

Российский физиологический журнал им. И.М. Сеченова, 2022, T. 108, № 5, стр. 594-625
Патофизиологические подходы к изучению дисфункции эндотелия и методологические аспекты определения ее критериев в контексте моделирования гистогематического барьера
А. Г. Кутихин 1, *, Д. К. Шишкова 1, Е. А. Великанова 1, М. Ю. Синицкий 1, А. В. Синицкая 1, В. Е. Маркова 1
1 Научно-исследовательский институт комплексных проблем сердечно-сосудистых заболеваний
Кемерово, Россия
* E-mail: antonkutikhin@gmail.com
Поступила в редакцию 05.03.2022
После доработки 04.04.2022
Принята к публикации 05.04.2022
- EDN: CBJJQK
- DOI: 10.31857/S0869813922050077
Аннотация
Данная проблемная статья посвящена обсуждению методологических аспектов определения различных критериев дисфункции эндотелия (провоспалительной активации эндотелия, нарушений эндотелиальной механотрансдукции, развития эндотелиально-мезенхимального перехода, нарушения биосинтеза и выделения монооксида азота (NO), нарушения целостности эндотелия и его протромботической активации) при экспериментальном моделировании in vitro и in vivo. Классическое определение дисфункции эндотелия включает в себя патофизиологически значимое снижение выделения эндотелиальными клетками вазодилататоров, частичную потерю эндотелием своих атромбогенных свойств, провоспалительную активацию эндотелиальных клеток и патологическое повышение проницаемости эндотелия. Клинически значимыми последствиями дисфункции эндотелия являются артериальная гипертензия, макро- и микроангиопатия вследствие атерогенеза, а также возникающая по причине повышения сосудистой проницаемости микроальбуминурия. На основании результатов исследований последних лет предлагается расширить определение дисфункции эндотелия, добавив в него нарушения эндотелиальной механотрансдукции и развитие эндотелиально-мезенхимального перехода. Чувствительными и надежными биомаркерами, обусловливающими паракринные эффекты дисфункциональных эндотелиальных клеток in vitro, являются интерлейкин-6, интерлейкин-8 и MCP-1/CCL2, однако in vivo данные цитокины также выделяются иммунными клетками и поэтому не обладают достаточной специфичностью для отражения дисфункции эндотелия. Концептуальные улучшения методологии моделирования дисфункции эндотелия, в том числе в контексте имитации гистогематического барьера, включают в себя культивирование эндотелиальных клеток в системе пульсирующего потока (в том числе на подложке из коллагена IV типа – основного компонента эндотелиальной базальной мембраны), а также выделение лизата эндотелия из кровеносных сосудов модельных лабораторных животных in situ с целью последующего профилирования генной и белковой экспрессии. В сочетании с моделированием паракринных эффектов дисфункциональных эндотелиальных клеток при помощи кондиционированной среды указанные модели анализа состояния эндотелия в условиях потока обладают высоким потенциалом для приближения экспериментального сценария дисфункции эндотелия к реальным физиологическим условиям.
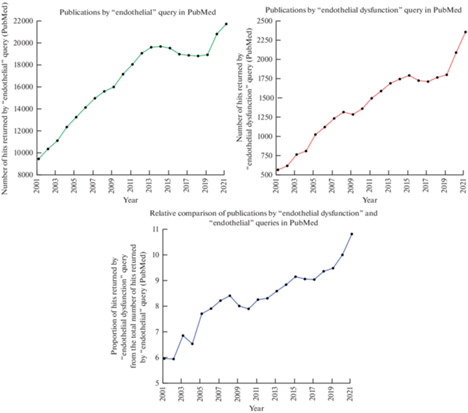
Исследование различных аспектов нормальной и патологической физиологии эндотелия продолжает оставаться одной из самых актуальных тем сердечно-сосудистой биологии, что подтверждается стабильным увеличением количества соответствующих публикаций за последние 20 лет (с 9512 в 2001 до 21748 в 2021 гг.). Особую актуальность при этом имеет изучение дисфункции эндотелия, поскольку доля посвященных этой проблеме публикаций от общего количества публикаций по физиологии эндотелия растет с каждым годом (с 5.99% в 2001 до 10.83% в 2021 гг.). Интерес к дисфункции эндотелия в последние два года, отраженный резким подъемом соответствующей кривой на графике (рис. 1), может быть объясним особенностями течения новой коронавирусной инфекции (COVID-19), которая сопровождается развитием дисфункции эндотелия у существенной доли пациентов.
Рис. 1.
Динамика публикаций по запросам “endothelial” и “endothelial dysfunction”, а также доля публикаций по запросу “endothelial dysfunction” от общего количества публикаций по запросу “endothelial” в базе данных PubMed с 2001 по 2021 гг.
Несмотря на существенное количество публикаций по дисфункции эндотелия в ведущих журналах, печатающих исследования, посвященных сердечно-сосудистым проблемам (Circulation Research, Cardiovascular Research, Arteriosclerosis, Thrombosis и Vascular Biology) за последние 20 лет (671), в которых была показана расшифровка целого ряда механизмов, обеспечивающих нормальную физиологию эндотелия (поддержание эндотелиального фенотипа, поддержание артериальной, венозной или лимфатической спецификации эндотелия, протективное влияние ламинарного потока с высоким напряжением сдвига) или способствующих его патологической активации (атерогенное воздействие турбулентного потока, эндотелиально-мезенхимальный переход, нарушения эндотелиальной механотрансдукции), определение дисфункции эндотелия с 1990-х годов остается статичным. Как в описательных, так и в критических обзорах дисфункция эндотелия определяется как “сниженный синтез, выделение или биодоступность монооксида азота (NO), которое может приводить к нарушению баланса между процессами вазоконстрикции и вазодилатации и сопровождаться провоспалительной и протромботической активацией эндотелия” [1–6]. Более краткие определения дисфункции эндотелия звучат как “нарушение эндотелий-зависимой вазодилатации в ответ на физиологическое воздействие” [7, 8] или “потеря эндотелием своей противовоспалительной, антитромботической и вазодилатирующей способности” [9]. Клинически релевантное определение дисфункции эндотелия звучит как “снижение вазодилатирующего ответа на воздействие эндотелий-зависимого вазодилататора (к примеру, ацетилхолина или брадикинина) или пульсирующего потока (flow-mediated vasodilation)” [10]. Пожалуй, наиболее полное определение дисфункции эндотелия на сегодняшний день принадлежит советскому и американскому патофизиологу М.С. Голигорскому, который, помимо патофизиологически значимого (приводящего к нарушению регуляции сосудистого тонуса гладкомышечными клетками) снижения выделения вазодилататоров и частичной потери эндотелием атромбогенности, предложил также включить в него нарушения барьерной функции эндотелия, сниженную способность к пролиферации и миграции и сниженный ангиогенный потенциал дисфункциональных эндотелиальных клеток, а также выделил клинические последствия дисфункции эндотелия: артериальную гипертензию, макро- и микроангиопатию, развивающуюся вследствие атерогенеза и воспалительной инфильтрации, и микроальбуминурию, возникающую вследствие повышения сосудистой проницаемости [11, 12]. Им были постулированы следующие признаки дисфункциональных эндотелиальных клеток: сниженное выделение биодоступного NO, повышенная способность к адгезии лейкоцитов, накопление холестерина и окисленных липопротеинов низкой плотности, нарушенная передача вазодилатирующих молекул сосудистым гладкомышечным клеткам, повышенный синтез компонентов внеклеточного матрикса (приобретение профибротического фенотипа) и склонность к преждевременному старению и апоптозу [11, 12].
Вместе с тем работа комплексной научной группы по определению ясных диагностических критериев дисфункции эндотелия, основанных на клинико-лабораторных биомаркерах, по собственному заключению ее членов не привела к удовлетворительному результату [13]. Практически все биомаркеры из 62 исследований, оцененных в данной работе, характеризовались отсутствием специфичности для эндотелиальных клеток [13]. К аналогичным результатам пришли и авторы масштабного протеомного исследования по идентификации маркеров, связанных с дисфункцией эндотелия у пациентов с сахарным диабетом [14]. Таким образом, в настоящее время можно констатировать отсутствие ясности в патофизиологическом и клиническом определении дисфункции эндотелия, а также ограниченность классического определения данного понятия нарушением эндотелий-зависимой вазорелаксации. Следует отметить, что некоторые авторы расширяют один из компонентов определения дисфункции эндотелия до “дисфункционального выделения молекул-мессенджеров”, не ограничиваясь NO [15]. Существенный объем новых знаний (с момента данного М.С. Голигорским определения прошло более 15 лет) о физиологии эндотелия требует анализа возможности введения новых критериев дисфункции эндотелия. Данная проблемная статья посвящена критическому обсуждению изучения различных аспектов нормальной и патологической физиологии эндотелия, в том числе важных для надлежащего моделирования гистогематического барьера, в методологическом контексте.
МЕТОДОЛОГИЧЕСКИЕ АСПЕКТЫ МОДЕЛИРОВАНИЯ ДИСФУНКЦИИ ЭНДОТЕЛИЯ
Культивирование эндотелиальных клеток в условиях пульсирующего потока
В отличие от большинства клеточных линий, трехмерное культивирование которых в статических условиях относительно соответствует испытываемым ими биомеханическим воздействиям в организме, эндотелиальные клетки требуют создания однонаправленного пульсирующего потока для адекватного моделирования своей физиологии, в том числе профиля генной и белковой экспрессии. Подходящим решением для культивирования эндотелиальных клеток в условиях потока является система, сочетающая в себе насос для создания пульсирующего потока, трубки-коннекторы, проточные камеры для культивирования клеток и программное обеспечение для регулирования задаваемых параметров потока (в том числе напряжения сдвига – shear stress). Преимуществами использования такой системы являются вариабельность доступных коннекторов и культуральных проточных камер, которые для более физиологичного моделирования базальной мембраны могут быть покрыты коллагеном IV типа. Прозрачность проточных камер позволяет в любое время осуществлять визуализацию жизнеспособных или фиксированных культур эндотелиальных клеток методами фазово-контрастной, эпифлуоресцентной или конфокальной микроскопии, в том числе после их мечения флуоресцентными зондами (для жизнеспособных культур) или антителами, конъюгированными с флуорофорами (для фиксированных культур). Возможность флуоресцентной визуализации жизнеспособных клеточных культур позволяет изучать различные аспекты функционирования лизосом (поскольку лизосомальные зонды являются сенсорами pH, градиент которого часто теряется при фиксации) и митохондрий, не прибегая к необходимости окрашивания данных органелл специфичными антителами. Наличие в системе четырех отдельных модулей, подключаемых к одному насосу и управляемых посредством специализированного программного обеспечения через ноутбук, позволяет проводить работу одновременно с четырьмя экспериментальными группами, что обеспечивает пространство для маневра при планировании дизайна эксперимента. Как правило, анализ производится после временной остановки системы пульсирующего потока и извлечения из нее проточных камер, однако в теории длина коннекторов позволяет визуализировать клетки и в условиях движущегося потока (то есть без остановки системы) в случае необходимости.
