

Российский физиологический журнал им. И.М. Сеченова, 2022, T. 108, № 12, стр. 1610-1626
TGN-020, ингибитор водного канала аквапорина-4, ускоряет процесс нейродегенерации в нигростриатной системе в модели болезни Паркинсона у крыс
К. В. Лапшина 1, *, Ю. Ю. Абрамова 1, М. А. Гузеев 1, И. В. Екимова 1
1 Институт эволюционной физиологии и биохимии им. И.М. Сеченова РАН
Санкт-Петербург, Россия
* E-mail: ksenia.lapshina@gmail.com
Поступила в редакцию 29.09.2022
После доработки 27.10.2022
Принята к публикации 27.10.2022
- EDN: HNMJNA
- DOI: 10.31857/S0869813922120081
Аннотация
Водный канал аквапорин-4 (aquaporin-4, (AQP-4)) является важнейшим звеном в механизмах обеспечения водного гомеостаза в головном мозге и участником процесса “очистки” паренхимы мозга от различных метаболитов, в том числе – от амилоидогенных белков, способствующих развитию нейродегенеративных заболеваний. Задача настоящего исследования состояла в том, чтобы выяснить, какое влияние оказывает фармакологическое ингибирование водного канала AQP-4 в головном мозге на темп развития нейродегенерации и компенсаторные процессы в нигростриатной системе в лактацистиновой модели болезни Паркинсона (БП) у крыс. Модель БП у самцов крыс популяции Вистар воспроизводили с помощью микроинъекций специфического ингибитора протеасом лактацистина в компактную часть черной субстанции (кчЧС). Для фармакологического подавления активности AQP-4 применяли ингибитор TGN-020, который вводили в ликвор бокового желудочка головного мозга. В работе применены методы иммуногистохимии и поведенческие тесты для оценки моторного дефицита. Созданная модель БП характеризовалась допороговым уровнем гибели дофаминергических нейронов в кчЧС (27%) и их аксонов в стриатуме (19%) по сравнению с уровнем клинической стадии, отсутствием изменений уровня ключевого фермента синтеза дофамина тирозингидроксилазы в нейронах кчЧС и моторной функции. Такие патофизиологические изменения характерны для доклинической стадии БП. Применение TGN-020 в модели БП ускоряло переход из доклинической в клиническую стадию БП, о чем свидетельствовало прогрессирование нейродегенерации в нигростриатной системе, ослабление компенсаторных процессов и развитие симптомов паркинсонизма. Полученные данные указывают, что AQP-4 играет важную роль в молекулярных механизмах, обеспечивающих защиту мозга от нейротоксических факторов, и снижение его активности может приводить к нарушению функционирования глимфатической системы. Водный канал AQP-4 может оказаться перспективной терапевтической мишенью для нейропротекции при БП и других нейродегенеративных заболеваниях.
Болезнь Паркинсона (БП) — это хроническое прогрессирующее нейродегенеративное заболевание. Ключевым патогенетическим признаком БП является гибель более половины популяции дофамин (ДА)-ергических нейронов в компактной части черной субстанции (кчЧС) головного мозга, что приводит к снижению уровня ДА в стриатуме на 70–80% и появлению характерных моторных нарушений – тремора покоя, гипокинезии, ригидности, постуральной неустойчивости и других симптомов [1, 2]. Особенностью этого заболевания является длительное развитие (до 30 лет) доклинической стадии без проявления характерных моторных дисфункций. БП до сих пор остается неизлечимым заболеванием, так как пациент в подавляющем большинстве случаев обращается к врачу при проявлении моторных нарушений (в клинической стадии), когда основная часть нейронов в кчЧС уже погибла, и достичь полного излечения уже невозможно. Прогресс в лечении БП связывается с разработкой ранней диагностики БП и развитием патогенетически значимых нейропротективных стратегий. Однако в настоящее время нет фармакологического препарата, который бы эффективно защищал клетки мозга от патологического процесса или способствовал их восстановлению при БП [2, 3]. Поэтому основной акцент в этом направлении делается на изучение механизмов патогенеза БП с целью поиска молекулярных мишеней для превентивного лечения этого заболевания.
Согласно современным представлениям, важнейшим звеном патогенеза БП является накопление в нейронах токсичных олигомеров белка α-синуклеина, возникающее из-за нарушения его конформации [4]. Известно, что деградацию аберрантных белков (в том числе и α-синуклеина) в клетке обеспечивают убиквитин-протеасомная (УПС) и аутофаголизосомная системы [5, 6]. На основе фармакологического угнетения активности УПС в кчЧС были созданы модели БП у грызунов, отражающие доклиническую и клиническую стадии БП [7–10].
Одним из важных достижений последнего десятилетия стало формирование представлений о глимфатической системе, осуществляющей очистку паренхимы головного мозга от различных метаболитов (в том числе – амилоидогенных белков), и активное исследование роли менингеальных лимфатических сосудов и глубоких шейных лимфатических узлов в обеспечении элиминации метаболитов [11–13]. Предполагается, что нарушение работы этих механизмов может являться одним из важнейших факторов развития нейродегенеративных заболеваний, и в связи с этим данная область исследований привлекает большое внимание ученых. Однако стоит отметить, что глимфатическая концепция остается довольно противоречивой и вызывает разногласия в научном сообществе, поскольку остается много открытых вопросов в отношении механизмов поступления жидкости в ткань мозга, функционально-анатомической организации и других аспектов [12, 13]. В настоящее время знания о взаимоотношениях между образованием, накоплением и клиренсом амилоидогенных белков из головного мозга по-прежнему остаются недостаточными. Считается, что одним из важнейших участников уникального механизма “самоочистки” мозга является водный канал аквапорин-4 (aquaporin-4, (AQP-4)) [11]. AQP-4 является трансмембранным белком и локализуется преимущественно на отростках астроцитов, окружающих кровеносные сосуды, что обеспечивает контакт с параваскулярным пространством. AQP-4, в отличие от других подтипов аквапоринов, наиболее распространен в головном мозге и считается основным каналом, поддерживающим водный гомеостаз и быстрое движение жидкости в глимфатической системе в ответ на осмотические изменения и другие особые требования в ЦНС [14–17]. Помимо своей основной функции в обеспечении водного гомеостаза, AQP-4 вовлечен и в другие процессы, такие как поглощение внеклеточного K+, клиренс глутамата, миграция астроцитов [17]. За последние 7 лет накоплены данные, указывающие на возможную взаимосвязь между функционированием водного канала AQP-4 и развитием БП. Обнаружено, что у пациентов с БП в ткани кчЧС происходит накопление свободной воды, усиливающееся с развитием болезни, что может свидетельствовать о нарушении водного гомеостаза и функционирования AQP-4 [18]. При этом показана отрицательная корреляция между отложением α-синуклеина и экспрессией AQP-4 [19]. Выяснено, что блокирование менингеального лимфатического оттока путем лигирования глубоких шейных лимфатических узлов у мышей усугубляет патологические и моторные симптомы, вызванные аберрантным α-синуклеином [20], а введение фибрилл α-синуклеина в дорсальный стриатум и кчЧС нокаутированным по AQP-4 мышам ускоряет патологическое отложение α-синуклеина и способствует гибели ДА-ергических нейронов и раннему проявлению моторных нарушений [21]. В МФТП (метил-4-фенил-1,2,3,6-тетрагидропиридин)-модели БП у мышей нокаутирование по AQP-4 приводило также к значительному усилению гибели ДА-ергических нейронов в кчЧС, сопряженному с усилением нейровоспаления [22–24 ]. Эти данные свидетельствуют о том, что снижение экспрессии белка AQP-4 может усугубить Паркинсон-подобную патологию. Вероятно, это происходит за счет сбоя в работе глимфатической системы. Учитывая то, что в астроцитах кроме AQP-4 экспрессируются другие подтипы аквапоринов, пока остается неясным, могут ли эти подтипы компенсировать недостаток AQP-4 и повлиять на патогенез БП.
