

Российский физиологический журнал им. И.М. Сеченова, 2022, T. 108, № 10, стр. 1243-1263
Механизмы физиологического и нейротоксического действия гипербарического кислорода
И. Т. Демченко 1, С. Ю. Жиляев 1, Т. Ф. Платонова 1, О. С. Алексеева 1, *
1 Институт эволюционной физиологии и биохимии им. И.М. Сеченова Российской академии наук
Санкт-Петербург, Россия
* E-mail: osa72@inbox.ru
Поступила в редакцию 30.06.2022
После доработки 25.08.2022
Принята к публикации 29.08.2022
- EDN: SIQZRM
- DOI: 10.31857/S0869813922100041
Аннотация
За период использования гипербарического кислорода в лечебной медицине и при подводных погружениях накоплены обширные знания о механизмах его биологического действия. В настоящей работе анализируются экспериментальные данные последних лет, относящиеся к клеточным и молекулярным механизмам физиологического и нейротоксического действия гипербарического кислорода. Новые данные о физиологическом действии гипербарического кислорода касаются механизмов гипероксической вазоконстрикции и активации барорефлекса в гипероксии. Токсическое действие гипербарического кислорода реализуется через интенсивную продукцию реактивных форм кислорода и азота, которые вызывают посттрансляционную модификацию белков, отвечающих за электрогенез нейронов и синаптическую передачу в ГАМК-ергической системе головного мозга.
Чистый кислород (~100% O2) под давлением выше атмосферного (гипербарический кислород или ГБО2) является техногенным фактором, который не встречается в природе, а используется для дыхания человека при решении множества медицинских и хозяйственных задач. Широкие потребности в ГБО2 для лечебной медицины и при подводных погружениях предопределили необходимость изучения механизмов его действия на организм человека с целью создания безопасных и эффективных технологий применения. За последние десятилетия накоплены обширные знания о механизмах биологического действии ГБО2, которые условно можно разделить на две части. Первую группу составляют данные о б интегративных физиологических, биохимических и патологических реакциях организма человека и животных на экстремальную гипероксию. Подавляющая часть этих данных касается реакций ЦНС, сердечно-сосудистой и дыхательной систем на гипербарическую гипероксию, а также изменений в системе крови и окислительном метаболизме в гипероксической среде. Патологические реакции на действие ГБО2 касаются в основном токсического действия ГБО2 на ЦНС и легкие. Интегративные физиологические и патологические реакции в ответ на дыхание ГБО2 имеют описательный характер и не раскрывают базисных (молекулярных) механизмов их реализации.
Вторую группу знаний составляют данные о молекулярных механизмах биологического действия ГБО2, которые накапливаются с конца прошлого столетия. Появление этой группы обязано развитию молекулярной биологии, при этом важным моментом является признание ключевой роли свободных радикалов кислорода и азота в реализации механизмов биологического действия кислорода под давлением. В данном кратком обзоре будут рассмотрены современные представления о путях и механизмах реализации физиологических и нейротоксических реакций организма на действие ГБО2, которые обобщены нами в виде гипотетической схемы (рис. 1). Отдельные элементы схемы и взаимодействие между ними будут объяснены по ходу изложения настоящего обзора. Основное внимание будет уделено механизмам вовлечения свободных радикалов кислорода и азота в реализацию физиологических реакций сердечно-сосудистой системы, таких, как гипероксическая вазоконстрикция и гипероксический барорефлекс, которые ранее подробно не анализировались в литературе. Из широкого спектра нейротоксического действия кислорода в рамках настоящего обзора будут проанализированы пути и механизмы действия свободных радикалов на ГАМК-ергическую передачу в мозге, нарушение которой формирует судорожную реакцию ЦНС, известную как “кислородная эпилепсия”.
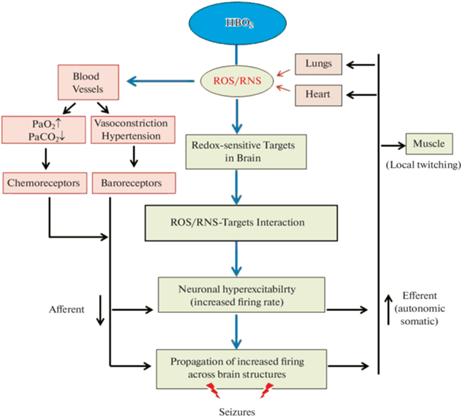
Рис. 1.
Физиологическое и нейротоксическое действие гипербарического кислорода реализуются через генерацию реактивных форм кислорода и азота (ROS/RNS). Экспериментальные данные допускают существование в гипербарической гипероксии по меньшей мере двух путей действия ROS/RNS на клеточные компоненты различных органов и тканей. По одному из них реализуются срочные адаптивные реакции организма на гипероксию, которые запускаются через угнетение хеморецепторов (chemoreceptors) при повышении напряжения кислорода в артериальной крови (РаО2) и активации барорецепторов (baroreceptors) в результате гипероксической вазоконстрикции (vasoconstriction) и острой гипертензии (hypertension). Афферентные (afferent) сигналы от рецепторов интегрируются в ЦНС, где формируются эфферентные (efferent) влияния на легкие и сердце (lungs, heart), которые реализуют адаптивные реакции на гипероксию через симпатические и парасимпатические отделы автономной нервной системы. Другой путь нейротоксического действия ROS/RNS направлен на редокс-чувствительные мишени в головном мозге (redox-sensitive targets in the brain). Взаимодействие ROS/RNS с редокс-чувствительными мишенями приводит к усилению возбудимости нейронов мозга (neuronal hyperexitability), проявляющейся в виде повышенной генерации спайковой активности. Локальные очаги высокой активности нейронов быстро распространяются по мозговым структурам, формируя генерализованную ЭЭГ-активацию, приводящую к судорогам. Подробные данные о генерации ROS/RNS и редокс-чувствительных мишенях в мозге представлены по ходу изложения обзора.
ФИЗИОЛОГИЧЕСКОЕ ДЕЙСТВИЕ ГИПЕРБАРИЧЕСКОГО КИСЛОРОДА
Кислород в составе атмосферного воздуха является, пожалуй, самым важным элементом для поддержания жизни на Земле. Геологические исследования указывают, что в атмосфере нашей планеты концентрация кислорода существенно колебалась и ее величина стабилизировалась на уровне 21% примерно 500 миллионов лет назад. Кислород имеет высокий окислительно-восстановительный потенциал, поэтому является сильным окислителем, который “отрывает” электроны от биологических макромолекул, вызывая внутриклеточное повреждение. Если нет адекватной защиты от кислорода и способов восстановления вызванных им повреждений, возникают токсические эффекты. Токсичность кислорода обусловлена его промежуточными формами, известными как активные формы кислорода (ROS), которые обычно удаляются клеточными антиоксидантными системами. Есть основания полагать, что искусственное повышение парциального давления кислорода в дыхательной среде через ROS активирует также эволюционно закрепленные адаптивные реакции сердечно-сосудистой системы, направленные на уменьшение доставки кислорода к органам и тканям при гипероксии. Такими физиологическими реакциями являются гипероксическая вазоконстрикция и гипероксический барорефлекс, которые направлены на ослабление или предотвращение токсического действия экстремальной гипероксии.
Гипероксическая вазоконстрикция. Как показано на рис. 1, первичными мишенями для гипербарического кислорода являются кровь и сосуды. Увеличение парциального давления кислорода в артериальной крови (РаО2) полностью подавляет генерацию импульсов с хеморецепторов, что приводит к подавлению функции симпатического отдела автономной нервной системы. Данный факт часто используется в экспериментах для выключения кислородных хеморецепторов вместо их химической или хирургической денервации [1]. Повышение РаО2 вызывает также сужение сосудов во всех органах у человека и животных [2–5]. Гипероксическая вазоконстрикция в меньшей степени выражена в коже, умеренно в кишечнике и отчетливо проявляется в сосудах скелетных мышц [6–8], легких [9], печени [10] и сетчатке глаза [11]. На-ибольший вазоконстрикторный эффект гипероксии наблюдается в миокарде [12, 13] и головном мозге [3].
Вазоконстрикторные эффекты зависят от парциального давления кислорода в дыхательной среде и продолжительности гипероксической экспозиции. Пороговой величиной для вазоконстрикции является ~60% кислорода в нормобарической дыхательной среде [14]. При дальнейшем повышении парциального давления кислорода вазоконстрикция усиливается, но ее продолжительность сокращается. Так, при дыхании кислородом под давлением 3 АТА (абсолютных атмосфер) снижение мозгового кровотока в результате вазоконстрикции у крыс наблюдается около 3 ч, при 4 АТА продолжается до 90 мин, а при 5 АТА выявляется только в первые 30 мин гипероксической экспозиции [3].
Хотя о гипероксической вазоконстрикции известно уже более 100 лет [15], молекулярные механизмы реализации этой сосудистой реакции во многом непонятны [14, 16]. Предполагается, что вазоконстрикция реализуется за счет прямого действия кислорода на сосуды путем изменения проницаемости калиевых и кальциевых каналов в мембране гладкомышечных клеток. В частности, коронарная гипероксическая вазоконстрикция опосредуется закрытием АТФ-зависимых К+-каналов [12, 17]. Гипероксия может вызывать вазоконстрикцию также действуя непосредственно на кальциевые каналы L-типа, присутствующие в гладкомышечных клетках сосудов [18]. За все время существования этой гипотезы молекулярный механизм прямого действия кислорода на сократительный аппарат гладкомышечных клеток сосудов не доказан.