Важным техническим нюансом работы с системой пульсирующего потока является равномерность предварительного заселения поверхности всех культуральных проточных камер для создания одинаковых экспериментальных условий во всех камерах и обеспечения сохранности целостности эндотелиального монослоя в процессе эксперимента. Поскольку прочность адгезии клеток является ключевым параметром для обеспечения исходной целостности эндотелиального монослоя и технической валидности проводимого эксперимента, с целью надежной адгезии эндотелиальных клеток к культуральному пластику проточной камеры (или подложки из коллагена IV типа, если таковая в камере имеется) после процедуры рассева следует оставлять аликвотированную в камеры суспензию клеток в течение 12–16 ч в статических условиях. Кроме того, для адаптации эндотелиальных клеток к условиям потока, необходимой для адекватной оценки изменения их генной и белковой экспрессии при каком-либо воздействии, рекомендуется предварительное поточное прекондиционирование адгезированных к культуральному пластику эндотелиальных клеток путем помещения проточной камеры в систему пульсирующего потока с последующим запуском системы (без каких-либо дополнительных воздействий) на 24–48 ч. Критерием адаптации клеток к потоку, косвенно отражающим соответствующие изменения исходной генной и белковой экспрессии, является элонгация (удлинение формы) эндотелиальных клеток.
Следует отметить, что полная конфлюэнтность эндотелиального монослоя препятствует надлежащей элонгации эндотелиальных клеток, что может снижать физиологичность моделирования механотрансдукции (трансформации эндотелиальными клетками механического воздействия, в данном случае потока, в биохимические сигналы), однако в то же время обеспечивает целостность эндотелиального монослоя, не менее важную для адекватной оценки его физиологии. Напротив, неполная конфлюэнтность эндотелиального монослоя позволяет эндотелиальным клеткам в достаточной степени изменить геометрию в соответствии с направлением потока, однако оставляет вероятность также и неполной целостности монослоя в момент запуска системы, что может привести к его размыванию и откреплению пласта адгезированных друг к другу эндотелиальных клеток от культурального пластика камеры. Оптимальной для физиологичного моделирования эндотелиальной механотрансдукции и, следовательно, профиля генной и белковой экспрессии, представляется степень конфлюэнтности эндотелиальных клеток в 75–80%.
С точки зрения молекулярного профилирования следует отметить, что площадь одной проточной камеры обеспечивает ее заполнение количеством эндотелиальных клеток, достаточным для анализа генной экспрессии методом количественной полимеразной цепной реакции после обратной транскрипции (ОТ-кПЦР) в случае лизиса тризолом. При необходимости проведения полнотранскриптомного секвенирования (RNA-seq), жидкостной хроматографии с тандемной масс-спектрометрией (LC-MS) или иммуноблоттинга рекомендуется объединять клеточный лизат с трех проточных камер внутри одной экспериментальной группы в один образец для получения достаточного для анализа количества материала. Иными словами, в случае анализа указанными методами для получения одного образца необходимо проведение трех последовательных запусков системы пульсирующего потока.
При лизисе клеток внутри проточной камеры тризолом (для выделения РНК) или RIPA-буфером (для выделения белка) важным является предварительно забрать из камеры максимальное количество фосфатно-солевого буфера для промывания клеток (оставив его только непосредственно внутри канала камеры). Вследствие различной плотности тризола, RIPA-буфера и фосфатно-солевого буфера тризол и RIPA-буфер практически не смешиваются с фосфатно-солевым буфером и в течение нескольких секунд (пока идет пипетирование) вытесняют его из канала наружу камеры, что позволяет не забирать изначально остающийся в канале фосфатно-солевой буфер во избежание воздушной эмболии канала и соответствующего воздействия на клетки (впрочем, при желании это тем не менее возможно сделать). После 5-минутного лизиса клеток в тризоле или RIPA-буфере рекомендуется еще два раза пропустить собранный из камеры тризол или RIPA-буфер через камеру, чтобы собрать остатки клеточного дебриса.
Как правило, для культивирования эндотелиальных клеток в условиях потока целесообразно использовать полную (содержащую сыворотку) культуральную среду. В частности, такая среда является наиболее подходящей при расселении эндотелиальных клеток в культуральные проточные камеры и при их прекондиционировании в условиях пульсирующего потока. В принципе, такая среда может быть использована и собственно при экспериментах в условиях потока, однако в соответствии с представлениями и желаниями экспериментатора может быть заменена на бессывороточную культуральную среду (поскольку сыворотка представляет из себя стохастическое сочетание гетерогенных молекул). С этой позиции представляют существенный интерес бессывороточные среды с заранее определенной концентрацией факторов роста (defined medium). Такие среды позволяют и повысить жизнеспособность эндотелиальных клеток в процессе эксперимента и минимизировать стохастичность влияния отдельных компонентов гетерогенной сыворотки на результаты. В настоящее время на рынке представлен широкий выбор сред для культивирования первичных эндотелиальных клеток человека, в число которых входят и сывороточная, и бессывороточная, и бессывороточная с факторами роста среда. Личный опыт авторов данной статьи также позволяет говорить о взаимозаменяемости сред различных производителей и соответствующих первичных эндотелиальных культур.
Что касается самих культур эндотелиальных клеток, то следует подчеркнуть важность использования именно первичных, а не иммортализованных культур, физиология которых необратимо изменена вне зависимости от способа иммортализации. При этом и с исследовательской, и с финансовой стороны совершенно целесообразно приобретение коммерческих культур эндотелиальных клеток у соответствующих производителей. Данные культуры обладают способностью к быстрой пролиферации (скорость которой, безусловно, варьирует в зависимости от донора и от клеточной линии, однако в ряде случаев не уступает иммортализованным эндотелиальным клеткам), неприхотливы в культивировании и выдерживают множественное пассажирование (при разморозке на 2-м пассаже их удавалось успешно культивировать и до 12-го пассажа без существенного замедления пролиферации). Кроме того, такой подход к источнику эндотелиальных клеток для экспериментов часто позволяет прицельно выбрать линию клеток именно того сосуда, патология которого исследуется концептуально. В частности, для моделирования дисфункции эндотелия при атеросклерозе рекомендуются сравнительные и одновременные эксперименты на любой из линий атерочувствительных артерий (к примеру, коронарной артерии, сонной артерии или аорты) и линии атерорезистентной артерии (традиционно в качестве такой артерии рассматривается внутренняя грудная артерия). Следует подчеркнуть необходимость использования артериальных эндотелиальных клеток для исследования артериальных патологий, а венозных эндотелиальных клеток – для изучения венозных патологий. Отдельно стоит заострить внимание на изучении нормальной и патологической физиологии эндотелиальных клеток, заселяющих новые биоматериалы для производства тканеинженерных медицинских изделий или их готовые прототипы (к примеру, сосудистые протезы малого диаметра). Для моделирования (пато)физиологии эндотелия в таких условиях также следует применять те эндотелиальные клетки, которые соответствуют анатомической локализации медицинского изделия (к примеру, в том случае, если сосудистые протезы малого диаметра планируется использовать в качестве кондуитов для коронарного шунтирования, эксперименты по изучению физиологии эндотелия на таких кондуитах или биоматериалах для них следует ставить на первичных эндотелиальных клетках коронарной или внутренней грудной артерии).
Культивирование эндотелиальных клеток в статических условиях
Несмотря на всю привлекательность культивирования эндотелиальных клеток в условиях пульсирующего потока с точки зрения объективного моделирования их нормальной физиологии, достаточно большое количество экспериментальных приложений требует значительной клеточной массы, невозможной к получению на малой культуральной площади проточных камер. К примеру, анализ паракринных эффектов эндотелиальных клеток в обязательном порядке включает в себя измерение содержания провоспалительных цитокинов, концентрация которых должна быть достаточна для их детекции. Объем культуральной среды, загружаемой в один модуль пульсирующего потока (на примере системы, с которой работали авторы), составляет 15 мл, а площадь одной культуральной проточной камеры составляет ≈2.5–3.0 см2, что эквивалентно площади 8–9 лунок 96-луночного плоскодонного планшета или 0.25–0.3 × 106 (250–300 тысяч) клеток. Соответственно, количество культивируемых в системе пульсирующего потока клеток не соответствует объему среды с точки зрения даже высокоточного и количественного иммуноферментного анализа, не говоря уже о менее чувствительных скрининговых полуколичественных методах, таких, как дот-блоттинг. Искусственное испарение культуральной среды для концентрирования содержащихся в них молекул сопряжено с высоким риском контаминации, которая делает невозможной дальнейший объективный анализ. Кроме того, моделирование паракринных эффектов эндотелиальных клеток требует добавления кондиционированной (то есть инкубированной с дисфункциональными эндотелиальными клетками) среды к интактным культурам, что опять же представляется малореальным в условиях потока вследствие несоответствия количества клеток объему среды.
Поэтому эксперименты, результат которых напрямую зависит не от содержимого эндотелиальных клеток (к примеру, транскрипты, внутриклеточные белки и рецепторы), а от их секретома (совокупности молекул, выделяемых ими в микроокружение – к примеру, цитокинов, про- и антиангиогенных молекул), целесообразно проводить в статических условиях. При моделировании паракринных эффектов культур эндотелиальных клеток следует помнить о важности применения бессывороточной культуральной среды (как вариант – бессывороточной культуральной среды с определенной концентрацией факторов роста), поскольку сыворотка содержит ксеногенные (гетерологические) внеклеточные везикулы, а также цитокины и про-/антиангиогенные молекулы, которые могут взаимодействовать с поликлональными антителами для иммуноферментного анализа или дот-блоттинга. Использование бессывороточной культуральной среды с определенной концентрацией факторов роста способствует жизнеспособности клеток, подвергшихся воздействию триггера дисфункции эндотелия (что позволяет осуществлять более физиологичное моделирование в контексте последующего добавления кондиционированной среды к интактным эндотелиальным клеткам), однако может затруднять объективную оценку изменений в концентрации про- и антиангиогенных молекул в культуральной среде методами иммуноферментного анализа и дот-блоттинга, поскольку в их число также входят и факторы роста. Как правило, кондиционированная среда переносится к интактным культурам эндотелиальных клеток после не более чем 24 ч культивирования, поскольку далее даже в контрольных культурах эндотелиальных клеток (без добавления стимула дисфункции эндотелия) начинаются изменения физиологии, связанные с сывороточной депривацией. Кроме того, для объективной оценки ответа интактных культур эндотелиальных клеток на добавление кондиционированной среды (в том числе для полуколичественного и количественного анализа изменений их секреторного профиля) требуется их 24-часовая инкубация с кондиционированной средой, а истощение культуральной среды при ее добавлении к монослою эндотелиальных клеток (как дисфункциональных, кондиционирующих среду, так и интактных, инкубируемых уже с кондиционированной средой) происходит как раз через 48 ч (24 ч инкубации с дисфункциональными клетками и 24 ч инкубации с интактными клеточными культурами).
Также культивирование эндотелиальных клеток в статических условиях применимо в том случае, когда эксперимент подразумевает их фракционирование на органеллы с целью разделения белков различной локализации (к примеру, ядерных, цитозольных и митохондриальных или лизосомальных белков). Одной из целей такого экспериментального дизайна может быть дальнейшая жидкостная хроматография с тандемной масс-спектрометрией, которая требует предварительного обогащения анализируемых образцов для максимизации суммарного количества идентифицируемых клеточных белков. В этом случае для выделения достаточного количества белка для анализа требуется существенный объем клеточной массы, невозможный к получению на ограниченной площади культуральных проточных камер.