Следует отметить, что исследования, посвященные изучению роли AQP-4 в патогенезе БП, остаются малочисленными и проводились всего лишь на двух моделях БП у животных – α-синуклеиновой патологии и окислительного стресса, индуцированного нейротоксином МРТР. При этом использовались нокаутированные по AQP-4 животные, у которых может ожидаться компенсаторное вовлечение других подтипов аквапоринов. Для лучшего понимания взаимосвязи между функционированием водного канала AQP-4 и развитием нейродегенерации при БП необходимо использовать как другие патогенетически значимые модели БП, так и иные методические подходы для ингибирования активности AQP-4, например – фармакологический. Модель БП, основанная на дисфункции УПС, отражает основные патофизиологические признаки БП и является пригодной для решения поставленных задач [10]. Таких исследований ранее не проводилось.
Задача настоящего исследования состояла в том, чтобы выяснить, какое влияние оказывает фармакологическое ингибирование водного канала AQP-4 в головном мозге на темп развития нейродегенерации и компенсаторные процессы в нигростриатной системе в модели БП, созданной на основе ингибирования протеасом в кчЧС у крыс.
Животные
Эксперименты были выполнены на 30 взрослых самцах крыс популяции Вистар в возрасте 6–7 мес. и массой тела 350–400 г, выращенных в виварии Института эволюционной физиологии и биохимии им. И.М. Сеченова РАН. На протяжении всего исследования крыс содержали в одиночных клетках с неограниченным доступом к пище и воде при естественном освещении и температуре воздуха 21–23°C. Манипуляции с животными проводились в соответствии принципам Базельской декларации, этическим стандартам, утвержденным правовыми актами РФ и протоколу, утвержденным этическим комитетом Института эволюционной физиологии и биохимии им. И.М. Сеченова РАН.
Проведение операции по вживлению проводящих канюль
Перед проведением операции животных наркотизировали с помощью золетила (Zoletil®; Virbac, Франция), в дозе 60 мг/кг (внутримышечно). Установку канюль проводили с использованием стереотаксического устройства (Narishige, Япония) согласно атласу координат для крыс линии Вистар [25]: для кчЧС – 5 мм каудальнее брегмы и 2 мм слева и справа от саггитального шва, для левого бокового желудочка – 0.5 мм каудальнее брегмы, 1.5 мм латерально. Глубина погружения канюли на уровне кчЧС в головной мозг крысы составляла 5.5–6 мм, на уровне левого бокового желудочка – 4 мм. Такие длины нижних частей канюль исключали возможность механического повреждения интересующих нас структур мозга. Для закрепления канюль использовался зубной цемент “Акродент” (“Стома”, Украина). После застывания цемента в канюли помещали мандрены. Эксперименты начинали не раньше, чем через 7 дней после проведения операции.
Препараты и экспериментальные группы
Для выполнения микроинъекций наркотизированное золетилом (внутримышечно, 45 мг/кг) животное помещали в стереотаксическое устройство (Narishige, Япония). Микроинъекции препаратов осуществляли с помощью шприцов Hamilton с инъекционными иглами нужной длины и диаметра. Используемые в исследовании препараты растворяли в стерильном фосфатно-солевом буфере (phosphate buffered saline (PBS), пропущенном через фильтр-насадку на шприц PVDF 0.22 мкм (JET BIOFIL®, Корея).
Для исследования нами была выбрана хроническая модель БП у крыс, разработанная в лаборатории сравнительной термофизиологии, и созданная путем угнетения активности УПС в нигростриатной системе [8, 10, 26]. Данная модель позволяет моделировать основные патофизиологические признаки доклинической и клинической стадий БП у крыс. Для угнетения активности протеасом использовали специфический ингибитор их ферментативной активности лактацистин (ЛЦ) (Enzo Life Sciences, Великобритания). Введение ЛЦ осуществляли двукратно и билатерально в кчЧС в дозах 0.4 мкг/мкл (первая микроинъекция ЛЦ) и 4.0 мкг/мкл (вторая микроинъекция ЛЦ). Интервал между введениями составлял 7 дней.
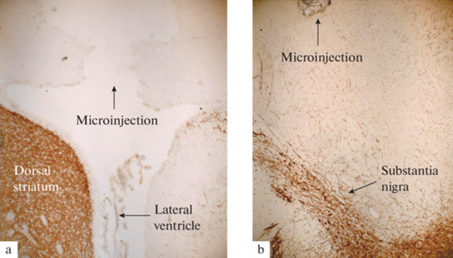
Для подавления функционирования белка AQP-4 был выбран фармакологический подход и применен ингибитор 2-(никотинамид)-1,3,4-тиадиазол (2-(nicotinamide)-1,3,4-thiadiazole, TGN-020. Введение TGN-020 выполняли в боковой желудочек мозга в дозе 40 мкг и в объеме 6 мкл за 15 мин до введения ЛЦ. Поскольку коммерчески доступный препарат TGN-020 имеет низкую растворимость и для приготовления рабочего раствора требуется диметилсульфоксид, применение которого нежелательно для микроинъекций в мозг, в нашем исследовании была использована водорастворимая форма TGN-020 в виде натриевой соли, синтезированная В.А. Полукеевым (АО “Вектон”, Санкт-Петербург) согласно методическим протоколам разработчиков [27 ] . Контрольные группы животных получали микроинъекции растворителя препаратов (PBS) в том же объеме, что и экспериментальные животные. Скорость введения препаратов в канюлю составляла 0.5 мкл/мин (для TGN-020) и 0.1 мкл/мин (для ЛЦ). В конце эксперимента проводился гистологический контроль места инъекции (рис. 1).