Согласно другой гипотезе, гипероксическая вазоконстрикция является результатом опосредованного действия кислорода на один или несколько эндотелиальных факторов, влияющих на сосудистый тонус, таких как эндотелин-1 [19–22] и вазоактивные простагландины [23]. Существование вазоактивных метаболитов в стенке сосудов экспериментально доказано, однако молекулярные механизмы их вазомоторного действия под влиянием ГБО2 не установлены. После открытия оксида азота (NO) и механизма его действия на сосуды высказана идея о возможности его инактивации с помощью супероксиданионов [24–26]. Согласно этой гипотезе, вазоконстрикция является следствием инактивации эндотелиального NO супероксидными анионами с последующей утратой вазодилататорной компоненты базального сосудистого тонуса [27–29]. Как показали исследования на модели, гипероксия вызывает повышенную продукцию в эндотелиальных клетках и окружающей их среде супероксиданионов, которые кардинально меняют биодоступность оксида азота при реализации им вазомоторной функции [30]. Оксид азота продуцируется в сосудистом эндотелии путем окисления L-аргинина с участием фермента – эндотелиальной NO-синтазы (eNOS). Из-зa большой площади эндотелиальной выстилки сосудов на ее долю приходится более 90% образуемого в организме оксида азота. Скорость синтеза NO в сосудистом эндотелии, измеренная in situ или в культуре, равна примерно 0.8 пмоль/мин/мг клеток, что в пересчете на 1.5 кг имеющихся в организме человека эндотелиальных клеток, составляет примерно 1728 мкмоль/сутки [31]. Данный расчет показывает, что в эндотелии образуется NO в тысячи раз больше, чем пороговая величина для реализации расслабления сосуда, равная 0.3 нM [32]. Установлена гетерогенность eNOS по ходу сосудистого русла: в артериолах она наиболее высока, а в венах существенно меньше [33]. Наблюдаемая неоднородность распределения эндотелиальной NO-синтазы, возможно, объясняет сосудистую специфику констрикции при гипероксии. Так, сокращение сосудов наиболее выражено в артериолах диаметром 15–25 мкм, где относительная плотность eNOS и базальный уровень синтеза NO выше, чем в венах или более крупных артериях [34].
NO является мощным вазодилататором, липофильные молекулы которого свободно проникают из места синтеза в эндотелии в гладкомышечные клетки сосудов, где активируют фермент гуанилатциклазу, вызывая образование циклического гуанозинмонофосфата (цГМФ). В свою очередь, цГМФ активирует протеинкиназу G, которая, стимулируя кальциевую АТФ-азу сарко/эндоплазматического ретикулума (SERCA), обеспечивает обратный захват цитозольного кальция в саркоплазматический ретикулум. Также цГМФ стимулирует выход кальция из клетки путем открытия активируемых кальцием калиевых каналов. Таким образом, внутриклеточная концентрация кальция снижается и киназа легких цепей миозина перестает фосфорилировать миозин. Результатом это процесса является расслабление гладкомышечных клеток. Благодаря непрерывному синтезу, свободной диффузии и мощной вазоактивной потенции, NO формирует базальную релаксацию сосудов [25, 27].
Сосудистый тонус определяется множеством различных конкурирующих сосудосуживающих и сосудорасширяющих влияний, действующих на кровеносный сосуд. Эти влияния можно разделить на внешние факторы, возникающие вне органа или ткани, в которой расположен кровеносный сосуд, и внутренние факторы, возникающие в самом сосуде или окружающей ткани. Как правило, внешние (нейрогуморальные) факторы, такие как симпатические нервы и циркулирующий ангиотензин II, повышают тонус (т. е. вызывают сужение) сосудов; однако некоторые циркулирующие факторы (например, предсердный натрийуретический пептид) снижают сосудистый тонус. К внутренним факторам относятся миогенный механизм, эндотелиальные факторы (оксид азота, эндотелин). Они также могут снижать или повышать тонус сосуда. Местные гормоны/химические вещества в стенке сосудов (например, метаболиты арахидоновой кислоты, гистамин и брадикинин) могут повышать или понижать тонус. Метаболические побочные продукты или гипоксия обычно снижают тонус сосудов.
Гипероксия и, в особенности, ГБО2 понижают уровень эндотелиального NО в головном мозге [35, 36]. Наиболее вероятным механизмом снижения эффективной концентрации NO является его инактивация, которая в норме осуществляется посредством химической реакции с молекулярным кислородом, супероксидным анионом, гемоглобином крови, веществами, содержащими металлы переменной валентности, и тканевыми тиолами. В условиях гипероксии концентрация гемоглобина и тканевых тиолов существенно не меняется. Напротив, тканевое рО2 и концентрация супероксид-анионов возрастают пропорционально уровню гипероксигенации и их реакционные взаимодействия с NO становятся наиболее важными. При реакции NO с кислородом (автоокисление) образуются нитраты и, в меньшей степени, нитриты. В условиях нормального атмосферного давления и концентрации кислорода такая реакция протекает сравнительно медленно и ее определяет короткое время биологической жизни NO. Расчеты показывают, что концентрация NO в тканях могла бы быть около 10 мкМ, если бы существовал только один путь деградации NO через автоокисление [37]. В действительности концентрация NO в норме составляет около 10 нМ, а время биологической жизни при физиологической концентрации кислорода в тканях около 50 с [37]. Однако период полураспада NO существенно снижается, когда концентрация кислорода в тканях повышается, по меньшей мере, в 6 раз. Поэтому автоокисление как механизм снижения эффективной концентрации NO не может быть игнорирован при гипербарической гипероксии, так как рО2 в ткани мозга при 3–4 АТА кислорода повышается в 15–20 раз [38]. Скорость реакции оксида азота с супероксиданионом почти в 1000 раз больше автоокисления NO [39]. Эта реакция протекает в 3.5 раза быстрее, чем процесс дисмутации супероксида, поэтому время биологической жизни NO в основном зависит от продукции свободного радикала кислорода.
Поскольку eNOS активна постоянно, непрерывная продукция NO формирует базальный дилататорный компонент сосудистого тонуса в противовес вазоконстрикторным миогенным и нейрогенным влияниям. При этом следует отметить также вклад оксида азота, продуцируемого в нервных клетках и нитроергических окончаниях, иннервирующих сосуды, в формирование сосудистого тонуса [30, 40]. Это подтверждается значительным ослаблением гипероксической вазоконстрикции у мышей-нокаутов по eNOS [41] или подавлением синтеза NO путем фармакологического ингибирования эндотелиальной NO-синтазы [3]. Следует добавить также, что дилататорные свойства NO-доноров утрачиваются в условиях гипероксии [35], а гипероксическая вазоконстрикция блокируется у животных с предварительным введением миметика супероксиддисмутазы – ловушки супероксидных радикалов [42]. Повышение экстраклеточной супероксиддисмутазы (SOD3) в мозговой ткани понижает уровень супероксиданионов, сохраняя эндогенно продуцируемый NO на уровне, достаточном для поддержания тонуса и реактивности мозговых сосудов.
Хотя кислород требуется для синтеза NO и гипероксия стимулирует его продукцию в эндотелии, сужение кровеносных сосудов происходит из-за того, что усиленно генерируемые в их стенке супероксидные анион-радикалы связывают (инактивируют) NO, в результате чего снижается его биодоступность и утрачивается NO-опосредованный базальный вазорелаксирующий компонент сосудистого тонуса [43]. Потенциальными источниками продукции супероксида в сосудах служат НАД(Ф)Н-зависимые оксидазы, ксантиноксидаза, липоксигеназа, митохондриальные оксидазы и различные синтазы [44]. Следовательно, гипероксическая вазоконстрикция является следствием утраты NO-опосредованного вазодилататорного компонента базального сосудистого тонуса путем быстрого связывания свободного радикала NO интенсивно генерируемыми супероксид-анионами с образованием пероксинитрита, не обладающего выраженным вазомоторным действием [39].
При гипероксии ограниченное поступление NO к мышцам сосудов приводит к утрате сосудорасширяющей компоненты базального тонуса, в результате чего сохраняются только вазоконстрикторные влияния. Сосудосуживающая реакция значительно усиливается при гипербарической гипероксии в силу более интенсивной продукции супероксид-анионов и соответственно повышенной нейтрализации оксида азота. Следует подчеркнуть, что синтез NO при гипероксии также повышается, поэтому появление и выраженность гипероксической вазоконстрикции зависит от скорости продукции супероксид-анионов и оксида азота и, следовательно, баланса этих свободных радикалов. Нарушение баланса за счет повышенной продукции супероксид-анионов приводит к вазоконстрикции, а его сдвиг в сторону увеличения концентрации NO вызывает вазодилатацию [43].
В 1997 г. был открыт новый механизм вовлечения NO в гипероксическую вазоконстрикцию [45]. Этот механизм основан на способности эритроцитов регулировать доступность NO гладкомышечным клеткам сосудов в зависимости от градиента концентрации кислорода в крови. После синтеза в эндотелии NO диффундирует как к гладким мышцам сосудов, так и в поток крови, где взаимодействует с гемоглобином эритроцитов через высокоаффинные Fe2+-связывающие участки гема, образуя нитрозилгемоглобин (FeNO-Hb). Реакции NO с гемоглобином происходят преимущественно в деоксигенированной крови, поэтому уровень FeNO-Hb всегда выше в венозной, чем в артериальной крови [46]. При оксигенации крови в легких часть связанного с гемом NO соединяется с остатками цистеина в бета-цепях гемоглобина (Cysβ93), образуя S-нитрозогемоглобин (SNO-Hb) [46, 47]. Некоторая часть SNO-Hb может образовываться также в результате реакций Hb с низкомолекулярным SNO и со свободным NO в насыщенной кислородом крови левого желудочка и крупных артерий.
Оксигенированная (R) и деоксигенированная (T) формы Hb находятся в динамическом равновесии. Обе формы всегда присутствуют в крови, но их количество изменяется в зависимости от концентрации кислорода и аллостерических факторов, влияющих на сродство Hb к кислороду (pH, углекислый газ и 2,3-бисфосфоглицерат). Синтез SNO-гемоглобина благоприятен в R-структуре (кровь с высоким содержанием кислорода) [48]. SNO-Hb образуется при воздействии на гемоглобин NO, нитритов или низкомолекулярных (LMW) SNO в реакциях, которые часто включают в себя нитрозил железа (FeNO-Hb) [48–51]. NO, связанный с гемовым железом в бета-цепях гемоглобина, переходит на тиолы Cysβ93 при оксигенации (образование SNO-Hb обычно сопровождается продукцией небольшого количества metHb, который обеспечивает окислительно-восстановительное взаимодействие гемового железа (III) NO/CysNO). Эти вновь образованные SNO скрыты глубоко внутри оксигенированной молекулы гемоглобина [48]. Их отсоединение от гемовой группы требует переключения гемоглобина в Т-состояние, после чего группы NO перемещаются от Cysβ93 к тиолам, находящимся в глутатионе и/или мембранном белке эритроцитов AE1 [52, 53]. S-нитрозилированный AE1 и SNO-глутатион могут усиливать выделение NO, вызывая вазодилатацию.