Исследование биологии эндотелия in vivo
Помимо экспериментов на культурах эндотелиальных клеток в условиях однонаправленного пульсирующего потока in vitro, изучение патологических эффектов и механизмов действия их пусковых факторов (триггеров) также подразумевает исследования in vivo на лабораторных животных. Наиболее подходящими моделями для этого можно считать мелких лабораторных животных (крыс и мышей), поскольку относительная простота их содержания и размножения позволяет проводить масштабные эксперименты с достаточным количеством биологических повторов (репликат) для обеспечения достаточной степени доказательности экспериментов.
Рассматривая возможные животные модели для изучения дисфункции эндотелия, следует подчеркнуть, что для каждого эксперимента может потребоваться индивидуализация экспериментальной модели (к примеру, определенные линии трансгенных мышей или крыс). Данный постулат особенно применим к анализу влияния различных коморбидных (сопутствующих) патологий, основными из которых являются сахарный диабет, хроническая болезнь почек и хроническая обструктивная болезнь легких. Каждое из данных состояний имеет свои характерные, классические и относительно новые, животные модели для исследования состояния органов и тканей, в том числе эндотелия. Если же рассматривать сценарий отсутствия выраженных коморбидных патологий, наиболее релевантный научной задаче поиска способов фармакологического и нефармакологического вмешательства для коррекции дисфункции эндотелия, стоит выделить три основных животных модели. Первой из них являются нормолипидемические и нормотензивные крысы Wistar, позволяющие анализировать состояние эндотелия в отсутствие каких-либо дополнительных факторов сердечно-сосудистого риска, второй – крысы с врожденной артериальной гипертензией (к примеру, линии SHR или НИСАГ), третьей – гиперлипидемические (к примеру, ApoE- или LDLR-нокаутные) мыши, с рождения склонные к гипер/дислипидемии, отложению липидов в крупных артериях и развитию атеросклероза. Использование трех данных моделей позволяет изучать развитие дисфункции эндотелия в отсутствие модифицируемых факторов сердечно-сосудистого риска, а также при двух наиболее распространенных и важных факторах сердечно-сосудистого риска – артериальной гипертензии и гипер/дислипидемии. Поскольку данные состояния являются скорее неизбежно ассоциированными с возрастом процессами, чем сопутствующими заболеваниями, целесообразно рассматривать данные животные модели как некий “нормальный патофизиологический сценарий”, обусловливающий релевантность исследования дисфункции эндотелия в дополнение к нормофизиологическому сценарию, наблюдаемому у крыс Wistar.
Наиболее подходящим сосудом для анализа дисфункции эндотелия у крыс и мышей in vivo представляется аорта, поскольку: 1) данный сосуд относится к артериальному руслу (что позволяет связывать дисфункцию эндотелия с развитием артериальных патологий, главным образом атеросклероза); 2) содержит неразрывно связанные сегменты с атерогенным турбулентным кровотоком (дуга и бифуркация аорты) и с атеропротективным ламинарным кровотоком (нисходящая аорта); 3) обладает максимальным диаметром среди всех остальных сосудов организма, что позволяет собрать достаточное количество материала для анализа. Из этого следует, что при изучении дисфункции эндотелия у лабораторных животных целесообразно исследовать состояние эндотелия в сосудистых сегментах как с турбулентным, так и с ламинарным кровотоком. Если в качестве сегмента с ламинарным кровотоком единственным возможным выбором является нисходящая аорта в силу своей длины и простоты своей геометрии, то для выбора сегмента с турбулентным кровотоком существуют два варианта (как указано выше, дуга и бифуркация аорты). Сравнивая данные сосудистые сегменты, стоит отметить крайне малые размеры собственно раздваивающегося фрагмента бифуркации (ее части с истинно турбулентным кровотоком) даже в сравнении с обладающей относительно небольшими размерами дугой аорты. Поэтому представляется разумным предпочесть для анализа состояния эндотелия при турбулентном кровотоке именно дугу аорты.
Анализ состояния эндотелия в дуге аорты и нисходящей аорте in situ (то есть в нативном состоянии, без дополнительного воздействия на эндотелиальные клетки) может быть выполнен как минимум тремя способами: при помощи быстрого промывания указанных аортальных сегментов тризолом с целью сбора эндотелиального лизата для последующего выделения РНК и профилирования методом ОТ-кПЦР, посредством аналогичного промывания RIPA-буфером с коктейлем ингибиторов протеаз и фосфатаз или, используя продольную диссекцию цельного сегмента и его окрашивание первичными и вторичными флуоресцентно меченными антителами с последующей en face-визуализацией, подразумевающей микроскопию уложенной интимой вверх аорты без приготовления срезов.
Сбор эндотелиального лизата тризолом или RIPA-буфером целесообразно проводить при помощи инсулинового шприца (в силу малого диаметра аорты крыс и мышей) после тщательного промывания физиологическим раствором хлорида натрия для очищения от крови. Как правило, для выделения достаточного количества РНК или белка из сегментов аорты крысы достаточно однократного, но медленного шприцевого промывания тризолом или RIPA-буфером (хотя также оправдан вариант с трехкратным относительно быстрым промыванием). При этом следует отметить, что для достижения достаточной для иммуноблоттинга концентрации белка применительно к сегментам крысиной аорты объем RIPA-буфера не должен превышать 200 мкл для дуги аорты и 300 мкл для нисходящей аорты; вероятно, он должен быть еще меньшим для обладающих меньшим диаметром аорт мышей. Гистологическим обоснованием применения такой методики у крыс и мышей является то, что их интима в норме состоит исключительно из монослоя эндотелиальных клеток, не содержит каких-либо других клеточных популяций и отделена от содержащей сосудистые гладкомышечные клетки мышечной оболочки внутренней эластической мембраной. Безусловно, данная методика не является абсолютно чистой и сопряжена с некоторой степенью контаминации лизатом гладкомышечных клеток (что подтверждается выявлением альфа-актина гладких мышц при помощи иммуноблоттинга и соответствующих транскриптов при ОТ-кПЦР), однако, во-первых, она может быть предварительно оптимизирована для каждого конкретного эксперимента, а во-вторых, в случае такой контаминации целесообразно внесение поправки на количество белка или транскриптов альфа-актина гладких мышц в контрольной и экспериментальной группах (по аналогии с принципом ΔCt при анализе данных ОТ-кПЦР).
Особенностью иммуноблоттинга белков крыс является то, что молекулярные массы детектируемых белков часто не совпадают с таковыми у человека, которые используются производителями антител в качестве референтных. Помимо того, что данная особенность может вызывать вопросы у требовательных к техническим деталям рецензентов, она ограничивает одновременное использование двух и более первичных антител, иногда практикуемое с целью экономии ресурсов при анализе белков человека в случае предварительного тестирования таких антител, расхождения их по молекулярным массам и отсутствия их неспецифического связывания с нецелевыми белками или их фрагментами. Тем не менее, приведенные ограничения не умаляют технической валидности иммуноблоттинга лизата эндотелия крыс.
Еще одним упомянутым выше способом анализа является конфокальная микроскопия эндотелия en face. Необходимость использования именно конфокальной микроскопии обусловлена сильным фоновым свечением от нижележащей внутренней эластической мембраны в зеленом флуоресцентном канале, что требует отсечения отдельных оптических срезов для визуализации окрашенных флуоресцентно меченными антителами эндотелиальных клеток. Вероятно, с целью минимизации аутофлуоресцентного зеленого свечения следует использовать сочетание Alexa Fluor 555- и Alexa Fluor 647-меченных вторичных антител, светящихся в красном и дальнем красном флуоресцентном каналах. Хотя данный способ анализа состояния эндотелия является допустимым, его доказательная мощность в большинстве случаев (за исключением оценки эндотелиально-мезенхимального перехода) представляется ограниченной в силу качественно-комплементарного характера полуколичественного анализа конфокальных изображений; кроме того, его использование приводит к довольно нерациональному использованию имеющейся ткани, поскольку достаточно большая часть аортального сегмента уходит на окрашивание всего лишь одной комбинацией антител.
МЕХАНИЗМЫ ДИСФУНКЦИИ ЭНДОТЕЛИЯ ПРИ РАССМОТРЕНИИ ФОРМУЛИРОВКИ ЕЕ КРИТЕРИЕВ
Провоспалительная активация эндотелия
Основными критериями провоспалительной активации эндотелия являются адгезия лейкоцитов к эндотелиальным клеткам вследствие связывания лейкоцитарных рецепторов с соответствующими рецепторами эндотелиальных клеток и повышенное выделение эндотелиальными клетками провоспалительных цитокинов [16–18]. К лейкоцитарным рецепторам, ответственным за прикрепление лейкоцитов к эндотелиальным клеткам, относятся VLA-4/CD49d (связь с VCAM1 эндотелиальных клеток), Mac-1 (комплекс CD11b и CD18, связь с ICAM1 эндотелиальных клеток), LFA-1/CD11a (связь с ICAM1 эндотелиальных клеток) и PSGL-1/CD162 (связь с Е-селектином эндотелиальных клеток) [19–22].
Объективный анализ адгезии клеток мононуклеарной фракции крови к эндотелию, частично отражающей провоспалительную активацию эндотелия, невозможен при культивировании эндотелиальных клеток в статических условиях по причине пассивного оседания и неминуемой адгезии моноцитов и лимфоцитов как к эндотелиальным клеткам, так и к культуральному пластику даже после многих (7–10) стадий интенсивной отмывки. Соответственно, даже в случае полной конфлюэнтности эндотелиального монослоя является невозможным отделить истинную адгезию клеток мононуклеарной фракции крови к эндотелию от “псевдоадгезии”, которая обусловлена механическим прикреплением осевших моноцитов и лимфоцитов к нижележащей поверхности независимо от ее покрытия клетками. В то же время добавление клеток мононуклеарной фракции крови в систему для культивирования эндотелиальных клеток в условиях пульсирующего потока (в количестве 1.25 × 105 клеток на 1 мл, что эквивалентно 1.5 × 106 клеток на модуль системы потока) позволяет исключить такую “псевдоадгезию” под действием силы тяжести и обеспечить достаточное количество адгезировавшихся к эндотелиальному монослою моноцитов и лимфоцитов. Визуализацию при таком методе анализа можно проводить при помощи флюоресцентной микроскопии на инвертированном микроскопе, используя любой альтернативный ядерный краситель (к примеру, CellTracker Green) для мечения клеток мононуклеарной фракции крови перед их добавлением в систему пульсирующего потока и стандартный ядерный краситель (к примеру, Hoechst 33342) для мечения клеток внутри камеры после остановки системы и ее извлечения непосредственно перед визуализацией. В таком дизайне клетки мононуклеарной фракции крови дают и синее, и зеленое свечение (если использовать CellTracker Green), а эндотелиальные клетки – исключительно синее свечение. Для большей наглядности рекомендуется сочетать фазово-контрастную и флуоресцентную визуализацию на инвертированном микроскопе.
