

Российский физиологический журнал им. И.М. Сеченова, 2021, T. 107, № 9, стр. 1126-1137
Ингибирование ГАМК-трансаминазы и ГАМК-транспортеров в головном мозге с помощью вигабатрина и тиагабина ослабляет развитие судорог у крыс при дыхании гипербарическим кислородом
И. Т. Демченко 1, С. Ю. Жиляев 1, Т. Ф. Платонова 1, О. С. Алексеева 1, *, Е. Р. Никитина 1
1 Институт эволюционной физиологии и биохимии
им. И.М. Сеченова РАН
Санкт-Петербург, Россия
* E-mail: osa72@inbox.ru
Поступила в редакцию 10.05.2021
После доработки 23.06.2021
Принята к публикации 05.07.2021
Аннотация
Гипербарический кислород (ГБО2) угнетает ГАМК-ергическую функцию в головном мозге, что приводит к развитию судорожных реакций в виде пароксизмальных разрядов на ЭЭГ и двигательных расстройств, аналогичных генерализованному эпилептическому припадку. Мы предполагаем, что дефицит ГАМК-ергической нейропередачи в ГБO2, возникающий за счет подавления синтеза ГАМК, может быть преодолен путем блокирования клиренса ГАМК из синаптической щели и замедления ее ферментативного расщепления. В настоящем исследовании изучали развитие судорожных реакций у крыс под давлением кислорода 5 АТА после блокады ГАМК-транспортеров с помощью тиагабина и ингибирования ГАМК-трансаминазы с помощью вигабатрина. В работе получены новые данные о том, что: (1) ингибирование ГАМК-трансаминазы с помощью вигабатрина или ГАМК-транспортеров с помощью тиагабина предотвращало развитие судорог у крыс под давлением кислорода 5 АТА; (2) кислородные судороги в ГБО2 проявлялись при снижении ГАМК в мозге на 30–40% от исходного уровня; (3) ингибирование ГАМК-трансаминазы с помощью вигабатрина предотвращало снижение ГАМК в стриатуме крыс под давлением 5 АТА. Следовательно, замедление синаптического клиренса ГАМК или ослабление ферментативного разрушения медиатора могут значительно компенсировать ГБО2-вызванное ослабление синтеза ГАМК и повысить концентрацию тормозного медиатора до уровня, достаточного для реализации ГАМК-ергической функции, предотвращающей развитие судорожного синдрома.
ВВЕДЕНИЕ
Кислород под давлением, превышающим атмосферное, или гипербарический кислород (ГБО2), широко используется в медицинской практике и при подводных погружениях для проведения работ в экономических и военных интересах. Несмотря на относительную безопасность, дыхание ГБО2 сопряжено с риском его токсического действия на ЦНС, клиническим проявлением которого являются пароксизмальная активность на ЭЭГ и тонико-клонические судороги, аналогичные генерализованному эпилептическому припадку [1–3].
Ключевая роль в инициировании кислородных судорог принадлежит интенсивно образующимся при гипероксии редокс-молекулам – реактивным формам кислорода (ROS) и азота (RNS), способным через окислительно-восстановительные реакции изменять молекулярную структуру клеток и нарушать межклеточное взаимодействие. Подтверждением этому является повышенное образование в мозге при ГБО2 компонентов ROS – супероксиданионов (${\text{O}}_{2}^{ - }$), гидроксильных радикалов (ОН–) и перекиси водорода (Н2О2), причем уровень их продукции возрастает с увеличением парциального давления вдыхаемого кислорода [4–6]. ГБО2 также стимулирует продукцию в мозге таких RNS, как оксид азота (NO) и пероксинитрит [7, 8], и их повышение в мозговой ткани предшествует появлению пароксизмальной активности на ЭЭГ [9, 10]. Вовлечение ROS–RNS в патогенез судорожного синдрома в ГБО2 подтверждается данными об ускорении судорог после введения NO-доноров (SNAP) или их задержки после подавления синтеза NO с помощью L-NAME [7]. Установлено также, что у мышей-нокаутов по нейрональной NOS судорожная реакция в ГБО2 развивалась значительно позже, чем у обычных животных [11].
Участие ROS–RNS в инициировании судорог предполагает наличие редокс-чувствительных мишеней в мозге, воздействие на которые приводит к нарушению электрогенеза в мозговых нейронах и синаптической передачи. Наиболее вероятная мишень для инициирования кислородных судорог определена в ГАМК-ергической нейропередаче. Установлено, что гипербарический кислород ингибирует синтез ГАМК в мозге, что приводит к ослаблению тормозной нейропередачи с последующим сдвигом баланса процессов возбуждения и торможения в ЦНС [12, 13]. Доказательством этого является снижение ГАМК в межклеточной среде мозга крыс, зарегистрированное in vivo во время ГБО2-экспозиции с помощью микродиализа, сопряженного с высокоэффективной жидкостной хроматографией [10, 12]. Причиной уменьшения внеклеточной ГАМК в ГБО2 явилось снижение активности глутаматдекарбоксилазы (GAD), катализирующей синтез тормозного медиатора в нервных клетках [14]. Угнетение ферментативной активности GAD происходит за счет ее S-нитрозилирования – присоединения группы оксида азота к тиоловым остаткам цистеина внутри белка-фермента [15].