Образовавшаяся группа (SNO-окси Hb) в насыщенной кислородом артериальной крови используется для транспорта NO в микроциркуляторное русло. Прочность связывания NO в составе группы (SNO-окси Hb) зависит от насыщения гемоглобина кислородом. Низкий уровень рО2 в крови на уровне артериол способствует высвобождению групп NO [48, 54] и, таким образом, прочность комплекса SNO-Hb обратно пропорциональна насыщению гемоглобина кислородом [53].
Реакции NO и его производных (нитрит и низкомолекулярный SNO), взаимодействующих с Hb, важно рассматривать в физиологическом контексте реализации NO-опосредованной гипероксической вазоконстрикции. Было показано, что при трансфузии донорской крови сосуды суживаются [55]. Позднее установили, что эта реакция является результатом захвата эндотелиального NO железом гема гемоглобина донорской крови, что снижает равновесную концентрацию NO в стенке сосуда ниже порога активации гуанилатциклазы и вызывает сокращения кровеносных сосудов [47, 56]. Данный факт послужил основанием для разработки новой технологии гемотрансфузии, основанной на насыщении донорской крови оксидом азота, что предотвращает вазоконстрикцию и усиливает уровень оксигенации скелетных мышц [57] и улучшает восстановление после субарахноидальной геморрагии [58].
Менее известен тот факт, что эритроциты также могут расширять кровеносные сосуды, тем самым увеличивая кровоток [47, 52, 59]. Вазодилатация в значительной степени осуществляется за счет S-нитрозотиолов (SNO), соединений NO, которые локализованы на остатках цистеина бета-цепей гемоглобина (Hb-Cysβ93-NO) или S-нитрозогемоглобина, (RSNO-Hb). Высвобождению NO из комплекса RSNO-Hb благоприятствует дезоксигенированная форма гемоглобина [48, 53]. Отсюда следует, что связывание и высвобождение групп NO из эритроцитов синхронизировано со связыванием и высвобождением ими кислорода. Сосуды сужаются, когда концентрация О2 в тканях повышается, и расширяются, когда концентрация О2 падает [48, 53, 60–62]. Регулируя биодоступность NO, тканевую перфузию можно согласовать с метаболической потребностью. Гипероксия и особенно ее гипербарическая форма повышает оксигенацию гемоглобина, что способствует большему захвату NO из эндотелия, но снижению выделения NO из эритроцитов [45]. Оба этих фактора снижают доступность NO гладкомышечным клеткам, что нарушает его баланс в сосудистой стенке и является важным элементом механизма вазоконстрикции.
Совокупность представленных данных свидетельствует, что NO-опосредованный механизм гипероксической вазоконстрикции основан на понижении биоактивности NO в стенке сосуда и периваскулярном пространстве. Такому снижению способствуют, по меньшей мере, три фактора: усиленная продукция супероксиданионов, инактивирующих NO, повышенный захват NO железом гема гемоглобина [47, 56] и блокирование выделения группы NO из SNO-Hb при высоком рО2 в артериальной крови, что совместно снижает равновесную концентрацию NO в стенке сосуда ниже порога активации гуанилатциклазы и реализует гипероксическую вазоконстрикцию. Важное значение в регулировании биоактивности NO в стенке сосудов и периваскулярном пространстве имеют также уровень нитратов и нитритов в крови [63].
Гипероксический барорефлекс. Нейрогенная регуляция сердечно-сосудистой системы у человека и животных представлена наиболее известным механизмом – артериальным барорецепторным рефлексом. В нормальных условиях артериальное давление достаточно лабильно и быстро меняется в ответ на разнообразные внутренние и внешние стимулы, такие, как появление вазоактивных веществ в крови во время стресса, физическая нагрузка, ортостатические изменения кровообращения и другие. Во всех этих случаях нейрогенный барорефлекс через контролируемые вегетативной нервной системой хронотропные и инотропные реакции сердца и сосудов возвращает острые изменения давления к начальному или близкому к нему уровню. Нормобарическая или гипербарическая гипероксия никогда не рассматривалась в качестве причинного фактора, запускающего нейрогенный барорефлекторный механизм регуляции артериального давления. Между тем известно, что сердечно-сосудистая система человека и позвоночных животных отвечает на гипероксию вазоконстрикторной реакцией, изменениями артериального давления, замедлением сердечного ритма и уменьшением ударного выброса крови за счет понижения сократимости миокарда левого желудочка сердца. Все эти реакции сердечно-сосудистой системы на гипероксию известны давно, но, как правило, изучались раздельно вне связи друг с другом.
Так, гипероксическая вазоконстрикция у человека и животных [3, 4, 64, 65] реализуется с помощью различных механизмов, подробно изложенных выше. Если говорить коротко, в ее реализации принимают участие эндотелий-производные метаболиты: эндотелин, простагландины и продукты метаболизма арахидоновой кислоты с выраженными вазомоторными свойствами [19, 23]. В вазоконстрикцию при гипероксии вовлекается также NO, доступность которого гладкомышечным клеткам снижается за счет его инактивации усиленно генерируемыми при гипероксии супероксиданионами [27, 42, 66]. Другой NO-опосредованный механизм вазоконстрикции основан на блокировании выделения NO эритроцитами артериальной крови [45, 48].
Давно замечено, что артериальное давление при дыхании нормобарическим или гипербарическим кислородом повышается как у человека [4, 67], так и у позвоночных животных [68], однако механизм развития острой гипертензии остается малопонятным. Брадикардия является самой известной реакцией на гипероксию [69]. Она ослабевает у животных при блокаде мускариновых холинергических рецепторов или ваготомии [67, 70]. Спектральный анализ вариабельности пульсового ритма показал, что кислород приводит к замедлению сердечных сокращений через повышение активности парасимпатической системы [70], однако причина возникновения брадикардической реакции остается неясной. Снижение сердечного выброса при гипероксии многократно демонстрировалось и связывалось с брадикардией, хотя понижение сократимости миокарда (уменьшение ударного объема) также не исключалось [71, 72].
Итак, если вазоконстрикцию представить как первичный ответ на гипероксию, приводящую к острой гипертензии, а брадикардию и снижение сердечного выброса – как ответные реакции, тогда все наблюдаемые сердечно-сосудистые реакции можно объединить в единый барорефлекторный механизм регуляции артериального давления при дыхании кислородом [73–75]. Высказанная гипотеза предполагает, что совокупность сердечно-сосудистых реакций, таких, как вазоконстрикция, острая гипертензия, брадикардия и снижение сердечного выброса является отражением единой нейрорефлекторной реакции – гипероксического барорефлекса. Системная вазоконстрикция при гипероксии неизбежно приводит к резкому подъему артериального давления, что, в свою очередь, стимулирует аортальные и каротидные барорецепторы, импульсация от которых интегрируется в головном мозге, а затем по нисходящим эфферентным симпатическим и парасимпатическим волокнам вызывает брадикардию и снижение сердечного выброса (см. рис. 1).
Гипероксический барорефлекторный механизм запускается в первые секунды ингаляции кислородом, поэтому его можно оценить только с помощью непрерывной регистрации артериального давления и показателей сердечной деятельности [74]. Благодаря барорефлекторной реакции артериальное давление возвращается к исходному уровню при умеренной гипероксии, но в случае высокого ГБО2 гипертензия не компенсируется даже при наличии брадикардии и пониженного сердечного выброса. Барорефлекс в таких случаях “перенастраивается” на более высокий уровень артериального давления, как это наблюдается в случае хронической гипертонии [65]. Такой гипероксический барорефлекс реализуется с участием барорефлексогенных зон аорты и каротидных синусов, исходящих от барорецепторов афферентных нервных волокон и эфферентных симпатических и парасимпатических нервов, регулирующих сосудистый тонус, частоту и силу сердечных сокращений.
Гипероксия снижает симпатическую и повышает парасимпатическую активность вегетативной нервной системы. Триггером сдвига симпато-вагального баланса в гипероксии является повышенная активность барорецепторов и утрата импульсации с периферических хеморецепторов [72]. Установлено также, что усиление афферентной импульсации периферических барорецепторов вызывает в ЦНС стойкое торможение за счет повышения активности ГАМК-ергической системы, на что указывает увеличение уровня ГАМК в головном мозге [65, 73]. Следовательно, релаксирующее действие умеренной гипероксии сходно с эффектами медитации и йоги и, несомненно, дополняет лечебное действие гипербарической оксигенации.
По структурным компонентам гипероксический рефлекторный механизм не отличается от классического барорефлекса, контролирующего быстрые колебания артериального давления. Однако существует несколько особенностей в реализации нейрогенного рефлекса при гипероксии. Во-первых, если в нормальных физиологических условиях триггером барорефлекса является повышение внутрисосудистого давления, вызываемое, в основном, сосудосуживающими симпатическими влияниями [76], то в гипероксии вазоконстрикция реализуется за счет потери (ослабления) NO-опосредованного базального вазодилататорного компонента сосудистого тонуса и не связана с нервными вазомоторными механизмами. Гипероксия вызывает усиленную продукцию в стенке сосудов и периваскулярном пространстве супероксиданионов, которые связывают (инактивируют) эндотелиальный NO, в результате чего утрачивается вазорелаксирующий компонент сосудистого тонуса [27, 42, 66]. Убедительным подтверждением этому является отсутствие гипероксической вазоконстрикции у мышей-нокаутов по эндотелиальной NOS [41].