Хотя анализ адгезии клеток мононуклеарной фракции крови к эндотелиальным клеткам и является функциональным анализом с высокой степенью доказательности провоспалительной активации эндотелия, современные критерии доказательности патофизиологических экспериментов (отражаемые требованиями соответствующих научных журналов) также включают в себя измерение экспрессии генов и белков, ответственных за адгезию лейкоцитов к эндотелиальным клеткам. К таким генам целесообразно относить VCAM1, ICAM1, SELE и SELP, кодирующие сосудистую молекулу клеточной адгезии, межклеточную молекулу клеточной адгезии, E-селектин и P-селектин соответственно. Оптимальным методом для измерения экспрессии данных генов являются различные варианты полимеразной цепной реакции (к примеру, ОТ-кПЦР или цифровая капельная ПЦР), а для измерения белков – иммуноблоттинг с использованием соответствующих антител (при этом количество молекул VCAM1 и ICAM1 существенно превышает количество E-селектина, что позволяет использовать именно их в качестве маркеров провоспалительной активации). Альтернативной мерой измерения данных белков может быть полуколичественный анализ полученных при конфокальной микроскопии изображений, однако множественное использование такого метода в различных экспериментах не привело к удовлетворительным результатам. В отношении молекул клеточной адгезии рекомендуется использовать конфокальную микроскопию исключительно для качественного анализа их локализации в клетке (к примеру, в цитозоле или на плазматической мембране), но не для количественного анализа их экспрессии. Другим вариантом включения подобного варианта анализа дисфункции эндотелия в доказательную базу экспериментальной работы является обязательное сочетание результатов полуколичественного анализа конфокальных изображений с данными ОТ-кПЦР и иммуноблоттинга. Таким образом, полуколичественный анализ конфокальных изображений используется либо для изучения качественных феноменов (локализация объекта интереса внутри клетки, в том числе в ассоциации с конкретными органеллами), либо в качестве комплементарного (дополнительного), но не самостоятельного аналитического метода.
В отношении повышенного выделения эндотелиальными клетками провоспалительных цитокинов следует отметить, что их спектр не столь велик и включает в себя шесть основных молекул: интерлейкин (ИЛ)-6, ИЛ-8, моноцитарную хемоаттрактантную молекулу 1 (MCP-1/CCL2), CXCL1 (связанный с ростом онкоген, GRO-α), фактор ингибирования миграции макрофагов (MIF) и ингибитор активатора плазминогена (PAI-1, серпин E1) [23, 24]. Намного шире спектр выделяемых эндотелиальными клетками про- и антиангиогенных молекул [23, 24], при этом к про-ангиогенным молекулам относятся ангиопоэтин-1 [25, 26], растворимая форма рецептора к сосудистому эндотелиальному фактору роста (VEGFR2) [27, 28], ангиогенин [29, 30], эпидермальный фактор роста (EGF) [31, 32], плацентарный фактор роста (PlGF) [33, 34], тромбоцитарный фактор роста (вернее, его субъединицы PDGF-AA и PDGF-BB) [35, 36], основной фактор роста фибробластов (bFGF-)[37, 38], эндотелин-1 [39, 40], пентраксин 3 [41, 42], дипептидилпептидаза 4 [43, 44], CD105/эндоглин [45, 46], фактор роста гепатоцитов (HGF) [47, 48], белки, связывающие инсулиноподобный фактор роста – IGFBP-1 [49, 50], IGFBP-2 [51, 52] и IGFBP-3 [53, 54], CXCL16 [55, 56], трансформирующий фактор роста (TGF-β1) [57, 58], урокиназный активатор плазминогена (uPA) и соответствующий растворимый рецептор (uPAR) [59, 60], матриксная металлопротеиназа-1 (ММП-1) [60, 61], ИЛ-6 [62, 63], ИЛ-8 [64, 65] и MCP-1/CCL2 [66, 67], а к антиангиогенным – ангиопоэтин-2 [68, 69], тромбоспондин-1 [70, 71], эндостатин [72, 73], тканевые ингибиторы металлопротеиназ (ТИМП-1 [74, 75] и ТИМП-2 [76, 77]) и ингибитор активатора плазминогена (PAI-1) [78, 79].
Методологически измерение цитокинов, про- и антиангиогенных факторов в культуральной среде с эндотелиальных клеток целесообразно осуществлять в первую очередь посредством скринингового полуколичественного профилирования при помощи дот-блоттинга (в настоящее время на рынке представлены специализированные наборы, позволяющие определение всех указанных выше цитокинов и абсолютного большинства из вышеуказанных про- и антиангиогенных белков). Скрининговое измерение молекул при помощи дот-блоттинга позволяет определить те из них, что являются дифференциально экспрессированными между экспериментальной и контрольной группами, и далее провести прицельный количественный анализ различий при помощи высокоточного иммуноферментного анализа. Если возможности провести дот-блоттинг нет (к примеру, вследствие отсутствия хемилюминесцентного сканера), для оценки провоспалительной активации эндотелиальных клеток следует измерить концентрацию ИЛ-6, ИЛ-8 и MCP-1/CCL2 в культуральной среде методом иммуноферментного анализа. Перед дот-блоттингом или иммуноферментным анализом следует провести низкоскоростное центрифугирование собранной среды (1000–3000 g) с целью очистки от клеточного дебриса.
Помимо собственно оценки выделения эндотелиальными клетками в микроокружение провоспалительных цитокинов как обязательного компонента провоспалительной активации эндотелия, для оценки патофизиологической значимости изменений секреторного профиля дисфункциональных эндотелиальных клеток также рекомендуется проведение экспериментов с кондиционированной (то есть инкубированной с эндотелиальными клетками и выбранным триггером дисфункции эндотелия) бессывороточной средой, которую не позднее чем через 24 ч следует добавить к интактным культурам эндотелиальных клеток не более чем на 24 ч для оценки ее патологического воздействия в сравнении с аналогичной средой, инкубированной с эндотелиальными клетками от того же донора без триггера дисфункции эндотелия. В том случае, если стоит задача по идентификации ответственного за патогенные эффекты паракринного компонента, после изначального эксперимента с полной кондиционированной средой целесообразно проведение повторного эксперимента с фракционированием кондиционированной среды (посредством ультрацентрифугирования) на внеклеточные везикулы и не содержащую внеклеточных везикул кондиционированную среду с их последующим раздельным добавлением к интактным культурам эндотелиальных клеток. Перед переносом кондиционированной среды от дисфункциональных эндотелиальных клеток к интактным следует провести ее низкоскоростное центрифугирование с целью очистки от клеточного дебриса для последующего объективного анализа паракринного воздействия кондиционированной среды.
Собственные исследования нашей группы показали, что добавление в систему пульсирующего потока кальципротеиновых частиц (также называемых в русскоязычной терминологии кальций-фосфатными бионами) приводит к их интернализации эндотелиальными клетками уже в течение часа, стабильно вызывает гибель небольшой, но статистически значимой доли (1–2%) эндотелиальных клеток в течение 4 ч вследствие патологической пермеабилизации лизосом и митохондрий и стимулирует выделение основных эндотелиальных провоспалительных цитокинов, указанных выше – ИЛ-6, ИЛ-8 и MCP-1/CCL2 [80, 81]. Это позволило рассматривать кальципротеиновые частицы как модельный триггер дисфункции эндотелия, пригодный для анализа основных патофизиологических звеньев ее развития.
Последующие эксперименты показали, что 4-часовое инкубирование первичных эндотелиальных клеток коронарной артерии и внутренней грудной артерии с кальципротеиновыми частицами вызывает адгезию мононуклеарных клеток периферической крови к артериальным эндотелиальным клеткам в потоке, которая сопровождается повышением уровня указанных выше молекул клеточной адгезии VCAM1 и ICAM1 [82–84]. Кроме того, было выявлено, что как полная, так и очищенная от внеклеточных везикул кондиционированная среда, полученная от первичных эндотелиальных клеток коронарной артерии в результате добавления к ним кальципротеиновых частиц, приводила к повышению экспрессии генов молекул клеточной адгезии VCAM1, ICAM1, SELE и SELP в сочетании с повышением экспрессии генов цитокинов IL6, CXCL8, CCL2, CXCL1 и MIF, увеличению экспрессии рецепторов эндотелиальных клеток к лейкоцитам VCAM1 и ICAM1, а также усиленному выделению ИЛ-6, ИЛ-8 и MCP-1/CCL2 при ее добавлении к интактным культурам эндотелиальных клеток коронарной артерии от этого же донора. В то же время добавление внеклеточных везикул от обработанных кальципротеиновыми частицами эндотелиальных клеток не приводило к таким эффектам, указывая на ведущую роль растворимых факторов в развитии паракринной провоспалительной активации эндотелия. Поскольку сдвига секретируемых эндотелиальными клетками в окружающую среду про- и антиангиогенных факторов выявлено не было, было предположено, что из растворимых факторов главную патогенетическую роль в данном патофизиологическом сценарии играют именно провоспалительные цитокины (ИЛ-6, ИЛ-8, MCP-1/CCL2).
Таким образом, первым и самым главным критерием дисфункции эндотелия следует считать его провоспалительную активацию, которая может быть определена по трем критериям: 1) повышенной адгезии лейкоцитов к эндотелиальным клеткам при их совместном культивировании в условиях пульсирующего потока, сопровождающейся повышением экспрессии белков VCAM1 и ICAM1, а также соответствующих генов; 2) повышенному выделению трех основных эндотелиальных провоспалительных цитокинов – ИЛ-6, ИЛ-8 и MCP-1/CCL2 в микроокружение; 3) доказанной патогенности паракринного воздействия эндотелиальных клеток (к примеру, способностью кондиционированной ими среды вызывать развитие дисфункции эндотелия в интактных клеточных культурах).
Нарушения эндотелиальной механотрансдукции
Еще одним аспектом патофизиологии эндотелия, невозможным к изучению в статических условиях культивирования, являются нарушения эндотелиальной механотрансдукции (трансформации механического воздействия, в данном случае потока, в биохимические сигналы). Механотрансдукция играет чрезвычайно важную роль в физиологии эндотелия in vivo, что отражается восприимчивостью сосудистых сегментов с турбулентным кровотоком (к примеру, дуга аорты) к атеросклерозу в сравнении с сосудистыми сегментами с ламинарным кровотоком (к примеру, нисходящая аорта) [16–18]. Помимо множества механосенсоров, в число которых входят рецепторы VEGFR2 [85, 86], Tie2 [87] и PECAM-1/CD31 [88, 89], интегрины и связанные с ними очаги адгезии [90–93], белки межклеточных контактов VE-кадгерин и окклюдин [85, 89, 94], активируемые растяжением ионные каналы [95, 96], трансмембранные протеогликаны [97], G-белок-связанные рецепторы [98], гетеротримерные G-белки [99, 100], гликокаликс [101, 102], кавеолин-1 и кавеолы [103, 104], первичные реснички [105, 106], промежуточные филаменты [107, 108] и актиновый цитоскелет [109–111], передача сигнала от механосенсоров осуществляется через ряд специализированных транскрипционных факторов: KLF2 [112–114], KLF4 [115–117], NRF2 [118, 119], YAP и TAZ [120, 121]. При этом три из данных факторов (KLF2, KLF4 и NRF2) являются атеропротективными [113–116, 118, 119, 122–131], а два (YAP и TAZ) – атерогенными [120, 121, 131–134].