Представленные данные допускают, что ингибирование синтеза ГАМК в головном мозге при гипероксии приводит к снижению ее выделения в синаптическую щель и, следовательно, к ослаблению тормозной медиаторной функции. В связи с этим возникает вопрос, можно ли с помощью каких-то воздействий сохранить содержание медиатора в синаптическом пространстве на уровне, достаточном для реализации полноценной тормозной нейропередачи. После квантового выделения ГАМК в синаптическую щель и активации постсинаптических рецепторов, большая часть неиспользованного медиатора активно удаляется из нее путем высоко аффинного Na+-зависимого захвата медиатора нервными и глиальными клетками при участии электрогенных ГАМК-транспортеров [16]. Часть медиатора пассивно диффундирует в межклеточное пространство, где реализуется его внесинаптическая тоническая медиация [17]. Критически значимым для поддержания необходимого уровня медиатора в синапсах является его ферментативное расщепление с участием ГАМК-трансаминазы (ГАМК-Т), локализованной преимущественно в астроцитах головного мозга.
Мы предполагаем, что блокирование захвата ГАМК и замедление ее расщепления может увеличить содержание медиатора в синаптической щели до уровня, достаточного для обеспечения тормозной нейропередачи и, таким образом, ослабить или предотвратить развитие судорожного синдрома. Для проверки этой гипотезы в настоящей работе изучали развитие судорог в ГБО2 у крыс, которым перед экспозицией в кислороде под давлением 5 АТА в боковой желудочек головного мозга вводили тиагабин – селективный ингибитор ГАМК-транспортеров и вигабатрин – селективный ингибитор ГАМК-трансаминазы. Для оценки содержания ГАМК в головном мозге при ГБО2 до и после введения ингибиторов, определяли ее концентрацию с помощью внутримозгового микродиализа, сопряженного с высокопроизводительной жидкостной хроматографией. Такая постановка задачи и подход к ее решению являются новыми, так как анализ существующих баз данных показал, что изучение противосудорожной эффективности вигабатрина и тиагабина в ГБО2 с учетом оценки динамики ГАМК в мозге до сих пор не проводилось.
МЕТОДЫ ИССЛЕДОВАНИЯ
Опыты проводили на крысах линии Sprague-Dawley массой 307 ± 9 г, полученных из питомника “Пущино” (Московская обл., г. Пущино, Россия). Протокол опытов одобрен Комиссией по этике Института эволюционной физиологии и биохимии им. И.М.Сеченова РАН в соответствии с Международными рекомендациями по проведению медико-биологических исследований с использованием животных (CIOMS, Geneva, 1985). За 5–7 дней до опытов каждому животному под наркозом (нембутал, 50 мг/кг) в боковой желудочек головного мозга вводили металлическую канюлю согласно стереотаксическим координатам [18]: АР = –1.0 мм, LМ = ±1.5 мм, V = 3.5 мм. Канюля укреплялась с помощью 2-х стальных винтов, ввернутых биполярно в теменные области кости черепа, и зубопротезного цемента. Перед опытами каждому животному через канюлю в боковой желудочек мозга с помощью микрошприца (Hamilton, 20 мкл) вводили раздельно искусственный ликвор (контроль), вигабатрин и тиагабин. Препараты, полученные из Sigma Aldrich, растворяли в искусственном ликворе, и их содержание в растворе объемом 10 мкл составляло: тиагабин – 0.05 мг, вигабатрин – 0.5 мг. Животным контрольной группы вводили искусственный ликвор в объеме 10 мкл. Указанные дозы ингибиторов не вызывали двигательных нарушений (дисбаланса) у животных, которых тестировали на вращающемся стержне (Rotarod test). После введения препаратов крыс помещали в барокамеру объемом 100 л по 1–2 животных в каждом опыте. Повышение давления кислорода в камере до 5 АТА осуществляли со скоростью 1 АТА/мин. Температуру в камере поддерживали в пределах 23–25°С, влажность – около 60%, содержание СО2 не превышало 0.05%.