Рис. 1.
Гистологический контроль мест микроинъекций. Световая микроскопия: (a) левый боковой желудочек, (b) черная субстанция. Срезы мозга окрашены антителами против тирозингидроксилазы. Стрелками обозначены места микроинъекций. Масштаб – 20 мкм.
Для выполнения исследования путем простой рандомизации были сформированы следующие группы животных:
Группа 1. Интактные животные (n = 6);
Группа 2. Контроль (n = 6; введение PBS (контрольного раствора) в левый боковой желудочек и в кчЧС);
Группа 3. TGN-020 (n = 6; введение TGN-020 в левый боковой желудочек и PBS в кчЧС);
Группа 4. ЛЦ (n = 6; введение PBS в левый боковой желудочек и ЛЦ в кчЧС);
Группа 5. ЛЦ + TGN-020 (n = 6; введение TGN-020 в левый боковой желудочек и ЛЦ в кчЧС).
Оценка изменений моторного поведения
Тесты на моторное поведение проводились через 19–20 дней после первого введения препаратов.
Тест с семенами подсолнечника (Sunflower seed test). Тест с семенами подсолнечника применяли для оценки изменений мелкой моторики передних конечностей, рта и языка [28]. В период содержания крыс в виварии в их рацион периодически добавляли небольшое количество семян подсолнечника. За сутки до тестирования крыс не кормили. В день эксперимента каждая крыса получала 10 семян подсолнечника, тестирование проводили с применением видеорегистрации. В ходе эксперимента определяли среднее время, затрачиваемое крысой на манипуляции, необходимые для поедания одного семени (взятие семечка передними конечностями, вскрытие кожуры, извлечение и поедание).
Тест “Открытое поле” (Оpen field test). Тест “Открытое поле” дает возможность выявить признаки гипокинезии и акинезии по изменению двигательной активности [29, 30]. Не менее чем за 1 ч до проведения теста животных переносили в экспериментальную комнату для адаптации. При проведении эксперимента соблюдали тишину. Крысу помещали в центр открытого поля (ООО “НПК Открытая Наука”, Россия), запускали программу видеотрекинга EthoVision (Noldus, США) и наблюдали за ее поведением в течение 5 мин. После тестирования каждого животного поверхности протирали влажной губкой и дезодорировали 3%-ным раствором перекиси водорода. Для того чтобы оценить изменения двигательной активности, выполняли автоматизированное вычисление общей длины пути, пройденного каждой крысой.
Тест “Сужающаяся дорожка” (Beam-walking test). Установку “Сужающаяся дорожка” (ООО “НПК Открытая Наука”, Россия) использовали для оценки моторной функции передних и задних конечностей, что позволяет выявить признаки развития сенсомоторного дефицита [31]. В данном тесте животное должно пройти по доске, которая постепенно сужается, и зайти в темный отсек. Доска состоит из 2 частей: верхней, по которой идет крыса, и более широкой нижней, позволяющей фиксировать соскальзывания лапок и ошибочные постановки конечностей. В течение 4–5 дней до финального тестирования проводили предварительную адаптацию животных к помещению и экспериментальной установке. В день эксперимента не менее чем за 1 ч до проведения теста животных переносили в экспериментальную комнату для адаптации. В тесте “Сужающаяся дорожка” регистрировали постановку лапы на верхнюю или нижнюю доску. Подсчитывали количество постановок конечности на нижнюю доску (ошибок), количество соскальзываний конечности с верхней доски на нижнюю и общее количество шагов животного от стартовой линии до попадания в темный отсек. Полученные по трем попыткам данные усредняли.
Степень выраженности моторного дефицита отдельно для передних и задних конечностей рассчитывали по формуле, рекомендованной производителем экспериментальной установки:
Иммуногистохимическое исследование
Крыс декапитировали через 21 день после первого введения препаратов. Животных наркотизировали с помощью хлоралгидрата (внутрибрюшинно, 400 мг/кг). Для дальнейшего иммуногистохимического исследования мозг быстро извлекали из черепной коробки и фиксировали в 10%-ном растворе формалина (Биовитрум, Россия) при температуре 4°С. Через 4–5 дней после декапитации мозг промывали в PBS (pH 7.4) и помещали в 20%-ный раствор сахарозы для криопротекции при температуре 4°С. Через 5–7 дней мозг замораживали в изопентане, охлажденном с помощью сухого льда до –45°С, и далее хранили в морозильной камере при температуре –80°С. Изготовление чередующихся серий фронтальных срезов головного мозга крыс толщиной 10 мкм осуществлялось на криостате Leica (Германия). Локализацию структур (кчЧС и дорсального стриатума) определяли с помощью анатомического атласа мозга крысы [25]. Предметные стекла со срезами высушивали в течение суток при комнатной температуре и далее хранили при температуре –20°С.
Для визуализации и последующего подсчета количества выживших ДА-ергических нейронов в кчЧС и их аксонов в стриатуме, а также оценки содержания в нейронах кчЧС скорость-лимитирующего фермента синтеза ДА тирозингидроксилазы (ТГ), использовали биотин-стрептавидиновый метод. После выполнения стандартных подготовительных этапов [10] срезы кчЧС и стриатума инкубировали с первичными поликлональными антителами кролика против ТГ (Abcam, Великобритания (1 : 700)) и оставляли на сутки при комнатной температуре во влажной камере. Затем срезы инкубировали со вторичными антителами козы против кролика, конъюгированными с биотином (Vector Laboratories, США (1 : 400)), в течение 1 ч при комнатной температуре, и далее – со стрептавидин-пероксидазой (Vector Laboratories, США (1 : 400)) в течение 1 ч при комнатной температуре. Визуализация осуществлялась с использованием 3,3'-диаминобензидина. После стандартной проводки в спиртах восходящей концентрации препараты помещали в среду Microclearing (Diapath, Италия) и затем заключали под покровное стекло с помощью среды Витрогель (Биовитрум, Россия). Далее стекла хранили при комнатной температуре.
Окрашивание нейронов по методу Ниссля
Известно, что на результат иммуногистохимического исследования может влиять ряд различных факторов, в частности – количество белка-мишени, качество взаимодействия антигена и антитела. Для того чтобы получить дополнительное свидетельство развития нейродегенеративного процесса в кчЧС, использовали стандартную методику окраски нейронов по методу Ниссля с использованием 1%-ного раствора толуидинового синего. Дифференцирование в 96%-ном этиловом спирте проводили под визуальным контролем с использованием светового микроскопа (Биомед, Россия) до проявления у окрашенных клеток ядра, ядрышка и светлой зернистой цитоплазмы. Затем стекла высушивали, помещали в среду Microclearing (Diapath, Италия) и заключали под покровное стекло с помощью среды Витрогель (Биовитрум, Россия). Окрашенные стекла со срезами хранили в затемненном месте при комнатной температуре.