Функция атерогенных транскрипционных факторов YAP и TAZ в значительной степени регулируется на посттрансляционном уровне посредством фосфорилирования (которое провоцирует их секвестрирование в цитозоле и деградацию данных белков посредством убиквитинирования с последующим транспортом в протеасомы), что отражает необходимость оценки не только общей, но и фосфорилированной формы данных молекул посредством иммуноблоттинга с их последующим сравнением относительно друг друга. Другим вариантом анализа активности атерогенных транскрипционных факторов YAP и TAZ могла бы быть оценка относительного содержания общей и фосфорилированной формы данных факторов в цитозоле и ядре методом иммуноблоттинга после фракционирования клеток. К сожалению, такой способ, хотя и является объективным, требует значительного количества клеточной массы, невозможной к получению в культуральных проточных камерах, а в статических условиях оценить нарушения эндотелиальной механотрансдукции возможным не представляется. Попытки оценки сравнительной интенсивности флуоресцентного сигнала общей и фосфорилированной формы YAP и TAZ в цитозоле и ядре при иммуноокрашивании также (по собственному опыту авторов) не привели к объективному результату; тем не менее, нельзя исключать их комплементарной (но не самостоятельной) доказательной мощности. При оценке нарушений эндотелиальной механотрансдукции, сопровождающей дисфункцию эндотелия, следует помнить об иррелевантности измерения генной экспрессии в том случае, если функциональность белка главным образом управляется посредством его посттрансляционных модификаций. К примеру, экспрессия генов YAP1 и WWTR1, кодирующих транскрипционные факторы YAP1 и TAZ соответственно, никак не связана с тем, подвергаются ли далее данные белки фосфорилированию и деградации в протеасомах.
Вполне естественно, что при отсутствии внешних биомеханических воздействий, за исключением контакта эндотелиальных клеток друг с другом, даже при полностью конфлюэнтном монослое надлежащее моделирование эндотелиальной механотрансдукции представляется затруднительным. В то же время при культивировании в системе пульсирующего потока соблюдаются сразу несколько условий для физиологичного моделирования эндотелиальной механотрансдукции: постоянное воздействие однонаправленного пульсирующего потока с регулируемым напряжением сдвига, постепенная элонгация эндотелиальных клеток вдоль направления потока и обеспечение контактов эндотелиальных клеток внутри монослоя друг с другом за счет их элонгации даже при изначально неполной конфлюэнтности.
Эксперименты с модельным триггером дисфункции эндотелия – кальципротеиновыми частицами показали, что интернализация КФБ приводила к снижению экспрессии механочувствительных атеропротективных транскрипционных факторов KLF2 и KLF4, также снижая степень фосфорилирования атерогенного транскрипционного фактора YAP1 и приводя таким образом к его активации, подавляемой в физиологических условиях. При этом данные эффекты наблюдались как в культурах первичных эндотелиальных клетках коронарной и внутренней грудной артерии, так и в эндотелии различных аортальных сегментов крыс (нисходящая аорта и дуга аорты). Особенно выражены указанные нарушения были в характеризующейся турбулентным кровотоком дуге аорты, где белок Yap1 был гипофосфорилирован в обоих изученных участках (серин-127 и серин-397), и также был гипофосфорилирован белок Taz, что отражало резистентность этих двух атерогенных транскрипционных факторов к деградации. Таким образом, и инкубированные с кальципротеиновыми частицами культуры эндотелиальных клеток, и эндотелий аорты подвергшихся внутривенному введению кальципротеиновых частиц крыс имели “молекулярные отпечатки” нарушенной механотрансдукции.
При попытке формулировки критериев дисфункции эндотелия следует упомянуть об отсутствии какого-либо функционального анализа (аналогичного анализу адгезии лейкоцитов к эндотелиальным клеткам) или какого-либо выделяемого в микроокружение биомаркера, позволяющего сделать однозначный вывод о развитии нарушений эндотелиальной механотрансдукции. Тем не менее, ее признаками при молекулярном профилировании эндотелиальных клеток могут являться снижение экспрессии атеропротективных транскрипционных факторов KLF2, KLF4 и NRF2 и кодирующих их генов (KLF2, KLF4, NFE2L2), а также гипофосфорилирование атерогенных транскрипционных факторов YAP1 и TAZ, приводящее к их активации. Вопрос о том, какие из этих изменений являются наиболее патофизиологически значимыми, является дискуссионным, поэтому представляется разумным определять нарушения эндотелиальной механотрансдукции как сочетание снижения экспрессии атеропротективных транскрипционных факторов и активирующего гипофосфорилирования атерогенных транскрипционных факторов. В соответствии с таким определением нарушение эндотелиальной механотрансдукции может быть обозначено как один из главных критериев дисфункции эндотелия.
Эндотелиально-мезенхимальный переход
Не менее важным аспектом дисфункции эндотелия является эндотелиально-мезенхимальный переход, сопровождающий развитие атеросклероза и способствующий потере ориентации эндотелиальных клеток, дезинтеграции межклеточных контактов, повышению экспрессии ими молекул клеточной адгезии и переходу на синтетический фенотип [135–139]. Эндотелиально-мезенхимальный переход сопровождается постепенным снижением экспрессии эндотелиальных маркеров (PECAM-1/CD31, VE-кадгерин/CD144, VEGFR2/KDR/CD309) и повышением экспрессии мезенхимальных маркеров (N-кадгерин/CD325, альфа-актин гладких мышц, виментин, кальпонин, SM22α, фибробласт-ассоциированный белок, фибробласт-специфичный белок) и регулируется транскрипционными факторами Snail, Slug, Twist1, Gata4 и Zeb1 [137, 139–141]. Экспрессия транскрипционного фактора Gata4 в эндотелиальных клетках человека вызывает вопросы, в то время как остальные четыре указанных транскрипционных фактора обнаруживаются в них в значительном количестве как на уровне транскриптов, так и на уровне белков.
Наиболее важными из них являются Snail и Slug, которые представляют собой два эволюционно родственных транскрипционных фактора (о чем говорят названия их генов SNAI1 и SNAI2), вследствие чего при иммуноблоттинге могут быть детектированы одним из представленных на рынке антител, распознающим характерный для обоих данных транскрипционных факторов эпитоп. Впрочем, данное антитело является поликлональным, в то время как современные требования к специфичности иммуноблотов все же рекомендуют использование моноклональных антител для минимизации их неспецифического связывания. Тем не менее, различие молекулярных масс транскрипционных факторов эндотелиально-мезенхимального перехода в теории позволяет одновременно оценивать уровень экспрессии Snail/Slug (или Twist1), которые, как правило, локализуются на молекулярных массах до 50 кДа, и транскрипционного фактора Zeb1, который может определяться на молекулярной массе в 200 кДа.
Помимо иммуноблоттинга, объективная оценка развития эндотелиально-мезенхимального перехода также может быть проведена при помощи анализа генной экспрессии (поскольку активность транскрипционных факторов или маркеров данного процесса не зависит от посттрансляционных модификаций). При этом в первую очередь наблюдается изменение экспрессии генов транскрипционных факторов, управляющих эндотелиально-мезенхимальным переходом, а именно SNAI1, SNAI2, TWIST1 и ZEB1, хотя дифференциальная экспрессия каждого из них существенно зависит от типа сосудистого эндотелия (клеточной линии в случае экспериментов in vitro и сосудистого сегмента в случае экспериментов in vivo). Изменение экспрессии самих маркеров эндотелиально-мезенхимального перехода происходит уже значительно позже и служит сигналом идущей трансдифференцировки. Наиболее убедительным доказательством такой трансдифференцировки, вероятно, является иммунофлуоресцентное окрашивание, которое качественно свидетельствует о приобретении экспрессии эндотелиальными клетками мезенхимальных маркеров (при условии аналогичного доказательства их отсутствия на начальном этапе эксперимента). Классическим примером такого окрашивания может служить сочетанное окрашивание на маркер эндотелиальных клеток PECAM-1/CD31 и маркер сосудистых гладкомышечных клеток альфа-актин гладких мышц, а также на другой маркер эндотелиальных клеток VE-кадгерин/CD144 и маркер мезенхимальных клеток N-кадгерин/CD325. Актуальность иммунофлуоресцентного окрашивания для определения эндотелиально-мезенхимального перехода, вероятно, также делает возможным доказательство его реализации in vivo при диссекции цельного сегмента аорты, его окрашивании первичными и вторичными флуоресцентно меченными антителами и последующей en face-визуализации (интимой вверх). Такой способ позволит отличить клетки с выраженным эндотелиальным фенотипом (CD31+αSMA– или CD144+CD325–) от клеток со смешанным (CD31+αSMA+ или CD144+CD325+) или чисто мезенхимальным (CD31–αSMA+ или CD144–CD325+) фенотипом и, в силу своего качественного характера, даст техническую возможность провести количественный подсчет клеток с переходным или мезенхимальным фенотипом. В отношении секретома подвергшихся или подвергающихся эндотелиально-мезенхимальному переходу (важно рассматривать данный процесс как континуум, а не дискретный переход между двумя состояниями) эндотелиальных клеток следует сказать, что практическая оценка выделяемых ими в этом состоянии в микроокружение растворимых факторов в клеточных культурах достаточно затруднительна в силу длительности процесса (1–2 нед.) и не является приоритетным способом анализа эндотелиально-мезенхимального перехода в сравнении с иммунофлуоресцентным окрашиванием.
В отношении искусственного моделирования дисфункции эндотелия было выявлено, что пролонгированное внутривенное введение кальципротеиновых частиц крысам Wistar инициирует эндотелиально-мезенхимальный переход, повышая экспрессию его транскрипционных факторов Snail, Slug и Twist1 в эндотелии различных аортальных сегментов (нисходящая аорта и дуга аорты). Динамика генной и белковой экспрессии позволила сделать вывод о том, что эндотелиально-мезенхимальный переход при воздействии КФБ развивается постепенно, а повышенная экспрессия генов его транскрипционных факторов (SNAI1, SNAI2 и ZEB1) после интернализации КФБ предшествует последующей гиперэкспрессии белков Snail, Slug и Twist1, регулирующих смену эндотелиального фенотипа на мезенхимальный.
Таким образом, к критериям эндотелиально-мезенхимального перехода можно отнести: 1) приобретение экспрессии маркеров мезенхимального фенотипа (к которым в первую очередь можно отнести N-кадгерин и альфа-актин гладких мышц); 2) повышение экспрессии не менее трех из четырех соответствующих транскрипционных факторов (Snail, Slug, Twist1 и Zeb1). Собственно эндотелиально-мезенхимальный переход также следует определять как континуум событий, а не как уже свершившийся факт приобретения эндотелиальными клетками мезенхимального фенотипа. Поскольку эндотелиально-мезенхимальный переход ассоциирован с обоими звеньями провоспалительной активации эндотелия (адгезией лейкоцитов к эндотелиальным клетками и выделением провоспалительных цитокинов [83, 142, 143]) и индуцируется хроническим воспалением [144, 145], турбулентным потоком [135] и нарушениями эндотелиальной механотрансдукции [146, 147] эндотелиально-мезенхимальный переход также можно рассматривать в качестве обязательного критерия дисфункции эндотелия.