Во время ГБО2-экспозиции проводили непрерывную видеорегистрацию животных в барокамере. Экспозицию продолжали до появления генерализованных клонических или тонических судорог, а при их отсутствии, максимально до 90 мин. Время декомпрессии составляло 8 мин. Для анализа результатов фиксировали время (в минутах) появления моторных судорожных реакций от начала экспозиции животных в камере при давлении кислорода 5 АТА. Для анализа результатов фиксировали время (в минутах) появления различных двигательных нарушений у животных от начала кислородной экспозиции под давлением 5 АТА. Для количественной оценки развития судорог у крыс в барокамере мы адаптировали известную шкалу судорожных состояний, позволяющую разделить тяжесть приступов на стадии: движение рта и лица, кивание головой, клонус передних конечностей, подъем с клонусом передних конечностей и подъем и падение с клонусом передних конечностей [19]. В случае отсутствия судорожных проявлений за латентный период принимали время кислородной экспозиции равное 90 мин. Всего в опытах использовано 38 крыс, разделенных на 3 группы. В контрольной группе 1 (n = 14) животным перед кислородной экспозицией при давлении 5 АТА в желудочек мозга вводили искусственный ликвор. Животным группы 2 (n = 12) перед гипероксической экспозицией в мозговой желудочек вводили вигабатрин, а крысам группы 3 (n = 12) – тиагабин.
В другой серии опытов на наркотизированных крысах Sprague-Dawley с помощью внутримозгового микродиализа измеряли содержание ГАМК в стриатуме во время кислородной экспозиции при 5 АТА. Эта работа выполнена И.Т. Демченко на базе Гипербарического центра университета Дьюка (Durham, NC, США). Подробное изложение метода измерений ГАМК в мозге с использованием микродиализа в ГБО2 представлено в публикациях [12, 20]. Наркотизированным животным (уретан + хлоралоза, 750 + 250 мг/кг) в стриатум (координаты по [18]: АР = +1.0 мм, LМ ± 2.5 мм, V = 5.8 мм) вводили микродиализные канюли (CMA/11, CMA/Microdialysis AB, Sweden). Во время ГБО2-экспозиции канюли перфузировали искусственным ликвором со скоростью 1.0 мкл/мин, а пробы диализата автоматически отбирали каждые 15 мин с помощью коллектора, расположенного в барокамере рядом с животным (CMA 142 Microfraction Collector, AB, Швеция). Измерения ГАМК в пробах проводили сразу после декомпрессии с помощью высокопроизводительной жидкостной хроматографии (HPLC) с электрохимической детекцией ГАМК (ESA model 5100A). Содержание ГАМК в пробах диализата определяли в нмоль/л по калибровочным стандартам. Параметры ГБО2-экспозиции были такими же, как и в первой части исследований. В опытах использовано 22 животных, разделенных на 3 группы. У 7-ми животных (группа 1) и 8-ми крыс (группа 2) микродиализные канюли после имплантации в стриатум перфузировали искусственным ликвором в течение 75 мин при дыхании атмосферным воздухом (контроль), а затем под давлением кислорода 3 и 5 АТА соответственно. У животных группы 3 (n = 7) перфузию мозговой микроканюли при дыхании воздухом и гипербарическим кислородом 5 АТА осуществляли искусственным ликвором с добавлением вигабатрина (0.7 мМ).
Статистическую оценку полученных результатов проводили с помощью дисперсионного анализа SigmaPlot 13.0 (Systat Software, Inc., San Jose, CA, США). Однофакторный дисперсионный анализ использовали для сравнения латентных периодов судорожных реакций в ГБО2 у животных при введении искусственного ликвора и ингибиторов. Двухфакторный дисперсионный анализ применяли для сравнения изменения содержания ГАМК в стриатуме крыс контрольной и экспериментальных групп. Для выявления достоверности отличий использовали парный t - критерий. Все данные представлены как М ± SEM, при этом в качестве статистически значимых принимали значения p < 0.05.
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
У бодрствующих крыс контрольной группы во время ГБО2-экспозиции наблюдались характерные паттерны моторной судорожной активности, которые проявлялись в определенной стадийной последовательности. В наших исследованиях мы адаптировали известную шкалу судорожных состояний [19] для количественной оценки тяжести стадий кислородных судорог. После окончания компрессии животные в течение 10–15 мин оставались неподвижными. Далее у крыс появлялся интенсивный груминг, отдельные сокращения мышц рта, легкое потряхивание головы и передних лап, переходящие у части животных в кратковременные встряхивания всего тела, типа эффекта “мокрой собаки”, длительностью 1–3 с (стадия 1). На стадии 2 наблюдались повторяющиеся локальные сокращения мышц мордочки, головы, передних конечностей, а также всего тела. Проявлялись непродолжительные кивания головой. Миоклонии продолжались от 5 до 15 с и могли повторяться через несколько минут. На стадии 3 у животных появлялись ритмические сокращения мышц всего тела продолжительностью от 10 до 25 с, при этом животные могли вставать на задние лапы, пятиться назад, сильно бить хвостом. Стадия 4 у животных характеризовалась наличием генерализованных клонических или тонических конвульсий. Судорожный припадок сопровождался тахикардией, гипервентиляцией и другими признаками расстройства вегетативной нервной системы.