Во-вторых, если в нормальных условиях исполнительным компонентом барорефлекса является уменьшение периферического сосудистого сопротивления из-за ослабления вазомоторного симпатического тонуса, то в гипероксии механизм снижения артериального давления базируется на вагус-опосредованной брадикардии и снижении сердечного выброса [73].
В-третьих, барорефлекторный механизм в гипероксии понижает возросшее артериальное давление, но полностью не возвращает его к исходному уровню. По всей вероятности, барорефлекс в гипероксии может адаптироваться или “перезапускаться”, как это происходит в условиях хронической гипертензии или после острого гипертонического криза [77]. Такая “перезагрузка” обеспечивает долговременный контроль артериального давления путем изменения афферентных, центральных или эфферентных компонентов артериального барорефлекса [76].
Эффективность или чувствительность барорефлекса, оцениваемая методом, предложенным Stauss [78], при повышенном давлении кислорода 1 и 3 АТА достоверно увеличивается по отношению к аналогичным показателям, полученным на тех же животных при дыхании атмосферным воздухом [74]. При 5 АТА наблюдается 2-фазное изменение чувствительности барорефлекса и соответствующих ему сердечно-сосудистых реакций. В первые 30–40 мин кислородной экспозиции эффективность барорефлекторной регуляции возрастает и отмечается умеренная брадикардия и снижение активности симпатической нервной системы. Продолжение гипербарической оксигенации приводит к развитию второй фазы, характеризующейся острым повышением артериального давления, переключением брадикардии на тахикардию и резким увеличением симпатической активности. В этот период отмечаются положительные хронотропный и инотропный эффекты деятельности сердца, а также значительное увеличение содержания в плазме крови адреналина и норадреналина [74]. Чувствительность барорефлекса сначала снижается, а затем полностью утрачивается на фоне появления кислородных судорог. Следовательно, устойчивость барорефлекса зависит от уровня и продолжительности гипероксического воздействия. При дыхании кислородом под давлением до 3 АТА чувствительность барорефлекса возрастает, что позволяет более эффективно реализовать адаптивные гемодинамические реакции, ограничивающие доставку токсической дозы кислорода в организм. Это выражается в прогрессирующей брадикардии и уменьшении сердечного выброса, связанных со снижением симпатических и увеличением парасимпатических влияний на сердце. Существенный вклад в краткосрочную адаптацию к гипероксии вносит также церебральная вазоконстрикция, благодаря которой уменьшается доставка кислорода в головной мозг и замедляется скорость генерации активных форм кислорода и азота ROS/RNS.
В норме барорецепторный рефлекс регулирует артериальное давление в пределах нормальных значений через петлю отрицательной обратной связи. Изменения артериального давления контролируются рецепторами растяжения, расположенными в каротидном синусе и дуге аорты. Одиночное ядро солитарного тракта, расположенное в сердечно-сосудистом центре продолговатого мозга, получает импульсы от этих рецепторов через афференты языкоглоточного и блуждающего нервов. Сердечно-сосудистый центр мозга состоит из двух функционально различных областей: область, ответственная за повышение артериального давления, расположена латерально и рострально, тогда как область, ответственная за снижение артериального давления, расположена в центре и каудально. Последняя область также интегрирует импульсы от гипоталамуса и лимбической систем, направленные на снижение симпатической активности, что приводит к уменьшению сократительной способности сердца, частоты сердечных сокращений и тонуса сосудов [79]. Кроме того, активация парасимпатической системы еще больше снижает частоту сердечных сокращений и сократительную способность миокарда.
Центральные механизмы реализации гипероксического барорефлекса систематически не изучались, но существуют отдельные данные о причастности центральной адренергичекой [80] и ГАМК-ергической систем мозга к барорефлекторной регуляции гемодинамики в гипероксии. Как показали опыты, активация ГАМК-ергической системы мозга путем блокады ГАМК-транспортеров в синапсах, усиливает эффективность барорефлекторной регуляции при гипероксии через симпатические и парасимпатические пути автономной нервной системы [81]. ГАМК-ергическая система не исчерпывает всех медиаторных систем мозга, вовлекающихся в реализацию гипероксического барорефлекса, и она не является специфической для реализации только гипероксического барорефлекса. Барорефлекс в гипероксии отличается лишь модальностью стимула, повышающего артериальное давление. Все исполнительные механизмы рефлекса такие же, как и при других способах активации барорефлекторной регуляции артериального давления. Вместе с тем ГБО2, который вызывает вазоконстрикцию, являющуюся триггером рефлекса, вносит определенные особенности в его реализацию. В частности, установлены фазные изменения эффективности гипероксического барорефлекса, зависящие от уровня и продолжительности гипероксического воздействия. При дыхании кислородом под давлением 3 АТА продолжительностью до 3 ч чувствительность барорефлекса сохраняется на более высоком уровне, чем до гипероксической экспозиции, что позволяет более эффективно реализовать адаптивные гемодинамические реакции, ограничивающие доставку токсической дозы кислорода в организм [74]. Эти реакции в виде прогрессирующей брадикардии и уменьшения ударного объема крови обеспечиваются понижением симпатических и увеличением парасимпатических влияний на сосуды и сердечную деятельность. Следовательно, нейрогенно-управляемое снижение сердечного выброса совместно с NO-опосредованной вазоконстрикцией ограничивают доставку кислорода в головной мозг, уменьшают скорость развития окислительного стресса в мозге и отодвигают нейротоксический эффект ГБО2. Более высокий уровень гипероксии при 5 АТА подавляет гипероксический барорефлекс и существенно сокращает время развития кислородных судорог [74, 82]. Пока не совсем ясно, какие звенья рефлекторной дуги вовлечены в утрату рефлекса при развившихся кислородных судорогах, равно как неизвестны и молекулярные редокс-опосредованные механизмы, ответственные за потерю его чувствительности.
НЕЙРОТОКСИЧЕСКОЕ ДЕЙСТВИЕ ГИПЕРБАРИЧЕСКОГО КИСЛОРОДА
Лимитирующим фактором широкого применения ГБО2 является его токсическое действие на ЦНС, которое в клинике проявляется в виде эпилептиформной активности на электроэнцефалограмме и тонико-клонических нервно-мышечных судорог [83–86]. Ключевая роль в нейротоксическом действии ГБО2 принадлежит активным формам кислорода (ROS) и азота (RNS), которые в силу своей высокой биологической активности способны поражать различные компоненты клеток мозга, нарушать функционирование нейрональных сетей и инициировать судороги [86, 87].
ROS представляют собой короткоживущие и высоко реакционноспособные молекулы. Генерация ROS в клетках мозга находится в равновесии с имеющейся в организме системой антиоксидантной защиты. Считается, что в низких и умеренных дозах ROS необходимы для регуляции физиологических функций и процессов, таких как клеточный цикл, пролиферация, дифференцировка, миграция и гибель клеток. ROS также играют важную роль в иммунной системе, поддержании окислительно-восстановительного баланса, участвуют в активации различных клеточных сигнальных путей. Избыточный уровень внутриклеточных ROS вызывает повреждение белков, нуклеиновых кислот, липидов, мембран и органелл, что может привести к активации процессов гибели клеток, таких как апоптоз и некроз [88, 89].
Существует две группы экспериментальных доказательств причастности ROS/RNS к развитию кислородных судорог. Во-первых, показано повышенное образование в мозге компонентов ROS – супероксид-анионов (${\text{O}}_{2}^{ - }$), гидроксильных радикалов (ОН–) и перекиси водорода (Н2О2), причем скорость их продукции пропорциональна парциальному давлению вдыхаемого кислорода [85, 90–92]. ГБО2 также стимулирует в мозге синтез оксида азота [36, 93], который является компонентом пула RNS, а происходящая с высокой скоростью химическая реакция между ${\text{O}}_{2}^{ - }$ и NO приводит к образованию пероксинитрита (ONOO–), мощного эндогенного оксиданта [39]. Накопление в мозге NO и 3-нитротирозина, маркера ONOO–, показано у крыс после кислородной экспозиции [94] или непосредственно в кислородной барокамере [93]. Как показали опыты на нокаутных мышах, основным источником NO-опосредованного образования ONOO– при гипероксии является нейрональная NO-синтаза (nNOS) [41, 95]. Скорость образовании ${\text{O}}_{2}^{ - }$ и NO в мозге различна, поэтому нарушение их баланса является критическим фактором для развития судорожной активности в ГБО2. Трансгенные мыши с повышенной экспрессией экстраклеточной супероксиддисмутазы (SOD3) показали более высокую восприимчивость к кислородным судорогам, чем обычные мыши, что демонстрирует ключевую роль баланса ${\text{O}}_{2}^{ - }$/NO в нейротоксичности экстремальной гипероксии [96].
Вторая группа фактов свидетельствует о тесной связи между накоплением ROS/RNS и судорожной реакцией ЦНС в ГБО2. Так, появлению пароксизмальной активности на ЭЭГ предшествует повышение продукции NO и ONOO, а также NO-опосредованного усиления мозгового кровотока [93]. Экспериментальные воздействия, лимитирующие продукцию или утилизацию ROS/RNS, предохраняют от развития кислородных судорог. Так, подавление биосинтеза NO путем системного ингибирования NOS с помощью L-NAME предотвращает появление кислородных судорог [97].