Нарушение биосинтеза и выделения монооксида азота (NO)
Одним из ключевых аспектов дисфункции эндотелия является снижение способности эндотелиальных клеток вырабатывать монооксид азота (NO), являющийся ключевым вазодилататором. С методологической точки зрения можно выделить четыре этапа, на которых возможна косвенная оценка выделения NO: транскрипция гена ключевого фермента этого процесса эндотелиальной NO-синтазы (NOS3), синтез данного белка в эндотелиальных клетках в результате трансляции на рибосомах, функционально значимое посттрансляционное модифицирование данного фермента (в число таких посттрансляционных модификаций входят ингибирующее активность данного фермента фосфорилирование в позиции треонин-495 и повышающее его активность фосфорилирование в позициях серин-113 и серин-1177), а также собственно синтез NO, отражаемый количеством продуктов его метаболизма – нитратов (NO3) и нитритов (NO2), поскольку собственно молекула NO существует после своего биосинтеза лишь несколько секунд. Помимо оценки компонентов цепи синтеза NO, при оценке дисфункции эндотелия важным является также анализ функциональной активности активных форм азота (reactive nitrogen species), в частности, пероксинитрита (ONOO−), образующегося при расцеплении сигнального пути эндотелиальной NO-синтазы, когда данный фермент “переключается” на синтез супероксида (${\text{O}}_{2}^{{ \bullet - }}$) вместо NO. Пероксинитрит (ONOO−) в таком случае образуется из супероксида, генерируемого при расцеплении сигнального пути эндотелиальной NO-синтазы, и синтезируемого свободного NO. Другими активными формами азота являются •NO2 и N2O3, образующиеся при реакциях пероксинитрита (ONOO−) с другими химическими соединениями. Все активные формы азота также имеют очень короткий период жизни, и маркером вызываемого ими нитрозирующего стресса (по аналогии с индуцируемым активными формами кислорода окислительным стрессом) является 3-нитротирозин, который может быть детектирован при иммуноблоттинге при помощи соответствующего антитела. Кроме того, поскольку нитрозирующий стресс неразрывно связан с окислительным стрессом через избыточный синтез супероксида при расцеплении сигнального пути NO-синтазы, при комплексном анализе дисфункции эндотелия также целесообразен анализ функциональной активности активных форм кислорода посредством измерения концентрации продуктов реакции с тиобарбитуровой кислотой (TBARS), включающих в себя малондиальдегид, 8-изопростан и 4-гидроксиноненаль и являющихся продуктами метаболизма свободных радикалов (в первую очередь супероксида ${\text{O}}_{2}^{{ \bullet - }}$, пероксида водорода H2O2, гидроксил-радикала •OH и синглетного кислорода 1[O2]).
При измерении транскрипции гена NOS3 у человека важно помнить о четырех изоформах данного фермента, которые должны детектироваться при помощи единого праймера. При оценке посттрансляционных модификаций данного белка принципиальную значимость имеет сравнение относительного количества его общей формы, а также ингибирующих и активирующих фосфорилированных форм. В отличие от транскрипционных факторов эндотелиальной механотрансдукции YAP и TAZ, фосфорилированные формы которых относительно легко определяются в лизате эндотелиальных клеток, детекция фосфорилированных форм NO-синтазы в лизате клеточных культур in vitro (в отличие от лизата эндотелия in vivo) достаточно затруднительна и требует большого количества загружаемого для иммуноблоттинга белка в сочетании с малым разведением соответствующих первичных антител. Измерение нитратов и нитритов как продуктов метаболизма выделяемого эндотелиальными клетками NO в культуральной среде, а также измерение продуктов реакции с тиобарбитуровой кислотой как продуктов метаболизма выделяемых ими в культуральную среду активных форм кислорода может быть проведено посредством колориметрического анализа при помощи соответствующих наборов. Поскольку детекция нитратов, нитритов и продуктов реакции с тиобарбитуровой кислотой в культуральной среде также требует их определенной концентрации в ней (по аналогии с белковыми или пептидными растворимыми факторами), подобные эксперименты по моделированию дисфункции эндотелия рекомендуется проводить в статических условиях, с бессывороточной культуральной средой (для предотвращения контаминации нитратами, нитритами и активными формами кислорода из сыворотки), после не менее чем 24 ч инкубации эндотелиальных клеток с триггером дисфункции эндотелия для накопления достаточного количества молекул и после низкоскоростного центрифугирования для очищения анализируемой культуральной среды от клеточного дебриса.
Добавление модельного триггера дисфункции эндотелия – кальципротеиновых частиц к культурам эндотелиальных клеток в системе пульсирующего потока in vitro вызывало снижение экспрессии эндотелиальной NO-синтазы и соответствующего гена NOS3. Также было выявлено, что эндотелий нормолипидемических и нормотензивных крыс Wistar при регулярном внутривенном введении кальципротеиновых частиц характеризовался относительным повышением экспрессии фосфорилированной формы эндотелиальной NO-синтазы в позиции треонин-495 (отражающей ингибирование активности данного фермента), а также 3-нитротирозина (отражающего возникающий при “расцеплении” сигнального пути эндотелиальной NO-синтазы нитрозирующий стресс) в нисходящей аорте. Данные результаты свидетельствовали о выраженных нарушениях функционирования эндотелиальной NO-синтазы в результате интернализации кальципротеиновых частиц, подтверждая справедливость их использования для моделирования дисфункции эндотелия.
Определение нарушений биосинтеза и выделения NO достаточно сложно вследствие многокомпонентности его составляющих (к примеру, измерение уровня нитратов, нитритов и продуктов реакции с тиобарбитуровой кислотой in vivo не будет отражать дисфункцию эндотелия в каком-либо конкретном сосуде), при этом отсутствие патологических изменений в каком-либо из компонентов цепи биосинтеза NO (к примеру, ингибирующих посттрансляционных модификаций) не обязательно говорит об отсутствии снижения выделения NO эндотелиальными клетками. Дополнительную сложность придает возможность расцепления сигнального пути NO-синтазы, при котором даже при нормальном уровне и функционировании данного фермента выделение NO эндотелиальными клетками является сниженным и сопровождается нитрозирующим и окислительным стрессом. Наиболее разумным представляется вариант, при котором нарушение биосинтеза NO определяется как по собственно детектируемому снижению NO в микроокружении эндотелиальных клеток (более актуально для экспериментов in vitro), так и по снижению экспрессии гена NOS3 и кодируемой им эндотелиальной NO-синтазы, а также повышению соотношения ее фосфорилированной формы в позиции треонин-495 к общей фракции данного фермента. Сопутствующими при этом являются полуколичественное или количественное измерение содержания 3-нитротирозина и продуктов реакции с тиобарбитуровой кислотой. Как было указано выше, нарушение биосинтеза и выделения NO является обязательным компонентом дисфункции эндотелия.
Нарушение целостности эндотелия
Завершающим компонентом дисфункции эндотелия, предлагаемым к рассмотрению, является патологическое повышение проницаемости эндотелиального монослоя в результате нарушения его целостности, следствием чего in vivo наблюдается проникновение атерогенных липопротеинов, цитокинов и иммунных клеток в интиму. Обязательным условием для моделирования данного патологического процесса, наиболее тесно относящегося к церебральному эндотелию (вследствие важности обеспечения полупроницаемости гематоэнцефалического барьера для целостности тканей центральной нервной системы), также является воздействие на эндотелиальные клетки пульсирующего потока.
Патологическое повышение проницаемости эндотелия можно смоделировать при помощи добавления плазмы крови от пациентов с изучаемой патологией или культуральной среды, насыщенной триггером дисфункции эндотелия, в систему пульсирующего потока, связанную с заселенной эндотелиальными клетками (и при необходимости покрытой коллагеном IV типа для имитации базальной мембраны) культуральной проточной камерой. После необходимого времени инкубации эндотелиальные клетки внутри контрольной и экспериментальной камер могут быть окрашены антителами к VE-кадгерину (маркер не только эндотелиальных клеток, но и межклеточных контактов, окрашивание на который позволяет оценить целостность эндотелиального монослоя) и F-актину (маркеру ориентации цитоскелета, по которому можно судить о физиологических и патологических изменениях геометрии клетки). Соответственно нарушение целостности эндотелия при конфокально-микроскопической визуализации будет определено как потеря межклеточных контактов (по окрашиванию антителами к VE-кадгерину) и потеря элонгации эндотелиальных клеток (по окрашиванию антителами к F-актину). Автоматизированный анализ изменений клеточной геометрии может быть проведен при помощи плагинов Directionality и OrientationJ в программе ImageJ.
Классическим способом для оценки нарушения целостности эндотелия in vivo является прижизненное окрашивание синим Эванса (Evans Blue) – красителем, связывающимся с альбумином, высокомолекулярным белком, в норме не проникающим в ткани через сохраняющий свою целостность эндотелий. При нарушении целостности эндотелия альбумин может проникать в интиму (либо через гематоэнцефалический барьер) и связываться с белками и протеогликанами межклеточного матрикса, что отражается синим окрашиванием сосудов после их эксплантации (в норме они остаются нативного цвета, поскольку окрашенный синим Эванса альбумин не в состоянии проникнуть через интактный эндотелий).
Несмотря на очевидные патофизиологические последствия нарушения целостности эндотелия, она является скорее следствием, чем обязательным компонентом дисфункции эндотелия. К примеру, при моделировании дисфункции эндотелия посредством добавления кальципротеиновых частиц в систему пульсирующего потока или их внутривенного введения лабораторным животным было показано, что, несмотря на их определенную токсичность (статистически значимая гибель 1–2% эндотелиальных клеток) и способность вызывать все вышеуказанные звенья дисфункции эндотелия (его провоспалительную активацию, нарушения эндотелиальной механотрансдукции, эндотелиально-мезенхимальный переход и нарушения биосинтеза NO), выраженности данных патологических эффектов при таком сценарии недостаточно для нарушения целостности эндотелиального монослоя в культуральных проточных камерах. Кроме того, несмотря на свою способность индуцировать формирование неоинтимы и развитие адвентициального и околососудистого воспаления, регулярное внутривенное введение кальципротеиновых частиц также не приводило к нарушениям целостности эндотелия по результатам прижизненного окрашивания альбумина синим Эванса (и наличии соответствующего положительного контроля в результате баллонной ангиопластики).
Протромботическая активация эндотелия
Следует отметить также важность протромботической активации эндотелия, особенно важной в контексте COVID-19 [148–150 ], основными маркерами которой являются патологически повышенное выделение фактора фон Виллебранда и ингибитора активатора плазминогена (PAI-1) в сочетании со сниженным выделением различных активаторов плазминогена (uPA и tPA). Безусловно, она также может (и должна) быть отнесена к признакам дисфункции эндотелия, однако в силу отсутствия подобных экспериментов у авторов более подробное обсуждение в данной статье возможным не представляется, поскольку ее целью является методологическое и концептуальное обсуждение критериев дисфункции эндотелия с целью ее четкого определения именно на основе практического опыта.