Выделенные стадии развития кислородных судорог в целом совпадают с известной шкалой пяти судорожных стадий [19], однако мы не нашли четких различий между стадиями 3 и 4 указанной классификации, поэтому ограничились выделением 4-х стадий в развитии судорог у крыс в ГБО2.
Стадийное развитие судорожного синдрома в ГБО2 сохранялось у животных, которым предварительно вводились тиагабин или вигабатрин, однако используемые препараты задерживали появление каждой стадии по отношению к временным параметрам развития аналогичных судорожных признаков у контрольных животных. Более выраженным противосудорожным эффектом обладал тиагабин, при введении которого 4-я стадия не развивалась у 34% животных (рис. 1).
Рис. 1.
Латентное время развития судорог у бодрствующих крыс под давлением кислорода 5 АТА после введения в мозговой желудочек искусственного ликвора (CSF), вигабатрина (VGB) и тиагабина (TGB).
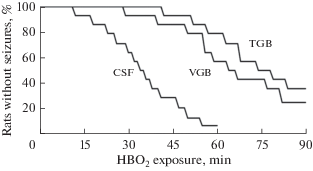
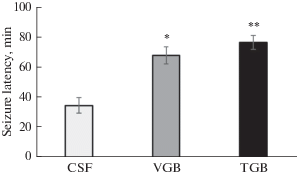
Величины латентного времени появления судорог у животных с ингибированными ГАМК-транспортерами или ГАМК-Т были достоверно больше, чем у крыс контрольной группы (рис. 2).
Рис. 2.
Средние значения латентного периода появления судорог у крыс под давлением кислорода 5 АТА после введения в мозговой желудочек искусственного ликвора (CSF), вигабатрина (VGB) и тиагабина (TGB), * p < 0.05 и ** p < 0.01 по сравнению с CSF.
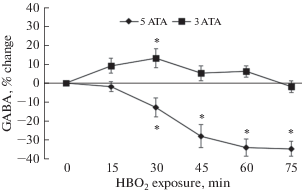
У наркотизированных животных признаков двигательных нарушений в ГБО2 не наблюдалось, но на ЭЭГ появлялись судорожные спайки через 68 ± 7.4 мин после начала экспозиции под давлением кислорода 5 АТА. Концентрация ГАМК в стриатуме наркотизированных крыс при дыхании атмосферным воздухом составляла 58 ± 8 нмоль/л. Под давлением кислорода 3 АТА уровень ГАМК в стриатуме не изменился, а при 5 АТА базовое значение медиатора снизилось на 34 ± 5.3% через 60 мин гипероксической экспозиции (рис. 3).
Рис. 3.
Временной профиль изменений ГАМК в стриатуме крыс под давлением кислорода 3 и 5 АТА. * p < 0.05 по сравнению с контролем (воздух).
Предварительное введение крысам вигабатрина повышало уровень ГАМК в стриатуме животных при дыхании воздухом, а в ГБО2 содержание медиатора снижалось достоверно меньше, чем у крыс без ингибирования ГАМК-трансаминазы (рис. 4).
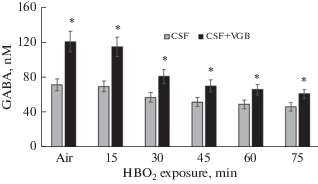
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
В настоящем исследовании получено несколько новых данных: (1) ингибирование ГАМК-трансаминазы с помощью вигабатрина или ГАМК-транспортеров с помощью тиагабина ослабляло развитие судорог у крыс под давлением кислорода 5 АТА; (2) кислородные судороги в ГБО2 появлялись при снижении ГАМК в стриатуме на 30–40% от исходного уровня; (3) ингибирование ГАМК-трансаминазы с помощью вигабатрина предотвращало снижение ГАМК в стриатуме крыс при их экспозиции в кислороде под давлением 5 АТА и значительно задерживало развитие судорог. Полученные данные позволяют проанализировать ГАМК-зависимый механизм кислородных судорог и оценить эффективность противоэпилептических препаратов для предотвращения судорожного синдрома в ГБО2.
Причастность ГАМК-ергической передачи к кислородным судорогам была установлена еще в 70-х годах прошлого столетия, когда было показано ингибирование в ГБО2 ферментативной активности GAD и снижение ГАМК в головном мозге животных после их экспозиции в гипербарическом кислороде [14, 21, 22]. В опытах in vivo был установлен пороговый уровень понижения мозгового ГАМК в ГБО2, ниже которого появлялись судороги. Так, при дыхании кислородом под давлением 5 АТА судорожная активность на ЭЭГ у крыс появлялась, когда содержание межклеточного ГАМК, зарегистрированное с помощью внутримозгового микродиализа, снижалось на 37% [12], а в других опытах на 29% [10]. В настоящей работе уровень ГАМК перед кислородными судорогами понижался на 34%. Для сравнения, в нормальных условиях судороги у крыс появлялись, когда ингибирование GAD с помощью 3-меркаптопропионовой кислоты приводило к снижению ГАМК в разных структурах мозга крыс на 20–35% [23].