Как показано на рис. 1, участие ROS/RNS в инициировании судорог предполагает наличие в мозге редокс-чувствительных мишеней, воздействие на которые приводит к нарушению электрогенеза нейронов и синаптической передачи. Наиболее вероятная мишень для инициирования кислородных судорог найдена в ГАМК-ергической нейропередаче. Установлено, что ГБО2 ингибирует синтез ГАМК в мозге, что приводит к ослаблению тормозной нейропередачи с последующим сдвигом баланса процессов возбуждения и торможения в ЦНС [85, 98]. Доказательством этому является снижение ГАМК в межклеточной среде мозга крыс, зарегистрированное in vivo во время ГБО2-экспозиции с помощью микродиализа, сопряженного с высокоэффективной жидкостной хроматографией [98]. Установлено, что причиной уменьшения внеклеточной ГАМК в ГБО2 является понижение активности глутаматдекарбоксилазы (GAD), катализирующей синтез тормозного медиатора в нервных клетках [99]. Если ROS/RNS угнетают синтез ГАМК в ГБО2 путем инактивации GAD, возникает вопрос о молекулярном механизме повреждения. Короткий латентный период появления кислородных судорог при высоких значениях ГБО2 позволяет исключить из рассмотрения синтез биомолекул de novo через генную экспрессию и оставить только две возможные реакции взаимодействия: окисление липидов и посттрансляционную модификацию белков. Атака ROS на жирные кислоты мембраны нервных клеток может заканчиваться полимеризацией последних, приводящей к нарушению ионных механизмов генерации вызванных потенциалов нейронов. Белки, представленные в ферментах, рецепторах, ионных каналах и транспортерах нейромедиаторов, содержат в своем составе аминокислоты, концевые остатки которых легко поддаются окислению или нитрозилированию при действии ROS/RNS. Особое внимание привлекает реакция белков, содержащих цистеин, с оксидом азота (S-nitrosilation). Показано, что угнетение ферментативной активности GAD в ГБО2 происходит за счет ее S-нитрозилирования – присоединения группы оксида азота к тиоловым остаткам цистеина внутри белка-фермента [99]. Снижение ферментативной активности в ГБО2 при 5 АТА наблюдается также у глутаминсинтетазы, катализирующей катаболизм глутамата [100]. Однако ГАМК-трансаминаза, участвующая в деградации тормозного нейромедиатора, и ГАМК-транспортеры, обеспечивающие удаление тормозного нейромедиатора из синаптической щели, не теряют своей активности в гипербарической кислородной среде [81, 99]. К указанным ферментам – мишеням для ROS/RNS следует добавить также мембранную Nа/K-ATФазу, обеспечивающую активный трансмембранный транспорт ионов в нервных клетках при генерации электрических потенциалов.
Кислородные судороги, возникающие из-за угнетения синтеза ГАМК, могут быть предотвращены путем увеличения уровня медиатора в синаптическом пространстве с помощью фармакологических препаратов, таких как вигабатрин [101]. Недавние исследования показали, что ингибиторы ГАМК-трансаминазы и ГАМК-транспортеров, вигабатрин и тиагабин соответственно, повышают концентрацию ГАМК-медиатора в головном мозге и предотвращают развитие кислородных судорог [81, 102]. Следовательно, при подавлении синтеза ГАМК в ГБО2 замедление клиренса медиатора из синапсов или ослабление его разрушения могут повысить концентрацию тормозного медиатора до уровня, достаточного для реализации полноценной ГАМК-ергической функции и, тем самым, предотвратить развитие судорожного синдрома. Поэтому вполне оправдано использование противоэпилептических препаратов для предотвращения развития гипербарических кислородных судорог при экстремальной гипероксии [103].
ЗАКЛЮЧЕНИЕ
При нормальных условиях в здоровом организме продукция и клиренс ROS хорошо сбалансированы. Дыхание гипербарическим кислородом смещает этот баланс в сторону увеличения количества ROS, которое выходит за пределы возможности системы антиоксидантной защиты и способствует развитию окислительного стресса. Однако не всякий оксидативный стресс вреден для функционирования физиологических систем. Увеличение активности молекулярных процессов может возникать при усилении окислительного стресса, хотя невозможно точно охарактеризовать его параметры. Примером сказанному может служить усиление биогенеза митохондрий в головном мозге крыс после их экспозиции в экстремальной гипероксической среде [104, 105]. Другим примером является усиление регуляции функций ионных каналов при повышенной продукции ROS [106]. Выявлено существенное подавление нейронального апоптоза после сеансов гипербарической оксигенации [107, 108]. Известно также, что во время физических упражнений, индуцирующих выработку ROS, происходит активация специфических антиоксидантных ферментов, таких как SOD, и усиление их влияния на функцию печени [109], наблюдается также и улучшение функций верхних конечностей после экспозиции в ГБО2 [110]. В последние годы неоднократно продемонстрировано существенное улучшение состояния пациентов с ишемическими поражениями мозга после нескольких сеансов гипербарической оксигенации [111–113].
Идея о том, что воздействие небольшой дозы опасного вещества или фактора может вызвать благоприятную биологическую реакцию давно известна, получила название “гормезис” и является характеристикой многих биологических процессов, а именно двухфазной реакцией на воздействие возрастающих концентраций вещества или фактора среды [114]. В пределах гормезисной зоны биологическая реакция на низкое воздействие стрессора, как правило, благоприятна, а в большей дозе наблюдается токсический эффект.
ГБО2 относится к терапевтическим веществам и, как многие лекарственные препараты, может оказывать физиологическое и токсическое действие. Биологические эффекты зависят от скорости продукции и продолжительности влияния ROS/RNS, определяемых величиной парциального давления вдыхаемого кислорода [115]. Основным терапевтическим эффектом ГБО2 является устранение дефицита кислородного снабжения тканей для обеспечения полноценного уровня окислительного метаболизма. Дополнительно к этому, умеренная гипероксия вызывает физиологические реакции, усиливающие терапевтическое действие кислорода. Кроме того, ROS и RNS участвуют в каскадах и путях активации различных факторов роста, мобилизации стволовых клеток [116], удлинении теломер хромосом [117] и других молекулярных процессах, определяющих лечебный эффект ГБО2. Высокий уровень продукции ROS/RNS токсичен для клеток, особенно для нейронов мозга. Клеточные мишени для действия ROS/RNS локализуются в местах продукции этих редокс-молекул, а биохимическое взаимодействие приводит к посттрансляционным изменениям структуры белков, приводящих к нарушению или полной утрате их функции. Нарушения нейропередачи, приводящие к развитию кислородных судорог, становятся понятнее при использовании фармакологических средств с известным механизмом действия. К ним относятся противоэпилептические препараты, допущенные и широко используемые в лечении заболеваний, в патогенезе которых присутствует повышенная судорожная готовность. Их тестирование на животных подтвердило высокую эффективность для предотвращения гипербарических кислородных судорог [118], что открывает перспективу трансляции результатов в практику использования ГБО2 в медицине и при подводных погружениях.
Список литературы
Fitzgerald RS, Rocher A (2021) Physiology and Pathophysiology of Oxygen Sensitivity. Antioxidants 10: 1114. https://doi.org/10.3390/antiox10071114
Brown MM, Wade JP, Marshall J (1985) Fundamental importance of arterial oxygen content in the regulation of cerebral blood flow in man. Brain 108 (Pt1): 81–93. https://doi.org/10.1093/brain/108.1.81
Demchenko IT, Boso AE, O’Neill TJ, Bennett PB, Piantadosi CA (2000) Nitric oxide and cerebral blood flow responses to hyperbaric oxygen. J Appl Physiol (1985) 88(4): 1381–1389. https://doi.org/10.1152/jappl.2000.88.4.1381
Molénat F, Boussuges A, Grandfond A, Rostain JC, Sainty JM, Robinet C, Galland F, Meliet JL (2004) Haemodynamic effects of hyperbaric hyperoxia in healthy volunteers: an echocardiographic and Doppler study. Clin Sci (Lond) 106(4): 389–395. https://doi.org/10.1042/CS20030293
Smit B, Smulders YM, van der Wouden JC, Oudemans-van Straaten HM, Spoelstra-de Man AME (2018) Hemodynamic effects of acute hyperoxia: systematic review and meta-analysis. Crit Care 22(1): 45. https://doi.org/10.1186/s13054-018-1968-2
Pries AR, Heide J, Ley K, Klotz KF, Gaehtgens P (1995) Effect of oxygen tension on regulation of arteriolar diameter in skeletal muscle in situ. Microvasc Res 49(3): 289–299. https://doi.org/10.1006/mvre.1995.1025
Smith KM, Moore LC, Layton HE (2003) Advective transport of nitric oxide in a mathematical model of the afferent arteriole. Am J Physiol Renal Physiol 284(5): F1080–F1096. https://doi.org/10.1152/ajprenal.00141.2002
Milstein DM, Helmers R, Hackmann S, Belterman CN, van Hulst RA, de Lange J (2016) Sublingual microvascular perfusion is altered during normobaric and hyperbaric hyperoxia. Microvasc Res 105: 93–102. https://doi.org/10.1016/j.mvr.2016.02.001
Ariyaratnam P, Loubani M, Bennett R, Griffin S, Chaudhry MA, Cowen ME, Guvendik L, Cale AR, Morice AH (2013) Hyperoxic vasoconstriction of human pulmonary arteries: a novel insight into acute ventricular septal defects. ISRN Cardiol 2013: 685735. https://doi.org/10.1155/2013/685735
Eshmuminov D, Becker D, Hefti ML, Mueller M, Hagedorn C, Dutkowski P, Rudolf von Rohr P, Halbe M, Segerer S, Tibbitt MW, Bautista Borrego L, Schuler MJ, Clavien PA (2020) Hyperoxia in portal vein causes enhanced vasoconstriction in arterial vascular bed. Sci Rep 10(1): 20966. https://doi.org/10.1038/s41598-020-77915-0
Mishra A, Hamid A, Newman EA (2011) Oxygen modulation of neurovascular coupling in the retina. Proc Natl Acad Sci U S A 108(43): 17827–17831. https://doi.org/10.1073/pnas.1110533108
Mouren S, Souktani R, Beaussier M, Abdenour L, Arthaud M, Duvelleroy M, Vicaut E (1997) Mechanisms of coronary vasoconstriction induced by high arterial oxygen tension. Am J Physiol 272(1Pt2): H67–H75. https://doi.org/10.1152/ajpheart.1997.272.1.H67
Farquhar H, Weatherall M, Wijesinghe M, Perrin K, Ranchord A, Simmonds M, Beasley R (2009) Systematic review of studies of the effect of hyperoxia on coronary blood flow. Am Heart J 158(3): 371–377. https://doi.org/10.1016/j.ahj.2009.05.037
Wolin MS, Ahmad M, Gupte SA (2005) Oxidant and redox signaling in vascular oxygen sensing mechanisms: basic concepts, current controversies, and potential importance of cytosolic NADPH. Am J Physiol Lung Cell Mol Physiol 289(2): L159–L173. https://doi.org/10.1152/ajplung.00060.2005
Benedict FG, Higgins HL (1911) Effects on men at rest of breathing oxygen-rich gas mixtures. Am J Physiol 28: 1–28.
