы atrogin-1/MAFbx, но не MuRF-1 в m.soleus. 2. Контроль экспрессии атрогена-1 (наряду с фосфорилированием/дефосфорилированием сайта FOXO3a) осуществляется с помощью его ацетилирования/деацетилирования. 3. HDAC4/5 осуществляет контроль над экспрессией atrogin-1/MAFbx также и с помощью миогенина. 4. При этом изменяется внутриклеточная локализация гистондеацетилаз: HDAC1 при функциональной разгрузке m. soleus поступает в ядро, HDAC5 экспортируется из ядра.
Можно заключить, что на раннем этапе функциональной разгрузки мышц влияние на экспрессию Е3-убиквитинлигаз осуществляется комплексно (со стороны нескольких сигнальных путей). Кроме известного пути Akt/mTORC1, сигнальные пути, регулируемые HDAC1 и HDAC4/5 контролируют экспрессию atrogin-1, а внутриклеточный путь p38 MAPK контролирует экспрессию MuRF1. Ранее мы не обнаружили взаимосвязи между регуляцией NF-kB сигнального пути (IKKbeta) и экспрессией MAFbx и MuRF1 на раннем этапе функциональной разгрузки. Знания о ранней активации убиквитин-протеасомной системы мышц приводят к выводу о том, что применение систем профилактики при возникновении условий для функциональной разгрузки мышц не должно быть отставленным. В настоящее время фармакологическая коррекция таких состояний начинает использоваться только после появления явных признаков дисфункции мышц. Определение механизмов, контролирующих протеолиз мышечных белков при функциональной разгрузке, поможет разработать систему фармакологической коррекции атрофических процессов в мышце.
Список литературы
Bodine SC, Baehr LM (2014) Skeletal muscle atrophy and the E3 ubiquitin ligases MuRF1 and MAFbx/atrogin-1. Am J Physiol Endocrinol Metab 307 (6): E469–484. https://doi.org/10.1152/ajpendo.00204.2014
Baehr LM, West DWD, Marshall AG, Marcotte GR, Baar K, Bodine SC (2017) Muscle-specific and age-related changes in protein synthesis and protein degradation in response to hindlimb unloading in rats. J Appl Physiol 122 (5): 1336–1350. https://doi.org/10.1152/japplphysiol.00703.2016
Baldwin KM, Haddad F (2002) Skeletal muscle plasticity: cellular and molecular responses to altered physical activity paradigms. Am J Phys Med Rehabil 81 (11 Suppl): S40–S51. https://doi.org/10.1097/01.PHM.0000029723.36419.0D
Fitts RH, Riley DR, Widrick JJ (2000) Physiology of a microgravity environment invited review: microgravity and skeletal muscle. J Appl Physiol 89 (2): 823–839. https://doi.org/10.1152/jappl.2000.89.2.823
Fluck M, Hoppeler H (2003) Molecular basis of skeletal muscle plasticity–from gene to form and function. Rev Physiol Biochem Pharmacol 146: 159–216. https://doi.org/10.1007/s10254-002-0004-7
Glass DJ (2003) Signalling pathways that mediate skeletal muscle hypertrophy and atrophy. Nat Cell Biol 5(2): 87–90. https://doi.org/10.1038/ncb0203-87
Jackman RW, Kandarian SC (2004) The molecular basis of skeletal muscle atrophy. Am J Physiol Cell Physiol 287(4): C834–C843. https://doi.org/10.1152/ajpcell.00579.2003
Cohen S, Brault JJ, Gygi SP, Glass DJ, Valenzuela DM, Gartner C, Latres E, Goldberg AL (2009) During muscle atrophy, thick, but not thin, filament components are degraded by MuRF1-dependent ubiquitylation. J Cell Biol 185(6): 1083–1095. https://doi.org/10.1083/jcb.200901052
Baldwin KM, Herrick RE, Ilyina-Kakueva E, Oganov VS (1990) Effects of zero gravity on myofibril content and isomyosin distribution in rodent skeletal muscle. FASEB J 4(1): 79–83. https://doi.org/10.1096/fasebj.4.1.2136840
Solomon V, Goldberg AL (1996) Importance of the ATP-ubiquitin-proteasome pathway in the degradation of soluble and myofibrillar proteins in rabbit muscle extracts. J Biol Chem 271(43): 26690–26697. https://doi.org/10.1074/jbc.271.43.26690
Clarke BA, Drujan D, Willis MS, Murphy LO, Corpina RA, Burova E, Rakhilin SV, Stitt TN, Patterson C, Latres E, Glass DJ (2007) The E3 Ligase MuRF1 degrades myosin heavy chain protein in dexamethasone-treated skeletal muscle. Cell Metab 6(5): 376–385. https://doi.org/10.1016/j.cmet.2007.09.009