Какая из GAD-изоформ теряет каталитическую активность в гипербарической гипероксии остается до сих пор неясным. При этом одни авторы полагают, что это GAD67 [24], а другие – GAD65 [15]. У млекопитающих GAD существует в двух изоформах, GAD65 и GAD67, кодируемых различными генами [25]. В ГАМК-ергических нейронах мозга экспрессируются обе изоформы GAD, но их уровни экспрессии и субклеточная локализация различаются. GAD67 является цитоплазматическим ферментом, находящимся во всех телах и отростках ГАМК-ергических нейронов, тогда как GAD65 локализуется преимущественно в нервных терминалях и встроена в мембраны везикул, содержащих нейротрансмиттер. У мышей-нокаутов по GAD67 наблюдается снижение концентрации ГАМК в коре головного мозга на 93%, приводящее к смерти новорожденных, и их эпилептический фенотип остается в значительной степени неизвестным [26]. С другой стороны, у мышей-нокаутов по GAD65 не наблюдается значимых морфологических дефектов и не обнаруживается значительных изменений содержания ГАМК в мозге, однако у них развиваются спонтанные эпилептиформные припадки и выявлена повышенная чувствительность к химическим веществам, вызывающим судороги [27]. Показано также, что изоформа GAD65 непосредственно регулирует выделение ГАМК из пресинаптических терминалей [28].
Несмотря на имеющиеся доказательства ингибирования GAD и снижения содержания тормозного медиатора в ГБО2, механизм подавления активности этого фермента остается малоизученным. Активность GAD понижается в ГБО2 быстро, поскольку судороги при 5 АТА у большинства бодрствующих животных в настоящих опытах появлялись уже через 30–40 мин после начала кислородной экспозиции, а при 6 АТА – через 20–30 мин [7]. Очевидно, что за такой короткий период времени трудно ожидать блокирования внутриклеточного синтеза белка GAD. Наиболее вероятным механизмом потери каталитической активности фермента является посттрансляционное изменение его молекулярной структуры под действием RONS. При гипероксии генерация редокс-молекул достигает чрезмерных уровней, способных модифицировать GAD посредством окисления или нитрозилирования аминокислотных остатков [29]. Посттрансляционная модификация тиолсодержащих остатков цистеина (Cys) считается одним из основных драйверов редокс-сигнализации в клетках, так как они легко окисляются или нитрозилируются, причем эти реакции являются обратимыми и по скорости сопоставимы с внутриклеточной передачей сигналов [30]. Исследования показали, что механизм инактивации GAD состоит в S-нитрозилировании белка-фермента по цистеину путем присоединения к ее тиоловым остаткам группы оксида азота (NO) [15]. Такая реакция ведет к посттрансляционной модификации фермента и снижению его активности [31, 32]. Данный механизм ингибирования GAD подтверждают результаты исследований, в которых подавление синтеза NO путем системного ингибирования NO-синтаз (NOS) ослабляло развитие судорог у крыс [7], а у мышей-нокаутов по нейрональной NOS время наступления судорог в ГБО2 было значительно больше, чем у обычных животных [11].
В случае ингибирования GAD и снижения синтеза медиатора в ГБО2 потенциально существует несколько способов усиления ГАМК-ергической функции. Агонисты и миметики ГАМК (прогабид, диазепам, фенобарбитал, пропофол) могут усиливать ингибирующую функцию медиатора, действуя на проводимость ионов хлора или на рецепторы ГАМК-А. Стимуляция синтеза и высвобождение ГАМК с помощью вальпроата или габапентина способны повышать уровень синаптической ГАМК. Замедление деградации ГАМК с помощью вигабатрина или вальпроата могут повышать внеклеточное содержание ГАМК. И, наконец, блокаторы транспортеров, например, тиагабин, способны продлевать действие ГАМК в синаптической щели. В настоящей работе была изучена возможность усиления ГАМК-ергической функции в ГБО2 за счет снижения клиренса медиатора и замедления его разрушения. Полученные результаты свидетельствуют о том, что блокирование транспортеров ГАМК или ингибирование ГАМК-Т восстанавливает ослабленную тормозную нейротрансмиссию в ГБO2. Справедливость этого вывода подтверждается несколькими данными. Так, у крыс с предварительно введенными в мозговой желудочек тиагабином или вигабатрином, судороги в ГБO2 появлялись значительно позже, чем у контрольных животных, что может указывать на восстановление ГАМК-передачи из-за увеличения медиатора в межклеточной среде (рис. 1). Действительно, у крыс при нормоксии и атмосферном давлении ингибирование фермента ГАМК-Т с помощью вигабатрина увеличивало концентрацию ГАМК во внеклеточном пространстве более чем в 2 раза. ГАМК в стриатуме при 3 АТА кислорода достоверно не изменялся, что может свидетельствовать об относительно низкой продукции супероксид анионов и NO при таком уровне гипероксии [8, 13]. Далее, после ингибирования ГАМК-Т с помощью вигабатрина уровень медиатора в головном мозге не понижался в ГБО2 ниже исходного уровня, тогда как у животных без ингибирования фермента ГАМК в стриатуме снизился на 34% (рис. 3). Так как с помощью мозгового микродиализа измеряется межклеточное содержание ГАМК, которое на 50–70% состоит из синаптического нейромедиатора [33] и некоторой части ГАМК, выделяемой из глиальных клеток [34], можно полагать, что ингибирование ГАМК-Т или ГАМК-транспортеров может увеличивать ГАМК как в синаптической щели, так и во внеклеточной среде. Все вместе свидетельствует о том, что нарушение функции ГАМК в ГБО2 может быть восстановлено за счет увеличения фазовой синаптической и тонической внесинаптической ГАМК-ергической нейропередачи. Выраженный противосудорожный эффект применяемых препаратов свидетельствует о том, что ГБО2 не подавляет функцию транспортеров ГАМК и ферментативную активность ГАМК-трансаминазы. В противном случае вводимые в мозг препараты не вызывали бы столь выраженного противосудорожного эффекта.