Jackson WF (2016) Arteriolar oxygen reactivity: where is the sensor and what is the mechanism of action? J Physiol 594(18): 5055–5077. https://doi.org/10.1113/JP270192
Ngo AT, Riemann M, Holstein-Rathlou NH, Torp-Pedersen C, Jensen LJ (2013) Significance of K(ATP) channels, L-type Ca2+ channels and CYP450-4A enzymes in oxygen sensing in mouse cremaster muscle arterioles in vivo. BMC Physiol 13: 1–11. https://doi.org/10.1186/1472-6793-13-8
Welsh DG, Jackson WF, Segal SS (1998) Oxygen induces electromechanical coupling in arteriolar smooth muscle cells: a role for L-type Ca2+ channels. Am J Physiol 274(6): H2018–H2024. https://doi.org/10.1152/ajpheart.1998.274.6.H2018
Dallinger S, Dorner GT, Wenzel R, Graselli U, Findl O, Eichler HG, Wolzt M, Schmetterer L (2000) Endothelin-1 contributes to hyperoxia-induced vasoconstriction in the human retina. Invest Ophthalmol Vis Sci 41(3): 864–869.
Bourque SL, Davidge ST, Adams MA (2011) The interaction between endothelin-1 and nitric oxide in the vasculature: new perspectives. Am J Physiol Regul Integr Comp Physiol 300(6): R1288–R1295. https://doi.org/10.1152/ajpregu.00397.2010
Stow LR, Jacobs ME, Wingo CS, Cain BD (2011) Endothelin-1 gene regulation. FASEB J 25(1): 16–28. https://doi.org/10.1096/fj.10-161612
Nishiyama SK, Zhao J, Wray DW, Richardson RS (2017) Vascular function and endothelin-1: tipping the balance between vasodilation and vasoconstriction. J Appl Physiol (1985) 122(2): 354–360. https://doi.org/10.1152/japplphysiol.00772.2016
Rousseau A, Tesselaar E, Henricson J, Sjöberg F (2010) Prostaglandins and radical oxygen species are involved in microvascular effects of hyperoxia. J Vasc Res 47(5): 441–450. https://doi.org/10.1159/000282667
Ignarro LJ, Buga GM, Wood KS, Byrns RE, Chaudhuri G (1987) Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc Natl Acad Sci U S A 84(24): 9265–9269. https://doi.org/10.1073/pnas.84.24.9265
Iadecola C, Pelligrino DA, Moskowitz MA, Lassen NA (1994) Nitric oxide synthase inhibition and cerebrovascular regulation. J Cereb Blood Flow Metab 14(2): 175–192. https://doi.org/10.1038/jcbfm.1994.25
Czapski G, Goldstein S (1995) The role of the reactions of NO with superoxide and oxygen in biological systems: a kinetic approach. Free Radic Biol Med 19(6): 785–794. https://doi.org/10.1016/0891-5849(95)00081-8
Rubanyi GM, Vanhoutte PM (1986) Superoxide anions and hyperoxia inactivate endothelium-derived relaxing factor. Am J Physiol 250(5Pt2): H822–H827. https://doi.org/10.1152/ajpheart.1986.250.5.H822
Katusic ZS (1996) Superoxide anion and endothelial regulation of arterial tone. Free Radic Biol Med 20(3): 443–448. https://doi.org/10.1016/0891-5849(96)02116-8
Zhilyaev SYu, Moskvin AN, Platonova TF, Gutsaeva DR, Churilina IV, Demchenko IT (2003) Hyperoxic vasoconstriction in the brain is mediated by inactivation of nitric oxide by superoxide anions. Neurosci Behav Physiol 33(8): 783–787. https://doi.org/10.1023/a:1025145331149
Haselden WD, Kedarasetti RT, Drew PJ (2020) Spatial and temporal patterns of nitric oxide diffusion and degradation drive emergent cerebrovascular dynamics. PLoS Comput Biol 16(7): e1008069. https://doi.org/. pcbi.1008069https://doi.org/10.1371/journal
Kelm M (1999) Nitric oxide metabolism and breakdown. Biochim Biophys Acta 1411(2–3): 273–289. https://doi.org/10.1016/s0005-2728(99)00020-1
Yan O, Liu Q, Zweier JL, Liu X (2007) Potency of authentic nitric oxide in inducing aortic relaxation. Pharmacol Res 55(4): 329–334. https://doi.org/10.1016/j.phrs.2007.01.001
Heaps CI, Bray JF, McIntosh AI, Schroeder F (2019) Endothelial nitric oxide synthase protein distribution and nitric oxide production in endothelial cells along the coronary vascular tree. Microvasc Res 122: 34–40. https://doi.org/10.1016/j.mvr.2018.11.004
Andries LJ, Brutsaert DL, Sys SU (1998) Nonuniformity of endothelial constitutive nitric oxide synthase distribution in cardiac endothelium. Circ Res 82(2): 195–203. https://doi.org/10.1161/01.res.82.2.195
Demchenko IT, Boso AE, Bennett PB, Whorton AR, Piantadosi CA (2000) Hyperbaric oxygen reduces cerebral blood flow by inactivating nitric oxide. Nitric Oxide 4(6): 597–608. https://doi.org/10.1006/niox.2000.0313
Thom SR, Bhopale V, Fisher D, Manevich Y, Huang PL, Buerk DG (2002) Stimulation of nitric oxide synthase in cerebral cortex due to elevated partial pressures of oxygen: an oxidative stress response. J Neurobiol 51(2): 85–100. https://doi.org/10.1002/neu.10044
Lancaster JR Jr (1994) Simulation of the diffusion and reaction of endogenously produced nitric oxide. Proc Natl Acad Sci U S A 91: 8137–8141. https://doi.org/10.1073/pnas.91.17.8137
Demchenko IT, Luchakov YI, Moskvin AN, Gutsaeva DR, Allen BW, Thalmann ED, Piantadosi CA (2005) Cerebral blood flow and brain oxygenation in rats breathing oxygen under pressure. J Cereb Blood Flow Metab 25(10): 1288–1300. https://doi.org/10.1038/sj.jcbfm.9600110
Beckman JS, Koppenol WH (1996) Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am J Physiol 271(5Pt1): C1424–C1437. https://doi.org/10.1152/ajpcell.1996.271.5.C1424
Zhao Y, Vanhoutte PM, Leung SW (2015) Vascular nitric oxide: Beyond eNOS. J Pharmacol Sci 129(2): 83–94. https://doi.org/10.1016/j.jphs.2015.09.002
Atochin DN, Demchenko IT, Astern J, Boso AE, Piantadosi CA, Huang PL (2003) Contributions of endothelial and neuronal nitric oxide synthases to cerebrovascular responses to hyperoxia. J Cereb Blood Flow Metab 23(10): 1219–1226. https://doi.org/10.1097/01.WCB.0000089601.87125.E4
Demchenko IT, Oury TD, Crapo JD, Piantadosi CA (2002) Regulation of the brain’s vascular responses to oxygen. Circ Res 91(11): 1031–1037. https://doi.org/10.1161/01.res.0000043500.03647.81
Gryglewski RJ, Palmer RM, Moncada S (1986) Superoxide anion is involved in the breakdown of endothelium-derived vascular relaxing factor. Nature 320(6061): 454–456. https://doi.org/10.1038/320454a0
Channon KM, Guzik TJ (2002) Mechanisms of superoxide production in human blood vessels: relationship to endothelial dysfunction, clinical and genetic risk factors. J Physiol Pharmacol 53(4Pt1): 515–524.