ЗАКЛЮЧЕНИЕ
Говоря об определении и о критериях дисфункции эндотелия, представляется важным подчеркнуть многогранность данного феномена, затрагивающего не только биосинтез NO, а практически все аспекты физиологии эндотелия. В частности, критериями дисфункции эндотелия являются:
1. Провоспалительная активация, определяемая по выраженной адгезии лейкоцитов к эндотелиальным клеткам (сопровождающейся повышением экспрессии белков VCAM1 и ICAM1 и кодирующих их генов), повышенному выделению основных эндотелиальных провоспалительных цитокинов (ИЛ-6, ИЛ-8 и MCP-1/CCL2) и патогенности паракринного воздействия дисфункциональных эндотелиальных клеток в эксперименте.
2. Нарушения эндотелиальной механотрансдукции, определяемые как сочетание снижения экспрессии атеропротективных транскрипционных факторов (KLF2, KLF4 и NRF2) и активирующего гипофосфорилирования атерогенных транскрипционных факторов (YAP и TAZ).
3. Эндотелиально-мезенхимальный переход, определяемый как континуум приобретения экспрессии маркеров мезенхимального фенотипа (в первую очередь N-кадгерина и альфа-актина гладких мышц) на фоне повышения экспрессии не менее трех из четырех ответственных за этот процесс транскрипционных факторов (Snail, Slug, Twist1 и Zeb1).
4. Нарушение биосинтеза NO, определяемое как по собственно детектируемому снижению продуктов метаболизма NO в микроокружении эндотелиальных клеток (более актуально для экспериментов in vitro), так и по снижению экспрессии гена NOS3 и кодируемой им эндотелиальной NO-синтазы, а также повышению соотношения ее фосфорилированной формы в позиции треонин-495 к общей фракции данного фермента.
5. Протромботическая активация эндотелия, отражаемая патологическим повышением выделения фактора фон Виллебранда и ингибитора активатора плазминогена (PAI-1) в сочетании со сниженным выделением различных активаторов плазминогена (uPA и tPA).
6. В качестве дополнительного критерия, абсолютно подтверждающего развитие дисфункции эндотелия – нарушение его целостности, характеризующееся патологическим повышением его проницаемости.
Таким образом, дисфункцию эндотелия целесообразно определять как комплексное патологическое изменение его физиологии, включающее в себя патофизиологически значимую провоспалительную и протромботическую активацию эндотелиальных клеток, нарушения эндотелиальной механотрансдукции, эндотелиально-мезенхимальный переход и нарушение биосинтеза NO. В отношении системных биомаркеров дисфункции эндотелия in vivo стоит отметить, что до настоящего времени не было выявлено ни одного надежного биомаркера, который мог бы однозначно свидетельствовать о ее развитии. Доказательными и чувствительными маркерами дисфункции эндотелия in vitro являются ИЛ-6, ИЛ-8 и MCP-1/CCL2, однако данные провоспалительные цитокины также вырабатываются и другими клеточными популяциями (к примеру, различными типами лейкоцитов) и поэтому не являются специфичными для сценария in vivo.
В качестве молекул для скрининга дисфункции эндотелия в эксперименте рекомендуется использовать гены VCAM1, ICAM1, SELE и SELP (рецепторы эндотелиальных клеток для лейкоцитов), IL6, CXCL8, CCL2, CXCL1 и MIF (основные эндотелиальные провоспалительные цитокины), KLF2, KLF4 и NFE2L2 (механочувствительные атеропротективные транскрипционные факторы – исключительно в условиях потока), SNAI1, SNAI2, TWIST1, ZEB1, CDH5 и CDH2 (транскрипционные факторы и маркеры эндотелиально-мезенхимального перехода), NOS3 (ген эндотелиальной NO-синтазы), VWF, SERPINE1, PLAU и PLAT (факторы эндотелий-зависимого гемостаза), а также соответствующие белки, кодируемые данными генами (табл. 1). В таком экспериментальном дизайне повышенная экспрессия генов VCAM1, ICAM1, SELE и SELP в сочетании с повышенной экспрессией генов IL6, CXCL8, CCL2, CXCL1 и MIF свидетельствует о провоспалительной активации эндотелиальных клеток, сниженная экспрессия генов KLF2, KLF4 и NFE2L2 (в условиях потока) является индикатором нарушенной механотрансдукции, повышенная экспрессия генов SNAI1, SNAI2, TWIST1 и ZEB1 в сочетании со сниженной экспрессией гена VE-кадгерина CDH5 и повышенной экспрессией гена N-кадгерина CDH2 говорит об эндотелиально-мезенхимальном переходе, сниженная экспрессия гена NOS3 указывает на нарушенную способность эндотелиальных клеток выделять основной вазодилататор NO, а повышенная экспрессия генов VWF и SERPINE1 в сочетании со сниженной экспрессией генов PLAU и PLAT является индикатором протромботической активации эндотелия. Аналогичное утверждение справедливо и для кодируемых данными генами белков, однако оптимальным подходом представляется предварительный скрининг указанных генов при помощи ОТ-кПЦР в силу относительно низкой стоимости и технической сложности данного метода. В случае выявления вышеуказанных признаков дисфункции эндотелия возможна их верификация при помощи иммуноблоттинга (если речь идет о клеточных белках) или иммуноферментного анализа (в случае провоспалительных цитокинов) либо расширенное профилирование посредством полнотранскриптомного секвенирования или жидкостной хромато-масс-спектрометрии (табл. 2).
Таблица 1.
Предлагаемая панель для скрининга дисфункции эндотелия методом ОТ-кПЦР
| Ген, кодирующий молекулу-маркер дисфункции эндотелия | Молекула-маркер дисфункции эндотелия | Звено дисфункции эндотелия, отражаемое молекулой-маркером |
|---|---|---|
| VCAM1 | Сосудистая молекула клеточной адгезии | Провоспалительная активация эндотелия (рецепторы эндотелиальных клеток для лейкоцитов) |
| ICAM1 | Межклеточная молекула клеточной адгезии | |
| SELE | E-селектин | |
| SELP | P-селектин | |
| IL6 | Интерлейкин-6 | Провоспалительная активация эндотелия (эндотелиальные провоспалительные цитокины) |
| CXCL8 | Интерлейкин-8 | |
| CCL2 | Моноцитарный хемоаттрактантный белок | |
| CXCL1 | Связанный с ростом онкоген, GRO-α | |
| MIF | Фактор ингибирования миграции макрофагов | |
| KLF2 | Транскрипционный фактор KLF2 | Нарушения эндотелиальной механотрансдукции (только в условиях потока) |
| KLF4 | Транскрипционный фактор KLF4 | |
| NFE2L2 | Транскрипционный фактор NRF2 | |
| SNAI1 | Транскрипционный фактор Snail | Эндотелиально-мезенхимальный переход |
| SNAI2 | Транскрипционный фактор Slug | |
| TWIST1 | Транскрипционный фактор Twist1 | |
| ZEB1 | Транскрипционный фактор Zeb1 | |
| CDH5 | VE-кадгерин | |
| CDH2 | N-кадгерин | |
| NOS3 | Эндотелиальная NO-синтаза | Нарушение синтеза NO |
| VWF | Фактор фон Виллебранда | Протромботическая активация эндотелия |
| SERPINE1 | Ингибитор активатора плазминогена | |
| PLAU | Урокиназный активатор плазминогена | |
| PLAT | Тканевой активатор плазминогена |
Таблица 2.
Верификационная панель дисфункции эндотелия
| Молекула-маркер дисфункции эндотелия | Метод верификации | Звено дисфункции эндотелия, отражаемое молекулой-маркером |
|---|---|---|
| Сосудистая молекула клеточной адгезии | Иммуноблоттинг (клеточный лизат) |
Провоспалительная активация эндотелия (рецепторы эндотелиальных клеток для лейкоцитов) |
| Межклеточная молекула клеточной адгезии | ||
| E-селектин | ||
| P-селектин | ||
| Интерлейкин-6 | Иммуноферментный анализ (культуральная среда) | Провоспалительная активация эндотелия (эндотелиальные провоспалительные цитокины) |
| Интерлейкин-8 | ||
| Моноцитарный хемоаттрактантный белок | ||
| Связанный с ростом онкоген, GRO-α | ||
| Фактор ингибирования миграции макрофагов | ||
| Транскрипционный фактор KLF2 | Иммуноблоттинг (клеточный лизат) |
Нарушения эндотелиальной механотрансдукции (только в условиях потока) |
| Транскрипционный фактор KLF4 | ||
| Транскрипционный фактор NRF2 | ||
| Транскрипционный фактор YAP1 (общая фракция и активирующее фосфорилирование в позициях серин-109, серин-127 и серин-397) |
||
| Транскрипционный фактор TAZ (общая фракция и активирующее фосфорилирование в позиции серин-89) |
||
| Транскрипционный фактор Snail | Иммуноблоттинг (клеточный лизат) |
Эндотелиально-мезенхимальный переход |
| Транскрипционный фактор Slug | ||
| Транскрипционный фактор Twist1 | ||
| Транскрипционный фактор Zeb1 | ||
| VE-кадгерин | ||
| N-кадгерин | ||
| Эндотелиальная NO-синтаза (общая фракция, ингибирующее фосфорилирование в позиции треонин-495 и активирующее фосфорилирование в позициях серин-113 и серин-1177) |
Иммуноблоттинг (клеточный лизат) |
Нарушение синтеза NO и расцепление пути NO-синтазы, нитрозирующий стресс, окислительный стресс |
| 3-нитротирозин | ||
| Уровень нитратов и нитритов | Колориметрический анализ (культуральная среда) | |
| Уровень продуктов реакции с тиобарбитуровой кислотой | ||
| Фактор фон Виллебранда | Иммуноблоттинг (клеточный лизат), иммуноферментный анализ (культуральная среда) | Протромботическая активация эндотелия |
| Ингибитор активатора плазминогена | ||
| Урокиназный активатор плазминогена | ||
| Тканевой активатор плазминогена |
Список литературы
Scioli MG, Storti G, D’Amico F, Rodríguez Guzmán R, Centofanti F, Doldo E, Céspedes Miranda EM, Orlandi A (2020) Oxidative Stress and New Pathogenetic Mechanisms in Endothelial Dysfunction: Potential Diagnostic Biomarkers and Therapeutic Targets. J Clin Med 9(6): 1995. https://doi.org/10.3390/jcm9061995
Liao JK (2013) Linking endothelial dysfunction with endothelial cell activation. J Clin Invest 123(2): 540–541. https://doi.org/10.1172/JCI66843
Mauricio MD, Aldasoro M, Ortega J, Vila JM (2013) Endothelial dysfunction in morbid obesity. Curr Pharm Des 19(32): 5718–5729. https://doi.org/10.2174/1381612811319320007