Использованные в данной работе ингибиторы продемонстрировали примерно равную противосудорожную активность. Вигабатрин ранее тестировался в опытах на мышах [35], крысах [36] и мини-свиньях [37] и проявил себя как эффективный протектор ГБО2-вызываемых судорог. Тиагабин был синтезирован как ГАМК-миметик, который содержит лиофильную группу, позволяющую препарату беспрепятственно проникать в мозг и оказывать противосудорожное действие. Благодаря этому свойству тиагабин нашел широкое применение в клинике для лечения эпилепсии.
Оба препарата, вводимые в желудочки головного мозга, показали высокую противосудорожную активность, однако, когда вигабатрин доставлялся в стриатум посредством микродиализа, он не проявлял противосудорожного эффекта, несмотря на повышение локального уровня ГАМК. Это согласуется с нашими более ранними данными, когда судороги в ГБO2 предотвращались после понижения продукции NO в мозге за счет введения L-NAME в мозговые желудочки, тогда как локальное ингибирование NOS только в стриатуме не предотвращало токсического действия кислорода на ЦНС [38]. Это указывает на то, что кислородные судороги можно предотвратить только путем ингибирования ГАМК-Т или транспортеров ГАМК одновременно в различных структурах мозга, идентификация которых требует дополнительных исследований.
Таким образом, дефицит ГАМК-ергической нейропередачи в ГБO2, который возникает из-за угнетения синтеза ГАМК, может быть компенсирован путем увеличения уровня ГАМК в синаптическом пространстве и во внеклеточной среде с помощью фармакологических препаратов. Изучение такой возможности показало, что ингибиторы ГАМК-трансаминазы и ГАМК-транспортеров повышают концентрацию медиатора в головном мозге и предотвращают развитие кислородных судорог. Следовательно, в случае подавления синтеза ГАМК в ГБО2, замедление клиренса медиатора из синапсов или ослабление его разрушения могут повысить концентрацию тормозного медиатора до уровня, достаточного для реализации полноценной ГАМК-ергической функции, и, тем самым, предотвратить развитие судорожного синдрома. Полученные результаты свидетельствуют о возможности практического использования вигабатрина и тиагабина для предотвращения развития кислородных судорог.
Список литературы
Зальцман ГЛ (1968) Стадии развития кислородной эпилепсии и функциональное состояние нервной системы. В кн. Гипербарические эпилепсия и наркоз. Л. Наука. [Zaltsman GL (1968) Stages of formation of oxygen epilepsy and the functional state of the centres of the nervous system. In book: Hyperbaric epilepsy and narcosis. Ed. Zaltsman G.L. Leningrad. Nauka. (In Russ)].
Balentine JD (1982) Pathology of Oxygen Toxicity. New York: Academic.
Селивра АИ (1983) Гипербарическая оксигенация. Физиологические механизмы реакций центральной нервной системы на гипероксию. Л. Наука. [Selivra AI (1983) Hyperbaric oxygenation. Physiological mechanisms of central nervous system responses to hyperoxia. Leningrad. Nauka. (In Russ)].