Stamler JS, Jia L, Eu JP, McMahon TJ, Demchenko IT, Bonaventura J, Gernert K, Piantadosi CA (1997) Blood flow regulation by S-nitrosohemoglobin in the physiological oxygen gradient. Science 276(5321): 2034–2037. https://doi.org/10.1126/science.276.5321.2034
McMahon TJ, Moon RE, Luschinger BP, Carraway MS, Stone AE, Stolp BW, Gow AJ, Pawloski JR, Watke P, Singel DJ, Piantadosi CA, Stamler JS (2002) Nitric oxide in the human respiratory cycle. Nat Med 8(7): 711–717. https://doi.org/10.1038/nm718
Jia L, Bonaventura C, Bonaventura J, Stamler JS (1996) S-nitrosohaemoglobin: a dynamic activity of blood involved in vascular control. Nature 380(6571): 221–226. https://doi.org/10.1038/380221a0
Singel DJ, Stamler JS (2005) Chemical physiology of blood flow regulation by red blood cells: the role of nitric oxide and S-nitrosohemoglobin. Annu Rev Physiol 67: 99–145. https://doi.org/10.1146/annurev.physiol.67.060603.090918
Luchsinger BP, Rich EN, Gow AJ, Williams EM, Stamler JS, Singel DJ (2003) Routes to S-nitroso-hemoglobin formation with heme redox and preferential reactivity in the beta subunits. Proc Natl Acad Sci U S A 100(2): 461–466. https://doi.org/10.1073/pnas.0233287100
Angelo M, Singel DJ, Stamler JS (2006) An S-nitrosothiol (SNO) synthase function of hemoglobin that utilizes nitrite as a substrate. Proc Natl Acad Sci U S A 103(22): 8366–8371. https://doi.org/10.1073/pnas.0600942103
Nagababu E, Ramasamy S, Rifkind JM (2006) S-nitrosohemoglobin: a mechanism for its formation in conjunction with nitrite reduction by deoxyhemoglobin. Nitric Oxide 15(1): 20–29. https://doi.org/10.1016/j.niox.2006.01.012
Pawloski JR, Hess DT, Stamler JS (2001) Export by red blood cells of nitric oxide bioactivity. Nature 409(6820): 622–626. https://doi.org/10.1038/35054560
Doctor A, Platt R, Sheram ML, Eischeid A, McMahon T, Maxey T, Doherty J, Axelrod M, Kline J, Gurka M, Gow A, Gaston B (2005) Hemoglobin conformation couples erythrocyte S-nitrosothiol content to O2 gradients. Proc Natl Acad Sci U S A 102(16): 5709–5714. https://doi.org/10.1073/pnas.0407490102
Pezacki JP, Ship NJ, Kluger R (2001) Release of nitric oxide from S-nitrosohemoglobin. Electron transfer as a response to deoxygenation. J Am Chem Soc 123(19): 4615–4616. https://doi.org/10.1021/ja015716o
Keipert PE, Gonzales A, Gomez CL, MacDonald VW, Hess JR, Winslow RM (1993) Acute changes in systemic blood pressure and urine output of conscious rats following exchange transfusion with diaspirin-crosslinked hemoglobin solution. Transfusion 33(9): 701–708. https://doi.org/10.1046/j.1537-2995.1993.33994025016.x
Tsai AG, Cabrales P, Manjula BN, Acharya SA, Winslow RM, Intaglietta M (2006) Dissociation of local nitric oxide concentration and vasoconstriction in the presence of cell-free hemoglobin oxygen carriers. Blood 108(10): 3603–3610. https://doi.org/10.1182/blood-2006-02-005272
Reynolds JD, Bennett KM, Cina AJ, Diesen DL, Henderson MB, Matto F, Plante A, Williamson RA, Zandinejad K, Demchenko IT, Hess DT, Piantadosi CA, Stamler JS (2013) S-nitrosylation therapy to improve oxygen delivery of banked blood. Proc Natl Acad Sci U S A 110(28): 11529–11534. https://doi.org/10.1073/pnas.1306489110
Sheng H, Reynolds JD, Auten RL, Demchenko IT, Piantadosi CA, Stamler JS, Warner DS (2011) Pharmacologically augmented S-nitrosylated hemoglobin improves recovery from murine subarachnoid hemorrhage. Stroke 42(2): 471–476. https://doi.org/10.1161/STROKEAHA.110.600569
Helms CC, Gladwin MT, Kim-Shapiro DB (2018) Erythrocytes and Vascular Function: Oxygen and Nitric Oxide. Front Physiol 9: 125–128. https://doi.org/0.3389/fphys.2018.00125
Roach RC, Koskolou MD, Calbet JA, Saltin B (1999) Arterial O2 content and tension in regulation of cardiac output and leg blood flow during exercise in humans. Am J Physiol 276(2): H438–H445. https://doi.org/10.1152/ajpheart.1999.276.2.H438
Gonzalez-Alonso J, Richardson RS, Saltin B (2001) Exercising skeletal muscle blood flow in humans responds to reduction in arterial oxyhaemoglobin, but not to altered free oxygen. J Physiol 530(Pt2): 331–341. https://doi.org/10.1111/j.1469-7793.2001.0331l.x
González-Alonso J, Mortensen SP, Dawson EA, Secher NH, Damsgaard R (2006) Erythrocytes and the regulation of human skeletal muscle blood flow and oxygen delivery: role of erythrocyte count and oxygenation state of haemoglobin. J Physiol 572(Pt1): 295–305. https://doi.org/10.1113/jphysiol.2005.101121
DeMartino AW. Kim-Shapiro DB, Patel RP, Gladwin MT (2019) Nitrite and nitrate: cheмical biology and signalling. Br J Pharmacol 176: 228–245. https://doi.org/10.1111/bph.14484
Floyd TF, Clark JM, Gelfand R, Detre JA, Ratcliffe S, Guvakov D, Lambertsen CJ, Eckenhoff RG (2003) Independent cerebral vasoconstrictive effects of hyperoxia and accompanying arterial hypocapnia at 1 ATA. J Appl Physiol (1985) 95(6): 2453–2461. https://doi.org/10.1152/japplphysiol.00303.2003
Bulte DP, Chiarelli PA, Wise RG, Jezzard P (2007) Cerebral perfusion response to hyperoxia. J Cereb Blood Flow Metab 27(1): 69–75 https://doi.org/10.1038/sj.jcbfm.9600319
Pasgaard T, Stankevicius E, Jørgensen MM, Ostergaard L, Simonsen U, Frøbert O (2007) Hyperoxia reduces basal release of nitric oxide and contracts porcine coronary arteries. Acta Physiol (Oxf) 191(4): 285–296. https://doi.org/10.1111/j.1748-1716.2007.01745.x
Shibata S, Iwasaki K, Ogawa Y, Kato J, Ogawa S (2005) Cardiovascular neuroregulation during acute exposure to 40, 70, and 100% oxygen at sea level. Aviat Space Environ Med 76(12): 1105–1110.
Gole Y, Gargne O, Coulange M, Steinberg JG, Bouhaddi M, Jammes Y, Regnard J, Boussuges A (2011) Hyperoxia-induced alterations in cardiovascular function and autonomic control during return to normoxic breathing. Eur J Appl Physiol 111(6): 937–946. https://doi.org/10.1007/s00421-010-1711-4
Schipke JD, Muth T, Pepper C, Schneppendahl J, Hoffmanns M, Dreyer S (2022) Hyperoxia and the cardiovascular system: experiences with hyperbaric oxygen therapy. Med Gas Res 12(4): 153–157. https://doi.org/10.4103/2045-9912.337997
Larsson A, Uusijärvi J, Eksborg S, Lindholm P (2010) Tissue oxygenation measured with near-infrared spectroscopy during normobaric and hyperbaric oxygen breathing in healthy subjects. Eur J Appl Physiol 109(4): 757–761. https://doi.org/10.1007/s00421-010-1403-0
Neubauer B, Tetzlaff K, Staschen CM, Bettinghausen E (2001) Cardiac output changes during hyperbaric hyperoxia. Int Arch Occup Environ Health 74(2): 119–122. https://doi.org/10.1007/s004200000201
Savitt MA, Rankin JS, Elberry JR, Owen CH, Camporesi EM (1994) Influence of hyperbaric oxygen on left ventricular contractility, total coronary blood flow, and myocardial oxygen consumption in the conscious dog. Undersea Hyperb Med 21(2): 169–183.
Demchenko IT, Zhilyaev SY, Moskvin AN, Piantadosi CA, Allen BW (2011) Autonomic activation links CNS oxygen toxicity to acute cardiogenic pulmonary injury. Am J Physiol Lung Cell Mol Physiol 300(1): L102–L111. https://doi.org/10.1152/ajplung.00178.2010
Demchenko IT, Gasier HG, Zhilyaev SYu, Moskvin AN, Krivchenko AI, Piantadosi CA, Allen BW (2014) Baroreceptor afferents modulate brain excitation and influence susceptibility to toxic effects of hyperbaric oxygen. J Appl Physiol (1985) 117(5): 525–534. https://doi.org/10.1152/japplphysiol.00435.2014
Zhilyaev SYu, Platonova TF, Alekseeva OS, Nikitina ER, Demchenko IT (2019) Adaptive mechanisms of baroreflectory regulation of the cardiovascular system in extreme hyperoxia. J Evol Biochem Physiol 55(5): 365–371. https://doi.org/10.1134/S002209301905003X
Guyenet PG (2006) The sympathetic control of blood pressure. Nat Rev Neurosci 7(5): 335–346. https://doi.org/10.1038/nrn1902
Thrasher TN (2005) Baroreceptors, baroreceptor unloading, and the long-term control of blood pressure. Am J Physiol Regul Integr Comp Physiol 288(4): R819–R827. https://doi.org/10.1152/ajpregu.00813.2004
Stauss HM, Moffitt JA, Chapleau MW, Abboud FM, Johnson AK (2006) Baroreceptor reflex sensitivity estimated by the sequence technique is reliable in rats. Am J Physiol Heart Circ Physiol 291(1): H482–H483. https://doi.org/10.1152/ajpheart.00228.2006
Henderson LA, Richard CA, Macey PM, Runquist ML, Yu PL, Galons JP, Harper RM (2004) Functional magnetic resonance signal changes in neural structures to baroreceptor reflex activation. J Appl Physiol (1985) 96(2): 693–703. https://doi.org/10.1152/japplphysiol.00852.2003
Platonova TF, Alekseeva OS, Nikitina ER, Demchenko IT (2020) Blockade of Brain Adrenoreceptors Delays Seizure Development during Hyperbaric Oxygen Breathing. J Evol Biochem Phys 56(5): 425–433. https://doi.org/10.1134/S0022093020050051
Demchenko IT, Zhilyaev SYu, Platonova TF, Alekseeva OS, Nikitina ER (2021) Inhibition of GABA transaminase and GABA transporters in the brain by vigabatrin and tiagabine prevents seizure development in rats breathing hyperbaric oxygen. J Evol Biochem Physiol 57(5): 1101–1109. https://doi.org/10.1134/S0022093021050112
Жиляев СЮ, Москвин АН, Платонова ТФ, Демченко ИТ (2015) Электрическая стимуляция вагусного нерва модулирует развитие кислородной эпилепсии у кроликов. Рос физиол журн им ИМ Сеченова 101(11): 1279–1288. [Zhilyaev SYu, Moskvin AN, Platonova TF, Demchenko IT (2015) Electric stimulation of vagus nerve modulates a propagation of oxygen epilepsy in rabbits. Russ J Physiol 101(11): 1279–1288. (In Russ)].
Зальцман ГЛ (1968) Стадии развития кислородной эпилепсии и функциональное состояние нервной системы. В кн. Гипербарические эпилепсия и наркоз. Л. Наука. [Zaltsman GL (1968) Stages of formation of oxygen epilepsy and the functional state of the centres of the nervous system. In book: Hyperbaric epilepsy and narcosis. Ed Zaltsman GL Leningrad. Nauka. (In Russ)].
Селивра АИ (1983) Гипербарическая оксигенация. Физиологические механизмы реакций центральной нервной системы на гипероксию. Л. Наука. [Selivra AI (1983) Hyperbaric oxygenation. Physiological mechanisms of central nervous system responses to hyperoxia. Leningrad. Nauka. (In Russ)].