Морфологический анализ препаратов
Изображения исследуемых структур мозга получали с помощью микроскопа Karl Zeiss (Imager A1) со встроенной видеокамерой и программы Zen Blue для захвата изображения в формате [jpg]. Подсчет ТГ-иммунопозитивных нейронов кчЧС с видимым ядром и нейронов кчЧС, окрашенных по методу Ниссля и имеющих отчетливо видимое ядро и ядрышко, проводили с применением программы PhotoM (http://www.t_lambda.chat.ru/). Для каждого животного было обработано 8–10 снимков кчЧС. Оптическая плотность ТГ, используемая для анализа содержания этого фермента в нейронах кчЧС, рассчитывалась компьютерной программой PhotoM как средний десятичный логарифм отношения яркости фона, не содержащего иммунореактивное вещество, к яркости точки объекта (нейронов кчЧС, содержащих иммунореактивное вещество ТГ) при прохождении волны света длиной 650 нм на том же срезе. Данные по изменению оптической плотности ТГ выражали в условных единицах (у. е). Для подсчета количества ТГ-иммунореактивных аксонов в дорсальном стриатуме использовали программу ImageJ/FiJi (NIH, США), позволяющей полуавтоматически (под контролем экспериментатора) выделять интенсивно окрашенные аксоны и анализировать их количество на каждом снимке. У каждого животного было обработано 15–20 снимков.
Статистическая обработка результатов
Анализ всех полученных результатов проводился в программе GraphPad Prism 8. Нормальность распределения проверяли с помощью критерия Шапиро–Уилка. Для сравнения средних значений двух независимых выборок использовали t‑критерий Стьюдента. Статистическую обработку результатов, распределенных нормально, осуществляли с помощью двухфакторного дисперсионного анализа ANOVA, с последующим Tukey post-hoc анализом. Различия результатов между группами считали достоверными при уровне значимости p < 0.05, результаты представлены в виде mean ± SEM.
Микроинъекции контрольного раствора и TGN-020 не оказывают влияния на моторное поведение, показатели нейродегенерации в нигростриатной системе и содержание тирозингидроксилазы в ДА-ергических нейронах компактной части черной субстанции
Для того чтобы выяснить, оказывает ли операция по вживлению направляющих канюль и последующее введение PBS влияние на моторное поведение и развитие нейродегенеративного процесса в нигростриатной системе у крыс, был проведен сравнительный анализ этих показателей у двух групп животных – интактных и подвергшихся хирургическому вмешательству и микроинъекциям. Установлено, что проведение операции и выполнение микроинъекций PBS в ликвор бокового желудочка мозга и кчЧС не приводило к появлению моторных нарушений у крыс. У контрольных животных, по сравнению с интактными, не было обнаружено статистически значимого изменения количества иммунореактивных к ТГ нейронов в кчЧС (99.1 ± 1.9 vs 103.0 ± 2.6, p = 0.64) и их аксонов в дорсальном стриатуме (4.9 ± ± 0.2 × 103 vs 5.0 ± 0.1 × 103, p = 0.69), а также общего числа нейронов в кчЧС, окрашенных по методу Ниссля (114.3 ± 3.9 vs 123.4 ± 3.3, p = 0.10). Анализ оптической плотности ТГ в телах ДА-ергических нейронов в кчЧС не выявил изменений содержания этого фермента у контрольных животных по сравнению с интактными (0.21 ± 0.01 vs 0.22 ± 0.01, p = 0.85). Таким образом, полученные данные свидетельствуют о том, что проведение хирургической операции и введение контрольных растворов не оказывает значительного влияния на исследуемые показатели.
Далее нами было изучено влияние микроинъекций TGN-020 на вышеперечисленные показатели по сравнению с контролем (PBS). Выяснено, что применяемая доза TGN-020 не вызывает изменений показателей моторного поведения (рис. 2), не приводит к развитию нейродегенерации в нигростриатной системе и изменению содержания ТГ в ДА-ергических нейронах кчЧС (рис. 3–5).
Ингибитор TGN-020 вызывает развитие моторных нарушений, усиление нейродегенерации в нигростриатной системе и снижение содержания тирозингидроксилазы в ДА-ергических нейронах компактной части черной субстанции в модели БП у крыс
Рис. 2.
Влияние TGN-020 на показатели моторного поведения в лактацистиновой модели БП у крыс. a) общая пройденная дистанция в тесте “Открытое поле”(см); b) cредняя продолжительность поедания семечка подсолнечника (с) (Interaction F(1, 20) = 2.71, p = 0.11; “TGN-020” factor F (1, 20) = 10.89, p = 0.0036; “LC” factor F(1, 20) = 29.86, p < 0.001); с) сенсомоторный дефицит верхних конечностей (%); d) сенсомоторный дефицит нижних конечностей (%) (Interaction F(1, 20) = 10.55, p = 0.0040; “TGN-020” factor F(1, 20) = 9.35, p = 0.0062; “LC” F(1, 20) = 31.51, p < 0.001). ***р < 0.001 – достоверность различий по сравнению с контролем; # p < 0.05, ##p < 0.01 – достоверность различий между ЛЦ и TGN-020 + ЛЦ.
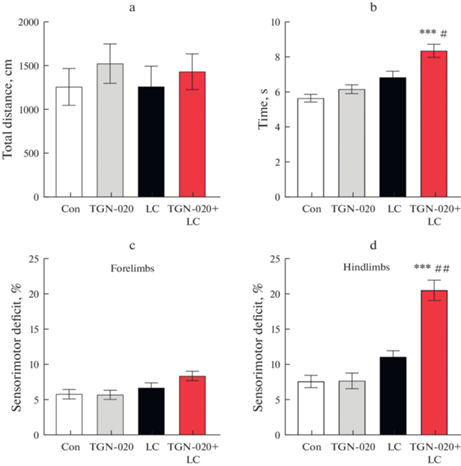
Рис. 3.
Влияние TGN-020 на число нейронов в кчЧС в лактацистиновой модели БП у крыс. Световая микроскопия: a) ТГ-иммунопозитивные нейроны кчЧС (масштаб 50 мкм); b) нейроны кчЧС, окрашенные по методу Ниссля (масштаб 50 мкм). c) число ТГ-иммунопозитивных нейронов в кчЧС (interaction F(1, 20) = 2.57, p = 0.12; “TGN-020” factor (F(1, 20) = 11.71, p = 0.0027; “LC” factor (F(1, 20)=124.2, p < 0.001); d) число нейронов кчЧС, окрашенных по методу Ниссля (interaction F(1, 20) = 2.02, p = 0.17; “TGN-020” factor F(1, 20) = 11.34, p = 0.0031; “LC” factor F(1, 20) = 75.54, p < 0.001). ***p < 0.001 – достоверность различий по сравнению с контролем; # p < 0.05, ## p < 0.01 – достоверность различий между ЛЦ и TGN-020 + ЛЦ.