Torbati D, Church DF, Keller JM, Pryor WA (1992) Free radical generation in the brain precedes hyperbaric oxygen-induced convulsions. Free Radic Biol Med 13(2): 101–106. https://doi.org/10.1016/0891-5849
Dean JB, Mulkey DK, Garcia AJ, Putnam RW, Henderson RA (2003) Neuronal sensitivity to hyperoxia, hypercapnia, and inert gases at hyperbaric pressures. J Appl Physiol (1985) 95(3): 883–909. https://doi.org/10.1152/japplphysiol.00920.2002
D'Agostino DP, Putnam RW, Dean JB (2007) Superoxide (*O2-) production in CA1 neurons of rat hippocampal slices exposed to graded levels of oxygen. J Neurophysiol 98(2): 1030–1041. https://doi.org/10.1152/jn.01003.2006
Demchenko IT, Boso AE, O’Neill TJ, Bennett PB, Piantadosi CA (2000) Nitric oxide and cerebral blood flow responses to hyperbaric oxygen. J Appl Physiol (1985) 88(4): 1381–1389. https://doi.org/10.1152/jappl.2000.88.4.1381
Thom SR, Bhopale V, Fisher D, Manevich Y, Huang PL, Buerk DG (2002) Stimulation of nitric oxide synthase in cerebral cortex due to elevated partial pressures of oxygen: an oxidative stress response. J Neurobiol 51(2): 85–100. https://doi.org/10.1002/neu.10044
Demchenko IT, Boso AE, Whorton AR, Piantadosi CA (2001) Nitric oxide production is enhanced in rat brain before oxygen-induced convulsions. Brain Res 917(2): 253–261. https://doi.org/10.1016/s0006-8993
Zhang S, Takeda Y, Hagioka S, Takata K, Aoe H, Nakatsuka H, Yokoyama M, Morita K (2005) Measurement of GABA and glutamate in vivo levels with high sensitivity and frequency. Brain Res Brain Res Protoc 14(2): 61–66. https://doi.org/10.1016/j.brainresprot.2004.03.005
Atochin DN, Demchenko IT, Astern J, Boso AE, Piantadosi CA, Huang PL (2003) Contributions of endothelial and neuronal nitric oxide synthases to cerebrovascular responses to hyperoxia. J Cereb Blood Flow Metab 23(10): 1219–1226. https://doi.org/10.1097/01.WCB.0000089601.87125.E4
Demchenko IT, Piantadosi CA (2006) Nitric oxide amplifies the excitatory to inhibitory neurotransmitter imbalance accelerating oxygen seizures. Undersea Hyperb Med 33(3): 169–174.
Ciarlone GE, Hinojo CM, Stavitzski NM, Dean JB (2019) CNS function and dysfunction during exposure to hyperbaric oxygen in operational and clinical settings. Redox Biol 27: 101159. https://doi.org/10.1016/j.redox.2019.101159
Wood JD, Watson WJ (1962) Protective action of gamma-aminobutyric acid against oxygen toxicity. Nature 195: 296. https://doi.org/10.1038/195296a0
Gasier HG, Demchenko IT, Tatro LG, Piantadosi CA (2017) S-nitrosylation of GAD65 is implicated in decreased GAD activity and oxygen-induced seizures. Neurosci Lett 653: 283–287. https://doi.org/10.1016/j.neulet.2017.05.067
Kersanté F, Rowley SC, Pavlov I, Gutièrrez-Mecinas M, Semyanov A, Reul JM, Walker MC, Linthorst AC (2013) A functional role for both -aminobutyric acid (GABA) transporter-1 and GABA transporter-3 in the modulation of extracellular GABA and GABAergic tonic conductances in the rat hippocampus. J Physiol 591(10): 2429–2441. https://doi.org/10.1113/jphysiol.2012.246298
Semyanov A, Walker MC, Kullmann DM, Silver RA (2004) Tonically active GABA A receptors: modulating gain and maintaining the tone. Trends Neurosci 27(5): 262–269. https://doi.org/10.1016/j.tins.2004.03.005
Paxinos G, Watson C (2005) The Rat Brain in Stereotaxic Coordinates. Boston, MA: Elsevier.
Racine RJ (1972) Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol 32(3): 281–294. https://doi.org/10.1016/0013-4694(72)90177-0
Donzanti BA, Yamamoto BK (1988) An improved and rapid HPLC-EC method for the isocratic separation of amino acid neurotransmitters from brain tissue and microdialysis perfusates. Life Sci 43(11): 913–922. https://doi.org/10.1016/0024-3205(88)90267-6
Щербакова ГВ (1962) Активность глютаматдекарбоксилазы и содержание γ-аминомасляной кислоты в мозге крыс при разных функциональных состояниях, вызванных повышенным давлением кислорода. ДАН СССР 146(5): 1213–1215. [Shcherbakova G.V. Glutamate decarboxylase activity and γ-aminobutyric acid content in rat brain at different functional states caused by high oxygen pressure. DAN USSR 146(5): 1213–1215. (In Russ)].
Mialon P, Gibey R, Bigot JC, Barthelemy L (1992) Changes in striatal and cortical amino acid and ammonia levels of rat brain after one hyperbaric oxygen-induced seizure. Aviat Space Environ Med 63(4): 287–291.