Dean JB, Mulkey DK, Garcia AJ 3rd, Putnam RW, Henderson RA 3rd (2003) Neuronal sensitivity to hyperoxia, hypercapnia, and inert gases at hyperbaric pressures. J Appl Physiol (1985) 95(3): 883–909. https://doi.org/10.1152/japplphysiol.00920.2002
Ciarlone GE, Hinojo CM, Stavitzski NM, Dean JB (2019) CNS function and dysfunction during exposure to hyperbaric oxygen in operational and clinical settings. Redox Biol 27: 101–159. https://doi.org/10.1016/j.redox.2019.101159
Poff AM, Kernagis D, D’Agostino DP (2017) Hyperbaric Environment: Oxygen and Cellular Damage versus Protection. Compr Physiol 7: 213–234. https://doi.org/10.1002/cphy.c150032
Zhang Y, Wang Z, Chen Y, Li R (2019) Intermittent hyperbaric oxygen exposure mobilizing peroxiredoxin 6 to prevent oxygen toxicity. J Physiol Sci 69: 779–790. https://doi.org/10.1007/s12576-019-00694-5
Zhang Y, You B, Chen Y, Yang J, Xie C, Huang G, Li R, Hu P (2020) Effect of Transcriptional Regulatory Factor FoxO3a on Central Nervous System Oxygen Toxicity. Front Physiol 11: 596326. https://doi.org/10.3389/fphys.2020.596326
Torbati D, Church DF, Keller JM, Pryor WA (1992) Free radical generation in the brain precedes hyperbaric oxygen-induced convulsions. Free Radic Biol Med 13(2): 101–106. https://doi.org/10.1016/0891-5849(92)90070-w
D’Agostino DP, Putnam RW, Dean JB (2007) Superoxide ($*O_{2}^{ - }$) production in CA1 neurons of rat hippocampal slices exposed to graded levels of oxygen. J Neurophysiol 98(2): 1030–1041.
Ciarlone GE, Dean JB (2016) Normobaric hyperoxia stimulates superoxide and nitric oxide production in the caudal solitary complex of rat brain slices. Am J Physiol Cell Physiol 311(6): C1014–C1026. https://doi.org/10.1152/ajpcell.00160.2016
Demchenko IT, Boso AE, Whorton AR, Piantadosi CA (2001) Nitric oxide production is enhanced in rat brain before oxygen-induced convulsions. Brain Res 917(2): 253–261. https://doi.org/10.1016/s0006-8993(01)03057-8
Chavko M, Auker CR, McCarron RM (2003) Relationship between protein nitration and oxidation and development of hyperoxic seizures. Nitric Oxide 9(1): 18–23. https://doi.org/10.1016/s1089-8603(03)00045-4
Allen BW, Demchenko IT, Piantadosi CA (2009) Two faces of nitric oxide: implications for cellular mechanisms of oxygen toxicity. J Appl Physiol (1985) 106(2): 662–667. https://doi.org/10.1152/japplphysiol.91109.2008
Oury TD, Ho YS, Piantadosi CA, Crapo JD (1992) Extracellular superoxide dismutase, nitric oxide, and central nervous system O2 toxicity. Proc Natl Acad Sci U S A 89(20): 9715–9719. https://doi.org/10.1073/pnas.89.20.9715
Bitterman N, Bitterman H (1998) L-arginine-NO pathway and CNS oxygen toxicity. J Appl Physiol (1985) 84(5): 1633–1638. https://doi.org/10.1152/jappl.1998.84.5.1633
Demchenko IT, Piantadosi CA (2006) Nitric oxide amplifies the excitatory to inhibitory neurotransmitter imbalance accelerating oxygen seizures. Undersea Hyperb Med 33(3): 169–174.
Gasier HG, Demchenko IT, Tatro LG, Piantadosi CA (2017) S-nitrosylation of GAD65 is implicated in decreased GAD activity and oxygen-induced seizures. Neurosci Lett 653: 283–287. https://doi.org/10.1016/j.neulet.2017.05.067
Alekseeva OS, Zhilyaev SYu, Platonova TF, Tsyba DL, Kirik OV, Korzhevskii DE, Demchenko IT (2022) Involvement of glutamine synthetase in the development of hyperbaric oxygen seizures. J Evol Biochem Physiol 58(1): 158–166. https://doi.org/10.1134/S0022093022010148
Hall AA, Young C, Bodo M, Mahon RT (2013) Vigabatrin prevents seizure in swine subjected to hyperbaric hyperoxia. J Appl Physiol (1985) 115(6): 861–867. https://doi.org/10.1152/japplphysiol.00221.2013
Moskvin AN, Platonova TPh, Zhilyaev SYu, Alekseeva OS, Nikitina ER, Demchenko IT (2020) Blockade of γ-Aminobutyric Acid Transporters in Brain Synapses Protects Against Hyperbaric Oxygen-Induced Convulsions. Neurosci Behav Physiol 50(4): 505–510. https://doi.org/10.1007/s11055-020-00930-1
Demchenko IT, Zhilyaev SYu, Moskvin AN, Krivchenko AI, Piantadosi CA, Allen BW (2017) Antiepileptic drugs prevent seizures in hyperbaric oxygen: A novel model of epileptiform activity. Brain Res 1657: 347–354. https://doi.org/10.1016/j.brainres.2016.12.032
Gutsaeva DR, Suliman HB, Carraway MS, Demchenko IT, Piantadosi CA (2006) Oxygen-induced mitochondrial biogenesis in the rat hippocampus. Neuroscience 137(2): 493–504. https://doi.org/10.1016/j.neuroscience.2005.07.061
Piantadosi CA, Suliman HB (2012) Redox regulation of mitochondrial biogenesis. Free Radic Biol Med 53(11): 2043–2053. https://doi.org/10.1016/j.freeradbiomed.2012.09.014
Matalon S, Hardiman KM, Jain L, Eaton DC, Kotlikoff M, Eu JP, Sun J, Meissner G, Stamler JS (2003) Regulation of ion channel structure and function by reactive oxygen-nitrogen species. Am J Physiol Lung Cell Mol Physiol 285(6): L1184–L1189. https://doi.org/10.1152/ajplung.00281.2003
He H, Li X, He Y (2019) Hyperbaric oxygen therapy attenuates neuronal apoptosis induced by traumatic brain injury via Akt/GSK3β/β-catenin pathway. Neuropsychiatr Dis Treat 15: 369–374. https://doi.org/10.2147/NDT.S183632
Cozene B, Sadanandan N, Gonzales-Portillo B, Saft M, Cho J, Park YJ, Borlongan CV (2020) An Extra Breath of Fresh Air: Hyperbaric Oxygenation as a Stroke Therapeutic. Biomolecules 10: 1279. https://doi.org/10.3390/biom10091279
Close GL, Kayani AC, Ashton T, McArdle A, Jackson MJ (2007) Release of superoxide from skeletal muscle of adult and old mice: an experimental test of the reductive hotspot hypothesis. Aging Cell 6(2): 189–195. https://doi.org/10.1111/j.1474-9726.2007.00277.x
Schiavo S, Richardson D, Santa Mina D, Buryk-Iggers S, Uehling J, Carroll J, Clarke H, Djaiani C, Gershinsky M, Katznelson R (2020) Hyperbaric oxygen and focused rehabilitation program: A feasibility study in improving upper limb motor function after stroke. Appl Physiol Nutr Metab 45(12): 1345–1352. https://doi.org/10.1139/apnm-2020-0124
Sankaran R, Radhakrishnan K, Sundaram KR (2019) Hyperbaric oxygen therapy in patients with hypoxic ischemic encephalopathy. Neurol India 67(3): 728–731. https://doi.org/10.4103/0028-3886.263236
Liang XX, Hao YG, Duan XM, Han XL, Cai XX (2020) Hyperbaric oxygen therapy for post-stroke depression: A systematic review and meta-analysis. Clin Neurol Neurosurg 195: 105910. https://doi.org/10.1016/j.clineuro.2020.105910
Golan H, Makogon B, Volkov O, Smolyakov Y, Hadanny A, Efrati S (2019) Imaging-based predictors for hyperbaric oxygen therapy outcome in post-stroke patients. Report 1. Med Hypotheses 136: 109510. https://doi.org/10.1016/j.mehy.2019.109510
Mattson MP (2008) Hormesis defined. Ageing Res Rev 7(1): 1–7. https://doi.org/10.1016/j.arr.2007.08.007
Brugniaux JV, Coombs GB, Barak OF, Dujic Z, Sekhon MS, Ainslie PN (2018) Highs and lows of hyperoxia: physiological, performance, and clinical aspects. Am J Physiol Regul Integr Comp Physiol 315(1): R1–R27. https://doi.org/10.1152/ajpregu.00165.2017
Thom SR, Bhopale VM, Velazquez OC, Goldstein LJ, Thom LH, Buerk DG (2006) Stem cell mobilization by hyperbaric oxygen. Am J Physiol Heart Circ Physiol 290(4): H1378–H1386. https://doi.org/10.1152/ajpheart.00888.2005
Hachmo Y, Hadanny A, Abu Hamed R, Daniel-Kotovsky M, Catalogna M, Fishlev G, Lang E, Polak N, Doenyas K, Friedman M, Zemel Y, Bechor Y, Efrati S (2020) Hyperbaric oxygen therapy increases telomere length and decreases immunosenescence in isolated blood cells: a prospective trial. Aging (Albany NY) 12(22): 22445–22456. https://doi.org/10.18632/aging.202188
Demchenko IT, Zhilyaev SYu, Alekseeva OS, Krivchenko AI, Piantadosi CA, Gasier HG (2019) Increased Antiseizure Effectiveness with Tiagabine Combined with Sodium Channel Antagonists in Mice Exposed to Hyperbaric Oxygen. Neurotox Res 36(4): 788–795. https://doi.org/10.1007/s12640-019-00063-5
Дополнительные материалы отсутствуют.
Инструменты
Российский физиологический журнал им. И.М. Сеченова