Рис. 4.
Влияние TGN-020 на количество ДА-ергических аксонов в дорсальном стриатуме в лактацистиновой модели БП у крыс. a) cветовая микроскопия: микрофотографии аксонов в дорсальном стриатуме (масштаб 50 мкм); b) число аксонов в дорсальном стриатуме (interaction F(1, 20) = 2.05, p = 0.17; “TGN-020” factor F(1, 20) = = 7.41, p = 0.01; “LC” factor F(1, 20) = 44.75, p < 0.001). **p < 0.01, ***p < 0.001 – достоверность различий по сравнению с контролем; #p < 0.05 – достоверность различий между ЛЦ и TGN-020 + ЛЦ.

Рис. 5.
Влияние TGN-020 на содержание тирозингидроксилазы в нейронах кчЧС в лактацистиновой модели БП у крыс. a) световая микроскопия: микрофотографии ТГ-иммунопозитивных нейронов кчЧС (масштаб 50 мкм); b) изменение оптической плотности ТГ в нейронах кчЧС (interaction F(1, 20) = 7.59, p = 0.01; “TGN-020” factor F(1, 20) = 9.54, p = 0.0058; “LC” factor F(1, 20) = 2.02, p = 0.17). *p < 0.05 – достоверность различий по сравнению с контролем; ##p < 0.01 – достоверность различий между ЛЦ и TGN-020 + ЛЦ.

Проведенное нами исследование показало, что введение ЛЦ не вызывает развития моторных нарушений (рис. 2). Проведение морфологического анализа позволило выяснить, что применение ЛЦ приводит к уменьшению числа иммунореактивных к ТГ нейронов в кчЧС на 27% (p < 0.001) (рис. 3a, c) и их аксонов в стриатуме на 19% (p < 0.01) (рис. 4). При подсчете числа нейронов в кчЧС на препаратах, окрашенных по методу Ниссля, было выявлено уменьшение их количества на 24% (p < 0.001) (рис. 3b, d). Этот результат согласуется с данными, полученными при подсчете ТГ-иммунопозитивных клеток в кчЧС. При изучении уровня ТГ в ДА-ергических нейронах кчЧС значимых изменений обнаружено не было (рис. 5). Таким образом, микроинъекции ЛЦ приводили к развитию нейродегенеративного процесса в нигростриатной системе, степень выраженности которого соответствовала допороговому уровню по сравнению с уровнем нейродегенерации при клинической стадии БП, при которой погибает более половины нейронов кчЧС. Наблюдаемые нами нейродегенеративные изменения в нигростриатной системе, не сопровождающиеся развитием моторных нарушений, соответствуют характеристикам доклинической стадии БП.
Нами впервые было апробировано введение TGN-020 в ликворную систему головного мозга с целью выявления взаимосвязи между снижением активности AQP-4 и развитием патологических признаков БП у модельных животных. При изучении моторного поведения не было выявлено достоверного изменения общей пройденной дистанции в тесте “Открытое поле” у животных, получавших TGN-20 перед микроинъекциями ЛЦ, по сравнению с группой животных, воспроизводивших модель доклинической стадии БП (рис. 2a). Однако и по сравнению с контролем, и по сравнению с доклинической моделью БП в тесте с семенами подсолнечника было отмечено увеличение среднего времени, затрачиваемого крысой на вскрытие и поедание семечка (рис. 2b), а в тесте “Сужающаяся дорожка” – появление признаков сенсомоторного дефицита нижних конечностей (рис. 2с, d).
Подсчет количества ДА-ергических нейронов в кчЧС и их аксонов в дорсальном стриатуме при введении TGN-020 в модели доклинической стадии БП позволил установить, что ингибитор AQP-4 приводит к увеличению числа погибших ДА-ергических нейронов кчЧС в 1.7 раз (р < 0.01) (рис. 3a, c), а их аксонов в дорсальном стриатуме – в 1.6 раз (р < 0.05) (рис. 4) по сравнению с действием одного ЛЦ. Сходные данные были получены при подсчете нейронов кчЧС на препаратах, окрашенных по методу Ниссля: применение TGN-020 в доклинической модели БП у крыс приводило к увеличению числа погибших нейронов в кчЧС в 1.56 раз (p < 0.05) по сравнению с действием ЛЦ (рис. 3b, d). Для того чтобы оценить влияние TGN-020 на уровень ТГ в ДА-ергических нейронах кчЧС в модели доклинической стадии БП, было проанализировано изменение оптической плотности ТГ. Установлено, что оптическая плотность ТГ в нейронах кчЧС в группе животных, получавших TGN-020 до ЛЦ, снижалась на 17% (р < 0.05) по сравнению с контролем и на 21% (р < 0.01) по сравнению с группой крыс с доклинической стадией БП (рис. 5)
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
В ходе проведенного исследования было установлено, что введение ЛЦ в кчЧС приводит к гибели 27% ДА-ергических нейронов в кчЧС и 19% их аксонов в дорсальном стриатуме. В этих условиях не было выявлено снижения уровня ТГ в выживших нейронах кчЧС и не наблюдалось признаков моторных нарушений (нарушений мелкой моторики передних конечностей, рта и языка, изменения общей пройденной дистанции, признаков сенсомоторного дефицита). Полученные нами результаты свидетельствуют о том, что примененный протокол введения ЛЦ позволяет моделировать доклиническую стадию БП. Отсутствие двигательных нарушений в этой модели можно объяснить сохранением компенсаторных процессов в нигростриатной системе, направленных на поддержание уровня дофамина на уровне нормы. Кроме того, не исключено, что маскированию моторной симптоматики при развитии нейродегенеративного процесса в нигростриатной системе может способствовать увеличение уровня везикулярного транспортера моноаминов 2 [10], уменьшение экспрессии мембранного переносчика дофамина, уменьшение активности моноаминоксидазы B, обеспечивающей деградацию дофамина [32].