Attaix D, Baracos VE (2010) MAFbx/Atrogin-1 expression is a poor index of muscle proteolysis. Curr Opin Clin Nutr Metab Care 13(3): 223–224. https://doi.org/10.1097/MCO.0b013e328338b9a6
Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, Poueymirou WT, Panaro FJ, Na E, Dharmarajan K, Pan ZQ, Valenzuela DM, DeChiara TM, Stitt TN, Yancopoulos GD, Glass DJ (2001) Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 294(5547): 1704–1708. https://doi.org/10.1126/science.1065874
Kachaeva EV, Shenkman BS (2012) Various jobs of proteolytic enzymes in skeletal muscle during unloading: facts and speculations. J Biomed Biotechnol 2012: 493618. https://doi.org/10.1155/2012/493618
Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, Walsh K, Schiaffino S, Lecker SH, Goldberg AL (2004) Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 117(3): 399–412. https://doi.org/10.1016/s0092-8674(04)00400-3
Shenkman BS, Belova SP, Lomonosova YN, Kostrominova TY, Nemirovskaya TL (2015) Calpain-dependent regulation of the skeletal muscle atrophy following unloading. Arch Biochem Biophys 584: 36–41. https://doi.org/10.1016/j.abb.2015.07.011
Belova SP, Shenkman BS, Kostrominova TY, Nemirovskaya TL (2017) Paradoxical effect of IKKbeta inhibition on the expression of E3 ubiquitin ligases and unloading-induced skeletal muscle atrophy. Physiol Rep 5(16): e13291. https://doi.org/10.14814/phy2.13291
McGee SL, Hargreaves M (2010) Histone modifications and skeletal muscle metabolic gene expression. Clin Exp Pharmacol Physiol 37(3): 392–396. https://doi.org/10.1111/j.1440-1681.2009.05311.x
Gaur V, Connor T, Sanigorski A, Martin SD, Bruce CR, Henstridge DC, Bond ST, McEwen KA, Kerr-Bayles L, Ashton TD, Fleming C, Wu M, Pike Winer LS, Chen D, Hudson GM, Schwabe JWR, Baar K, Febbraio MA, Gregorevic P, Pfeffer FM, Walder KR, Hargreaves M, McGee SL (2016) Disruption of the Class IIa HDAC CoRepressor Complex Increases Energy Expenditure and Lipid Oxidation. Cell Rep 16 (11): 2802–2810. https://doi.org/10.1016/j.celRep.2016.08.005
Астратенкова ИВ, Рогозкин ВА (2016) Сигнальные пути, участвующие в регуляции метаболизма белков скелетных мышц. Рос физиол журн им ИМ Сеченова 102 (7): 753–772. [Astratenkova IV, Rogozkin VA (2016) Signaling Pathways Involved in the Regulation of Protein Metabolism in Skeletal Muscle. Russ J Physiol 102(7): 753–772. (In Russ).]
Астратенкова ИВ, Рогозкин ВА (2017) Роль ацетилирования/деацетилирования гистонов и транскрипционных факторов в регуляции метаболизма в скелетных мышцах. Рос физиол журн им ИМ Сеченова 103(6): 593–605. [Astratenkova IV, Rogozkin VA (2017) The role of acetylation /deacetylation of histones and transcription factors in the regulation of skeletal muscle metabolism. Russ J Physiol 103(6): 593–605. (In Russ)].
Liu Y, Shen T, Randall WR, Schneider MF (2005) Signaling pathways in activity-dependent fiber type plasticity in adult skeletal muscle. J Muscle Res Cell Motil 26(1): 13–21. https://doi.org/10.1007/s10974-005-9002-0
Shenkman BS, Nemirovskaya TL (2008) Calcium-dependent signaling mechanisms and soleus fiber remodeling under gravitational unloading. J Muscle Res Cell Motil 29(6–8): 221–230. https://doi.org/10.1007/s10974-008-9164-7
Brocca L, Toniolo L, Reggiani C, Bottinelli R, Sandri M, Pellegrino MA (2017) FoxO-dependent atrogenes vary among catabolic conditions and play a key role in muscle atrophy induced by hindlimb suspension. J Physiol 595(4): 1143–1158. https://doi.org/10.1113/JP273097
Beharry AW, Sandesara PB, Roberts BM, Ferreira LF, Senf SM, Judge AR (2014) HDAC1 activates FoxO and is both sufficient and required for skeletal muscle atrophy. J Cell Sci 127 (Pt 7): 1441–1453. https://doi.org/10.1242/jcs.136390
Peris-Moreno D, Taillandier D, Polge C (2020) MuRF1/TRIM63, Master Regulator of Muscle Mass. Int J Mol Sci 21(18): 6663. https://doi.org/10.3390/ijms21186663
Bertaggia E, Coletto L, Sandri M (2012) Posttranslational modifications control FoxO3 activity during denervation. Am J Physiol Cell Physiol 302(3): C587–C596. https://doi.org/10.1152/ajpcell.00142.2011