Karlsson A, Fonnum F, Malthe-Sorenssen D, Storm-Mathisen J (1974) Effect of the convulsive agent 3-mercaptopropionic acid on the levels of GABA, other amino acids and glutamate decarboxylase in different regions of the rat brain. Biochem Pharmacol 23(21): 3053–3061. https://doi.org/10.1016/0006-2952(74)90281-0
Li Q, Guo M, Xu X, Xiao X, Xu W, Sun X, Tao H, Li R (2008) Rapid decrease of GAD 67 content before the convulsion induced by hyperbaric oxygen exposure. Neurochem Res 33(1): 185–193. https://doi.org/10.1007/s11064-007-9436-4
Soghomonian JJ, Martin DL (1998) Two isoforms of glutamate decarboxylase: why? Trends Pharmacol Sci 19(12): 500–505. https://doi.org/10.1016/s0165-6147(98)01270-x
Asada H, Kawamura Y, Maruyama K, Kume H, Ding RG, Kanbara N, Kuzume H, Sanbo M, Yagi T, Obata K (1997) Cleft palate and decreased brain gamma-aminobutyric acid in mice lacking the 67-kDa isoform of glutamic acid decarboxylase. Proc Natl Acad Sci U S A 94(12): 6496–6499. https://doi.org/10.1073/pnas.94.12.6496
Kash SF, Johnson RS, Tecott LH, Noebels JL, Mayfield RD, Hanahan D, Baekkeskov S (1997) Epilepsy in mice deficient in the 65-kDa isoform of glutamic acid decarboxylase. Proc Natl Acad Sci U S A 94(25): 14060–14065. https://doi.org/10.1073/pnas.94.25.14060
Tian N, Petersen C, Kash S, Baekkeskov S, Copenhagen D, Nicoll R (1999) The role of the synthetic enzyme GAD65 in the control of neuronal gamma-aminobutyric acid release. Proc Natl Acad Sci U S A 96(22): 12911–12916. https://doi.org/10.1073/pnas.96.22.12911
Davis K, Foos T, Wu JY, Schloss JV (2001) Oxygen-induced seizures and inhibition of human glutamate decarboxylase and porcine cysteine sulfinic acid decarboxylase by oxygen and nitric oxide. J Biomed Sci 8(4): 359–364. https://doi.org/10.1007/BF02258378
Schulman H (2014) Intracellular Signaling. From Molecules to Networks (Third Edition) An Introduction to Cellular and Molecular Neuroscience 119–148.
Stamler JS, Simon DI, Osborne JA, Mullins ME, Jaraki O, Michel T, Singel DJ, Loscalzo J (1992) S-nitrosylation of proteins with nitric oxide: synthesis and characterization of biologically active compounds. Proc Natl Acad Sci U S A 89(1): 444–448. https://doi.org/10.1073/pnas.89.1.444
Gould N, Doulias PT, Tenopoulou M, Raju K, Ischiropoulos H (2013) Regulation of protein function and signaling by reversible cysteine S-nitrosylation. J Biol Chem 288(37): 26473–26479. https://doi.org/10.1074/jbc.R113.460261
Van der Zeyden M, Oldenziel WH, Rea K, Cremers TI, Westerink BH (2008) Microdialysis of GABA and glutamate: analysis, interpretation and comparison with microsensors. Pharmacol Biochem Behav 90(2): 135–147. https://doi.org/10.1016/j.pbb.2007.09.004
Del Arco A, Segovia G, Fuxe K, Mora F (2003) Changes in dialysate concentrations of glutamate and GABA in the brain: an index of volume transmission mediated actions? J Neurochem 85(1): 23–33. https://doi.org/10.1046/j.1471-4159.2003.01692.x
Demchenko IT, Zhilyaev SY, Moskvin AN, Krivchenko AI, Piantadosi CA, Allen BW (2017) Antiepileptic drugs prevent seizures in hyperbaric oxygen: A novel model of epileptiform activity. Brain Res 1657: 347–354. https://doi.org/10.1016/j.brainres.2016.12.032
Tzuk-Shina T, Bitterman N, Harel D (1991) The effect of vigabatrin on central nervous system oxygen toxicity in rats. Eur J Pharmacol 202(2): 171–175. https://doi.org/10.1016/0014-2999(91)90291-w
Hall AA, Young C, Bodo M, Mahon RT (2013) Vigabatrin prevents seizure in swine subjected to hyperbaric hyperoxia. J Appl Physiol (1985)115(6): 861–867. https://doi.org/10.1152/japplphysiol.00221.2013
Gasier HG, Demchenko IT, Allen BW, Piantadosi CA (2015) Effects of striatal nitric oxide production on regional cerebral blood flow and seizure development in rats exposed to extreme hyperoxia. J Appl Physiol (1985)119(11): 1282–1288. https://doi.org/10.1152/japplphysiol.00432.2015
Дополнительные материалы отсутствуют.
Инструменты
Российский физиологический журнал им. И.М. Сеченова