Для того чтобы оценить влияние ослабления функции AQP-4 на развитие нейродегенеративного процесса в нигростриатной системе, нами был применен фармакологический ингибитор AQP-4 TGN-020 в модели доклинической стадии БП у крыс [27, 33 ] . Согласно данным литературы, этот препарат является одним из наиболее часто используемых для ингибирования AQP-4 в различных моделях неврологических нарушений [34–36]. Применение магнитно-резонансной томографии показало, что введение TGN-020 замедляло распространение контрастного вещества в тканях головного мозга и подавляло клиренс тау-белка [11, 37]. Эти данные свидетельствует о том, что применение TGN-020 может нарушать работу глимфатической системы. Однако следует отметить, что все вышеперечисленные исследования были выполнены с использованием внутрибрюшинного пути введения TGN-020. В исследовании на мышах было обнаружено, что меченый препарат [11C]TGN-020 после системного введения может обнаруживаться в сердце [27 ] . Кроме того, авторы высказали предположение, что TGN-020 также может оказывать ингибирующее действие на другой водный канал, AQP-1, который на 60% гомологичен AQP-4. Для того чтобы избежать влияния TGN-020 на работу AQP-4 и, возможно, AQP-1 в периферических органах, нами было принято решение использовать центральные микроинъекции этого препарата в ликворную систему головного мозга. Проведенное нами исследование показало, что применение TGN-20 в модели доклинической стадии БП не привело к развитию признаков гипокинезии, однако были обнаружены симптомы нарушения тонкой моторики (судя по увеличению времени, затрачиваемого крысой на вскрытие и поедание семечек подсолнечника) и признаки развития сенсомоторного дефицита задних конечностей. Появление моторных нарушений было сопряжено с увеличением числа погибших ДА-ергических нейронов в кчЧС в 1.7 раза и их аксонов в дорсальном стриатуме в 1.6 раза по сравнению действием одного ЛЦ, что свидетельствовало об ускорении процесса нейродегенерации в нигростриатной системе. Подобный уровень нейродегенерации сам по себе может быть недостаточно высоким, чтобы вызвать отчетливые моторные нарушения, однако нами было обнаружено снижение уровня ТГ в нейронах кчЧС на 21%, которое свидетельствует об ослаблении компенсаторных механизмов поддержания уровня дофамина в нигростриатной системе, что способствует проявлению симптомов паркинсонизма.
Результаты выполненного нами исследования, свидетельствующие об ускорении нейродегенеративного процесса в условиях действия ингибитора AQP-4, согласуются с данными, полученными другими исследователями в экспериментах на нокаутированных по AQP-4 мышах [21–23 ]. Установлено, что через 6 месяцев после введения фибрилл α-синуклеина в дорсальный стриатум число ДА-ергических нейронов в кчЧС у мышей AQP4+/– и мышей дикого типа уменьшалось на 55.2% и 31.8%, что свидетельствовало об усилении нейродегенерации в кчЧС при подавлении экспрессии AQP-4. Кроме того, у мышей AQP4+/– по сравнению с мышами дикого типа было отмечено более раннее развитие моторной дисфункции (ухудшение координации, уменьшение мышечной силы и снижение двигательной активности [21]. При использовании нокаутированных по AQP-4 мышей в острой и хронической МФТП-моделях БП также было обнаружено достоверное увеличение числа погибших ДА-ергических нейронов в кчЧС по сравнению с мышами дикого типа [23, 24 ] . Кроме того, было выявлено, что в острой и хронической МФТП-модели БП у AQP4–/– мышей отмечается более выраженная потеря ТГ-иммунопозитивных волокон не только в дорсальном, но и в вентральном стриатуме, и увеличение числа погибших ДА-ергических нейронов в вентральной тегментальной области [23 ] . Эти данные свидетельствуют о том, что снижение экспрессии AQP-4 приводит к увеличению уязвимости к МФТП и цитотоксичности олигомеров α-синуклеина.
Используемая нами ЛЦ-модель БП характеризуется развитием α-синуклеиновой патологии в кчЧС [9, 10, 38]. Это позволило нам предположить, что одним из ключевых механизмов, лежащих в основе TGN-020 – индуцированной прогрессии нейродегенерации в нигростриатной системе, может являться накопление токсичных олигомеров α-синуклеина во внеклеточной среде из-за снижения активности водного канала AQP-4, которое может приводить к нарушению водного обмена между цереброспинальной и интерстициальной жидкостями в головном мозге. Известно, что олигомерный α-синуклеин, обладающий прионными свойствами, может посредством различных механизмов выходить во внеклеточное пространство и затем интернализоваться соседними клетками (нейронами, астроцитами, микроглией) [39, 40]. Недостаточно эффективная “очистка” клеток мозга и межклеточного пространства от нейротоксичных форм этого белка может приводить к прогрессированию патологии БП. Было продемонстрировано, что введение патогенных фибрилл α-синуклеина в дорсальный стриатум частично нокаутированным по AQP-4 мышам (AQP4+/–) по сравнению с мышами дикого типа вызывает усиленное накопление патологического α-синуклеина в стриатуме, черной субстанции и моторной коре [21]. При этом отмечалось увеличение содержания нерастворимой формы α-синуклеина в различных областях коры и стриатуме. Введение в дорсальный стриатум рекомбинантного α-синуклеина с флуоресцентной меткой показало, что у мышей со сниженной экспрессией AQP-4 область флуоресценции была значительно больше, чем у мышей дикого типа, что могло свидетельствовать о нарушении глимфатического клиренса [21]. В другом исследовании было показано, что у мышей AQP4−/− по сравнению с мышами дикого типа также было отмечено замедление клиренса растворимых форм рекомбинантного человеческого α-синуклеина (A53T и α-syn A30P) из паренхимы мозга. Однако авторы исследования не обнаружили увеличения агрегации олигомеров α-синуклеина и предполагают, что AQP-4 участвует в клиренсе растворимых макромолекул из паренхимы головного мозга [20]. Применение фармакологического ингибирования AQP-4 с помощью TGN-020 в моделях болезни Альцгеймера на животных также способствовало развитию амилоидогенеза и прогрессии нейродегенерации [36, 37]. Совокупность представленных выше наших экспериментальных данных и данных литературы указывает на связь AQP-4 с развитием БП. Можно предположить, что дисфункция AQP-4 может приводить к нарушению функционирования глимфатической системы вследствие снижения эффективности водного обмена между интерстициальной и спинномозговой жидкостью в головном мозге, что, в свою очередь, вызывает усугубление α-синуклеиновой патологии и усиление нейродегенеративного процесса.