Mochalova EP, Belova SP, Mirzoev TM, Shenkman BS, Nemirovskaya TL (2019) Atrogin-1/MAFbx mRNA expression is regulated by histone deacetylase 1 in rat soleus muscle under hindlimb unloading. Sci Rep 9(1): 10263. https://doi.org/10.1038/s41598-019-46753-0
Senf SM, Sandesara PB, Reed SA, Judge AR (2011) p300 Acetyltransferase activity differentially regulates the localization and activity of the FOXO homologues in skeletal muscle. Am J Physiol Cell Physiol 300(6): C1490–C1501. https://doi.org/10.1152/ajpcell.00255.2010
Moresi V, Williams AH, Meadows E, Flynn JM, Potthoff MJ, McAnally J, Shelton JM, Backs J, Klein WH, Richardson JA, Bassel-Duby R, Olson EN (2010) Myogenin and class II HDACs control neurogenic muscle atrophy by inducing E3 ubiquitin ligases. Cell 143(1): 35–45. https://doi.org/10.1016/j.cell.2010.09.004
Tang H, Inoki K, Lee M, Wright E, Khuong A, Khuong A, Sugiarto S, Garner M, Paik J, DePinho RA, Goldman D, Guan KL, Shrager JB (2014) mTORC1 promotes denervation-induced muscle atrophy through a mechanism involving the activation of FoxO and E3 ubiquitin ligases. Sci Signal 7(314): ra18. https://doi.org/10.1126/scisignal.2004809
Dupre-Aucouturier S, Castells J, Freyssenet D, Desplanches D (2015) Trichostatin A, a histone deacetylase inhibitor, modulates unloaded-induced skeletal muscle atrophy. J Appl Physiol 119(4): 342–351. https://doi.org/10.1152/japplphysiol.01031.2014
Mochalova EP, Belova SP, Kostrominova TY, Shenkman BS, Nemirovskaya TL (2020) Differences in the Role of HDACs 4 and 5 in the Modulation of Processes Regulating MAFbx and MuRF1 Expression during Muscle Unloading. Int J Mol Sci 21(13): 4815. https://doi.org/10.3390/ijms21134815
Du Bois P, Pablo Tortola C, Lodka D, Kny M, Schmidt F, Song K, Schmidt S, Bassel-Duby R, Olson EN, Fielitz J (2015) Angiotensin II Induces Skeletal Muscle Atrophy by Activating TFEB-Mediated MuRF1 Expression. Circ Res 117(5): 424–436. https://doi.org/10.1161/CIRCRESAHA.114.305393
Wang J, Xia Y (2012) Assessing developmental roles of MKK4 and MKK7 in vitro. Commun Integr Biol 5(4): 319–324. https://doi.org/10.4161/cib.20216
Hilder TL, Baer LA, Fuller PM, Fuller CA, Grindeland RE, Wade CE, Graves LM (2005) Insulin-independent pathways mediating glucose uptake in hindlimb-suspended skeletal muscle. J Appl Physiol 99(6): 2181–2188. https://doi.org/10.1152/japplphysiol.00743.2005
Dupont E, Cieniewski-Bernard C, Bastide B, Stevens L (2011) Electrostimulation during hindlimb unloading modulates PI3K-AKT downstream targets without preventing soleus atrophy and restores slow phenotype through ERK. Am J Physiol Regul Integr Comp Physiol 300(2): R408–R417. https://doi.org/10.1152/ajpregu.00793.2009
Sharlo KA, Mochalova EP, Belova SP, Lvova ID, Nemirovskaya TL, Shenkman BS (2020) The role of MAP-kinase p38 in the m. soleus slow myosin mRNA transcription regulation during short-term functional unloading. Arch Biochem Biophys 695: 108622. https://doi.org/10.1016/j.abb.2020.108622
Belova SP, Mochalova EP, Kostrominova TY, Shenkman BS, Nemirovskaya TL (2020) P38alpha-MAPK Signaling Inhibition Attenuates Soleus Atrophy during Early Stages of Muscle Unloading. Int J Mol Sci 21(8): 2756. https://doi.org/10.3390/ijms21082756
Kawamoto E, Koshinaka K, Yoshimura T, Masuda H, Kawanaka K (2016) Immobilization rapidly induces muscle insulin resistance together with the activation of MAPKs (JNK and p38) and impairment of AS160 phosphorylation. Physiol Rep 4(15): e12876. https://doi.org/10.14814/phy2.12876
Yuasa K, Okubo K, Yoda M, Otsu K, Ishii Y, Nakamura M, Itoh Y, Horiuchi K (2018) Targeted ablation of p38alpha MAPK suppresses denervation-induced muscle atrophy. Sci Rep 8(1): 9037. https://doi.org/10.1038/s41598-018-26632-w
Clavel S, Siffroi-Fernandez S, Coldefy AS, Boulukos K, Pisani DF, Derijard B (2010) Regulation of the intracellular localization of Foxo3a by stress-activated protein kinase signaling pathways in skeletal muscle cells. Mol Cell Biol 30 (2): 470–480. https://doi.org/10.1128/MCB.00666-09
Дополнительные материалы отсутствуют.
Инструменты
Российский физиологический журнал им. И.М. Сеченова