Известно, что α-синуклеин может удаляться из внеклеточного пространства путем эндоцитоза или фагоцитоза астроцитами и микроглиоцитами [41]. Нарушение функционирования AQP-4 в результате фармакологического ингибирования могло негативно сказаться на способности астроцитов захватывать и разрушать патологические формы α-синуклеина. Кроме того, AQP-4 может быть вовлечен в механизмы развития нейровоспаления, являющегося одним из важнейших звеньев патогенеза БП [3, 23, 42 ] . Можно предположить, что подавление функционирования AQP-4 может способствовать усилению нейровоспаления и ускорению нейродегенеративного процесса, наблюдаемому в нашем исследовании. Перечисленные выше механизмы являются основными предполагаемыми факторами, лежащими в основе TGN-020-индуцированного усиления нейродегенеративного процесса, наблюдаемого в нашем исследовании. Однако, несмотря на то, что молекулярные механизмы очистки головного мозга от метаболитов чрезвычайно активно исследуются, в отношении концепции глимфатико-лимфатической системы по-прежнему остается много противоречий и вопросов, на которые еще только предстоит ответить.
Таким образом, полученные нами результаты указывают на то, что AQP-4 играет важную роль в молекулярных механизмах защиты нейронов от нейротоксических факторов при развитии Паркинсон-подобной патологии. В связи с тем, что AQP-4 является ключевым звеном в обеспечении водного гомеостаза головного мозга, этот канал представляется перспективной терапевтической мишенью. Новые данные, полученные в ходе поиска путей модуляции функционирования и локализации AQP-4, могут иметь большое значение для разработки новых подходов превентивной терапии БП и других нейродегенеративных патологий мозга.
Список литературы
Bernheimer H, Birkmayer W, Hornykiewicz O, Jellinger K, Seitelberger F (1973) Brain dopamine and the syndromes of Parkinson and Huntington. Clinical, morphological and neurochemical correlations. J Neurol Sci 20(4): 415–455. https://doi.org/10.1016/0022-510x(73)90175-5
Váradi C (2020) Clinical features of Parkinson’s disease: the evolution of critical symptoms. Biology (Basel) 9(5): 103. https://doi.org/10.3390/biology9050103
Devos D, Hirsch E, Wyse R (2021) Seven solutions for neuroprotection in Parkinson’s disease. Mov Disord 36(2): 306–316. https://doi.org/10.1002/mds.28379
Henderson MX, Trojanowski JQ, Lee VM-Y (2019) α-Synuclein pathology in Parkinson’s disease and related α-synucleinopathies. Neurosci Lett 709: 134316. https://doi.org/10.1016/j.neulet.2019
Ebrahimi-Fakhari D, Wahlster L, McLean PJ (2012) Protein degradation pathways in Parkinson’s disease: curse or blessing. Acta Neuropathol 124(2): 153–172. https://doi.org/10.1007/s00401-012-1004-6
Frankowska N, Lisowska K, Witkowski JM (2022) Proteolysis dysfunction in the process of aging and age-related diseases. Front Aging 3: 927630. https://doi.org/10.3389/fragi.2022.927630
McNaught KS, Perl DP, Brownell AL, Olanow CW (2004) Systemic exposure to proteasome inhibitors causes a progressive model of Parkinson’s disease. Ann Neurol 56(1): 149–162. https://doi.org/10.1002/ana.20186
Пастухов ЮФ, Екимова ИВ, Чеснокова АЮ (2014) Молекулярные механизмы патогенеза болезни Паркинсона и перспективы превентивной терапии. В: Нейродегенеративные заболевания – от генома до целостного организма. МВ Угрюмов (ред) М. Научный мир. 316–355. [Pastukhov YuF, Ekimova IV, Chesnokova AYu (2014) Molecular mechanisms of the pathogenesis of Parkinson’s disease and the prospects for preventive therapy. In: Neurodegenerative diseases: from genome to the whole organism. MВ Ugryumov (ed) M. Nauchnyi Mir. 316–355. (In Russ)].
Bentea E, Verbruggen L, Massie A (2017) The proteasome inhibition model of Parkinson’s disease. J Parkinsons Dis 7(1): 31–63. https://doi.org/10.3233/JPD-160921
Ekimova IV, Plaksina DV, Pastukhov YF, Lapshina KV, Lazarev VF, Mikhaylova ER, Polonik SG, Pani B, Margulis BA, Guzhova IV, Nudler E (2018) New HSF1 inducer as a therapeutic agent in a rodent model of Parkinson’s disease. Exp Neurol 306: 199–208. https://doi.org/10.1016/j.expneurol.2018.04.012
Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, Benveniste H, Vates GE, Deane R, Goldman SA, Nagelhus EA, Nedergaard M (2012) A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci Transl Med 4(147): 147ra111. https://doi.org/10.1126/scitranslmed.3003748
Semyachkina-Glushkovskaya O, Postnov D, Kurths J (2018) Blood brain barrier, lymphatic clearance, and recovery: Ariadne’s thread in labyrinths of hypotheses. Int J Mol Sci 19(12): 3818. https://doi.org/10.3390/ijms19123818
Natale G, Limanaqi F, Busceti CL, Mastroiacovo F, Nicoletti F, Puglisi-Allegra S, Fornai F (2021) Glymphatic system as a gateway to connect neurodegeneration from periphery to CNS. Front Neurosci 15: 639140. https://doi.org/10.3389/fnins.2021.639140
Kong H, Fan Y, Xie J, Ding J, Sha L, Xueru S (2008) AQP4 knockout impairs proliferation, migration and neuronal differentiation of adult neural stem cells. J Cell Sci 121: 4029–4036. https://doi.org/10.1016/j.brainresbull.2017.02.011
Hubbard JA, Szu JI, Binder DK (2018) The role of aquaporin-4 in synaptic plasticity, memory and disease. Brain Res Bull 136: 118–129.
Mader S, Brimberg L (2019) Aquaporin-4 water channel in the brain and its implication for health and disease. Cells 8(2): 90. https://doi.org/10.3390/cells8020090
Zhou Z, Zhan J, Cai Q, Xu F, Chai R, Lam K, Luan Z, Zhou G, Tsang S, Kipp M, Han W, Zhang R, Yu ACH (2022) The water transport system in astrocytes-aquaporins. Cells 11(16): 2564. https://doi.org/10.3390/cells11162564
Ofori E, Pasternak O, Planetta PJ, Burciu R, Snyder A, Febo M, Golde TE, Okun MS, Vaillancourt DE (2015) Increased free water in the substantia nigra of Parkinson’s disease: a single-site and multi-site study. Neurobiol Aging 36(2): 1097–1104. https://doi.org/10.1016/j.neurobiolaging.2014.10.029
Hoshi A, Tsunoda A, Tada M, Nishizawa M, Ugawa Y, Kakita A (2017) Expression of aquaporin 1 and aquaporin 4 in the temporal neocortex of patients with Parkinson’s Disease. Brain Pathol 27(2): 160–168. https://doi.org/10.1111/bpa.12369
Zou W, Pu T, Feng W, Lu M, Zheng Y, Du R, Xiao M, Hu G (2019) Blocking meningeal lymphatic drainage aggravates Parkinson’s disease-like pathology in mice overexpressing mutated α-synuclein. Transl Neurodegener 8: 7. https://doi.org/0.1186/s40035-019-0147-y
Cui H, Wang W, Zheng X, Xia D, Liu H, Qin C, Tian H, Teng J (2021) Decreased AQP4 expression aggravates ɑ-Synuclein pathology in Parkinson’s disease mice, possibly via impaired glymphatic clearance. J Mol Neurosci 71: 1–14. https://doi.org/10.1007/s12031-021-01836-4
Zhang J, Yang B, Sun H, Zhou Y, Liu M, Ding J, Fang F, Fan Y, Hu G (2016) Aquaporin-4 deficiency diminishes the differential degeneration of midbrain dopaminergic neurons in experimental Parkinson’s disease. Neurosci Lett 614: 7–15. https://doi.org/10.1016/j.neulet.2015.12.057
Sun H, Liang R, Yang B, Zhou Y, Liu M, Fang F, Hu G (2016) Aquaporin-4 mediates communication between astrocyte and microglia: Implications of neuroinflammation in experimental Parkinson’s disease. Neuroscience 317: 65–75. https://doi.org/10.1016/j.neuroscience.2016.01.003
Xue X, Zhang W, Zhu J, Chen X, Zhou S, Xu Z, Hu G, Su C (2019) Aquaporin-4 deficiency reduces TGF-β1 in mouse midbrains and exacerbates pathology in experimental Parkinson’s disease. J Cell Mol Med 23(4): 2568–2582. https://doi.org/10.1016/j.neuroscience.2016.01.003
Paxinos G, Watson C (2007) The rat brain in stereotaxic coordinates. 6th Edition. San Diego. Acad Press.
Plaksina DV, Ekimova IV (2018) Study of age changes in compensatory processes on the model of neurodegeneration of nigrostriatal system in rats. Adv Gerontol 31(2): 170–177.
Nakamura Y, Suzuki Y, Tsujita M, Huber VJ, Yamada K, Nakada T (2011) Development of a novel ligand, [C]TGN-020, for aquaporin 4 positron emission tomography imaging. ACS Chem Neurosci 2(10): 568–571. https://doi.org/10.1021/cn2000525
Kane JR, Ciucci MR, Jacobs AN, Tews N, Russell JA, Ahrens AM Ma ST, Britt JM, Cormack LK, Schallert T (2011) Assessing the role of dopamine in limb and cranial-oromotor control in a rat model of Parkinson’s disease. J Commun Disord 44: 529–537. http://dx.doi.org/10.1016/j. jcomdis.2011.04.005
Campos FL, Carvalho MM, Cristovão AC, Je G, Baltazar G, Salgado AJ, Kim YS, Sousa N (2013) Rodent models of Parkinson’s disease: beyond the motor symptomatology. Front Behav Neurosci 7: 175. https://doi.org/10.3389/fnbeh.2013.00175
Prasad EM, Hung SY (2020) Behavioral tests in neurotoxin-induced animal models of Parkinson’s disease. Antioxidants (Basel) 9(10): 1007. https://doi.org/10.3390/antiox9101007
Fleming SM, Ekhator OR, Ghisays V (2013) Assessment of sensorimotor function in mouse models of Parkinson’s disease. J Vis Exp (76): 50303. https://doi.org/10.3791/50303
Blesa J, Trigo-Damas I, Dileone M, Del Rey NL, Hernandez LF, Obeso JA (2017) Compensatory mechanisms in Parkinson’s disease: Circuits adaptations and role in disease modification. Exp Neurol 298: 148–161. https://doi.org/10.1016/j.expneurol.2017.10.002
Huber VJ, Tsujita M, Nakada T (2009) Identification of Aquaporin 4 inhibitors using in vitro and in silico methods. Bioorgan Med Chem 17: 411–417. https://doi.org/10.1016/j.bmc.2007.12.040
Igarashi H, Huber VJ, Tsujita M, Nakada T (2011) Pretreatment with a novel aquaporin 4 inhibitor, TGN-020, significantly reduces ischemic cerebral edema. Neurol Sci 32(1): 113–116. https://doi.org/10.1007/s10072-010-0431-1
Abir-Awan M, Kitchen P, Salman MM, Conner MT, Conner AC, Bill RM (2019) Inhibitors of mammalian aquaporin water channels. Int J Mol Sci 20(7): 1589. https://doi.org/10.3390/ijms20071589
Harrison IF, Ismail O, Machhada A, Colgan N, Ohene Y, Nahavandi P, Ahmed Z, Fisher A, Meftah S, Murray TK, Ottersen OP, Nagelhus EA, O’Neill MJ, Wells JA, Lythgoe MF (2020) Impaired glymphatic function and clearance of tau in an Alzheimer’s disease model. Brain 143(8): 2576–2593. https://doi.org/10.1093/brain/awaa179
Rosu GC, Catalin B, Balseanu TA, Laurentiu M, Claudiu M, Kumar-Singh S, Daniel P (2020) Inhibition of aquaporin 4 decreases amyloid Aβ40 drainage around cerebral vessels. Mol Neurobiol 57(11): 4720–4734. https://doi.org/10.1007/s12035-020-02044-8
Savolainen MH, Albert K, Airavaara M, Myöhänen TT (2017) Nigral injection of a proteasomal inhibitor, lactacystin, induces widespread glial cell activation and shows various phenotypes of Parkinson’s disease in young and adult mouse. Exp Brain Res 235(7): 2189–2202. https://doi.org/10.1007/s00221-017-4962-z
Uemura N, Uemura MT, Luk KC, Lee VM, Trojanowski JQ (2020) Cell-to-cell transmission of tau and α-Synuclein. Trends Mol Med 26(10): 936–952. https://doi.org/10.1016/j.molmed.2020.03.012
Choi YR, Park SJ, Park SM (2021) Molecular events underlying the cell-to-cell transmission of α-synuclein. FEBS J 288(23): 6593–6602. https://doi.org/10.1111/febs.15674
Stefanis L, Emmanouilidou E, Pantazopoulou M, Kirik D, Vekrellis K, Tofaris GK (2019) How is alpha-synuclein cleared from the cell? J Neurochem 150(5): 577–590. https://doi.org/10.1111/jnc.14704
Salman MM, Kitchen P, Halsey A, Wang MX, Törnroth-Horsefield S, Conner AC, Badaut J, Iliff JJ, Bill RM (2022) Emerging roles for dynamic aquaporin-4 subcellular relocalization in CNS water homeostasis. Brain 145(1): 64–75. https://doi.org/10.1093/brain/awab311
Дополнительные материалы отсутствуют.
Инструменты
Российский физиологический журнал им. И.М. Сеченова