Pomilio M, Mohn A, Verrotti A, Chiarelli F (2002) Endothelial dysfunction in children with type 1 diabetes mellitus. J Pediatr Endocrinol Metab 15(4): 343–361. https://doi.org/10.1515/jpem.2002.15.4.343
Totoson P, Maguin-Gaté K, Prati C, Wendling D, Demougeot C (2014) Mechanisms of endothelial dysfunction in rheumatoid arthritis: lessons from animal studies. Arthritis Res Ther 16(1): 202. https://doi.org/10.1186/ar4450
Ferroni P, Basili S, Paoletti V, Davì G (2006) Endothelial dysfunction and oxidative stress in arterial hypertension. Nutr Metab Cardiovasc Dis 16(3): 222–233. https://doi.org/10.1016/j.numecd.2005.11.012
Prati C, Demougeot C, Guillot X, Godfrin-Valnet M, Wendling D (2014) Endothelial dysfunction in joint disease. Joint Bone Spine 81(5): 386–391. https://doi.org/10.1016/j.jbspin.2014.01.014
Simonsen U, Rodriguez-Rodriguez R, Dalsgaard T, Buus NH, Stankevicius E (2009) Novel approaches to improving endothelium-dependent nitric oxide-mediated vasodilatation. Pharmacol Rep 61(1) :105–115. https://doi.org/10.1016/s1734-1140(09)70012-x
Bertani F, Di Francesco D, Corrado MD, Talmon M, Fresu LG, Boccafoschi F (2021) Paracrine Shear-Stress-Dependent Signaling from Endothelial Cells Affects Downstream Endothelial Function and Inflammation. Int J Mol Sci 22(24): 13300. https://doi.org/10.3390/ijms222413300
Ding H, Triggle CR (2010) Endothelial dysfunction in diabetes: multiple targets for treatment. Pflugers Arch 459(6): 977–994. https://doi.org/10.1007/s00424-010-0807-3
O'Riordan E, Chen J, Brodsky SV, Smirnova I, Li H, Goligorsky MS (2005) Endothelial cell dysfunction: the syndrome in making. Kidney Int 67(5): 1654–1658. https://doi.org/10.1111/j.1523-1755.2005.00256.x
Goligorsky MS (2005) Endothelial cell dysfunction: can’t live with it, how to live without it. Am J Physiol Renal Physiol 288(5): F871–F880. https://doi.org/10.1152/ajprenal.00333.2004
Pierce RW, Giuliano JS, Whitney JE, Ouellette Y; Pediatric Organ Dysfunction Information Update Mandate (PODIUM) Collaborative (2022) Endothelial Dysfunction Criteria in Critically Ill Children: The PODIUM Consensus Conference. Pediatrics 149(1 Suppl 1): S97–S102. https://doi.org/10.1542/peds.2021-052888O
Soerensen M, Debrabant B, Halekoh U, Møller JE, Hassager C, Frydland M, Hjelmborg J, Beck HC, Rasmussen LM (2021) Does diabetes modify the effect of heparin on plasma proteins? – A proteomic search for plasma protein biomarkers for diabetes-related endothelial dysfunction. J Diabetes Complications 35(6): 107906. https://doi.org/10.1016/j.jdiacomp.2021.107906
Le Brocq M, Leslie SJ, Milliken P, Megson IL (2008) Endothelial dysfunction: from molecular mechanisms to measurement, clinical implications, and therapeutic opportunities. Antioxid Redox Signal 10(9): 1631–1674. https://doi.org/10.1089/ars.2007.2013
Cahill PA, Redmond EM (2016) Vascular endothelium – Gatekeeper of vessel health. Atherosclerosis 248:97–109. https://doi.org/10.1016/j.atherosclerosis.2016.03.007
Jensen HA, Mehta JL (2016) Endothelial cell dysfunction as a novel therapeutic target in atherosclerosis. Expert Rev Cardiovasc Ther 14(9): 1021–1033. https://doi.org/10.1080/14779072.2016.1207527
Gimbrone MA Jr, García-Cardeña G (2016) Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ Res 118(4): 620–636. https://doi.org/10.1161/CIRCRESAHA.115.306301
Hyun YM, Lefort CT, Kim M (2009) Leukocyte integrins and their ligand interactions. Immunol Res 45(2–3): 195–208. https://doi.org/10.1007/s12026-009-8101-1
McEver RP (2015) Selectins: initiators of leucocyte adhesion and signalling at the vascular wall. Cardiovasc Res 107(3): 331–339. https://doi.org/10.1093/cvr/cvv154
Allen S, Moran N (2015) Cell Adhesion Molecules: Therapeutic Targets for Inhibition of Inflammatory States. Semin Thromb Hemost 41(6): 563–571. https://doi.org/10.1055/s-0035-1556588
Dustin ML (2019) Integrins and Their Role in Immune Cell Adhesion. Cell 177(3): 499–501. https://doi.org/10.1016/j.cell.2019.03.038
Shishkova D, Markova V, Sinitsky M, Tsepokina A, Frolov A, Zagorodnikov N, Bogdanov L, Kutikhin A (2020) Co-Culture of Primary Human Coronary Artery and Internal Thoracic Artery Endothelial Cells Results in Mutually Beneficial Paracrine Interactions. Int J Mol Sci 21(21): 8032. https://doi.org/10.3390/ijms21218032
Kutikhin AG, Tupikin AE, Matveeva VG, Shishkova DK, Antonova LV, Kabilov MR, Velikanova EA (2020) Human Peripheral Blood-Derived Endothelial Colony-Forming Cells Are Highly Similar to Mature Vascular Endothelial Cells yet Demonstrate a Transitional Transcriptomic Signature. Cells 9(4): 876. https://doi.org/10.3390/cells9040876
Suri C, Jones PF, Patan S, Bartunkova S, Maisonpierre PC, Davis S, Sato TN, Yancopoulos GD (1996) Requisite role of angiopoietin-1, a ligand for the TIE2 receptor, during embryonic angiogenesis. Cell 87(7): 1171–1180. https://doi.org/10.1016/s0092-8674(00)81813-9
Lee J, Kim KE, Choi DK, Jang JY, Jung JJ, Kiyonari H, Shioi G, Chang W, Suda T, Mochizuki N, Nakaoka Y, Komuro I, Yoo OJ, Koh GY (2013) Angiopoietin-1 guides directional angiogenesis through integrin alphavbeta5 signaling for recovery of ischemic retinopathy. Sci Transl Med 5(203): 203ra127. https://doi.org/10.1126/scitranslmed.3006666
Rezzola S, Di Somma M, Corsini M, Leali D, Ravelli C, Polli VAB, Grillo E, Presta M, Mitola S (2019) VEGFR2 activation mediates the pro-angiogenic activity of BMP4. Angiogenesis 22(4): 521–533. https://doi.org/10.1007/s10456-019-09676-y
Zarkada G, Heinolainen K, Makinen T, Kubota Y, Alitalo K (2015) VEGFR3 does not sustain retinal angiogenesis without VEGFR2. Proc Natl Acad Sci U S A 112(3): 761–766. https://doi.org/10.1073/pnas.1423278112
Kishimoto K, Liu S, Tsuji T, Olson KA, Hu GF (2005) Endogenous angiogenin in endothelial cells is a general requirement for cell proliferation and angiogenesis. Oncogene 24(3): 445–456. https://doi.org/10.1038/sj.onc.1208223
He T, Qi F, Jia L, Wang S, Wang C, Song N, Fu Y, Li L, Luo Y (2015) Tumor cell-secreted angiogenin induces angiogenic activity of endothelial cells by suppressing miR-542-3p. Cancer Lett 368(1): 115–125. https://doi.org/10.1016/j.canlet.2015.07.036
Karl E, Zhang Z, Dong Z, Neiva KG, Soengas MS, Koch AE, Polverini PJ, Núñez G, Nör JE (2007) Unidirectional crosstalk between Bcl-xL and Bcl-2 enhances the angiogenic phenotype of endothelial cells. Cell Death Differ 14(9): 1657–1666. https://doi.org/10.1038/sj.cdd.4402174
Falcon BL, Swearingen M, Gough WH, Lee L, Foreman R, Uhlik M, Hanson JC, Lee JA, McClure DB, Chintharlapalli S (2014) An in vitro cord formation assay identifies unique vascular phenotypes associated with angiogenic growth factors. PLoS One 9(9): e106901. https://doi.org/10.1371/journal.pone.0106901
Li B, Sharpe EE, Maupin AB, Teleron AA, Pyle AL, Carmeliet P, Young PP (2006) VEGF and PlGF promote adult vasculogenesis by enhancing EPC recruitment and vessel formation at the site of tumor neovascularization. FASEB J 20(9): 1495–1497. https://doi.org/10.1096/fj.05-5137fje
Gigante B, Morlino G, Gentile MT, Persico MG, De Falco S (2006) Plgf-/-eNos-/- mice show defective angiogenesis associated with increased oxidative stress in response to tissue ischemia. FASEB J 20(7): 970–972. https://doi.org/10.1096/fj.05-4481fje
Shikada Y, Yonemitsu Y, Koga T, Onimaru M, Nakano T, Okano S, Sata S, Nakagawa K, Yoshino I, Maehara Y, Sueishi K (2005) Platelet-derived growth factor-AA is an essential and autocrine regulator of vascular endothelial growth factor expression in non-small cell lung carcinomas. Cancer Res 65(16): 7241–7248. https://doi.org/10.1158/0008-5472.CAN-04-4171
Wu LW, Chen WL, Huang SM, Chan JY (2019) Platelet-derived growth factor-AA is a substantial factor in the ability of adipose-derived stem cells and endothelial progenitor cells to enhance wound healing. FASEB J 33(2): 2388–2395. https://doi.org/10.1096/fj.201800658R
Esser JS, Rahner S, Deckler M, Bode C, Patterson C, Moser M (2015) Fibroblast growth factor signaling pathway in endothelial cells is activated by BMPER to promote angiogenesis. Arterioscler Thromb Vasc Biol 35(2): 358–367. https://doi.org/10.1161/ATVBAHA.114.304345
Litwin M, Radwańska A, Paprocka M, Kieda C, Dobosz T, Witkiewicz W, Baczyńska D (2015) The role of FGF2 in migration and tubulogenesis of endothelial progenitor cells in relation to pro-angiogenic growth factor production. Mol Cell Biochem 410(1-2): 131–142. https://doi.org/10.1007/s11010-015-2545-5
Salani D, Taraboletti G, Rosanò L, Di Castro V, Borsotti P, Giavazzi R, Bagnato A (2000) Endothelin-1 induces an angiogenic phenotype in cultured endothelial cells and stimulates neovascularization in vivo. Am J Pathol 157(5): 1703–1711. https://doi.org/10.1016/S0002-9440(10)64807-9
Spinella F, Rosanò L, Di Castro V, Natali PG, Bagnato A (2002) Endothelin-1 induces vascular endothelial growth factor by increasing hypoxia-inducible factor-1alpha in ovarian carcinoma cells. J Biol Chem 277(31): 27850–27855. https://doi.org/10.1074/jbc.M202421200
Rodriguez-Grande B, Varghese L, Molina-Holgado F, Rajkovic O, Garlanda C, Denes A, Pinteaux E (2015) Pentraxin 3 mediates neurogenesis and angiogenesis after cerebral ischaemia. J Neuroinflam 12: 15. https://doi.org/10.1186/s12974-014-0227-y
Rajkovic I, Wong R, Lemarchand E, Rivers-Auty J, Rajkovic O, Garlanda C, Allan SM, Pinteaux E (2018) Pentraxin 3 promotes long-term cerebral blood flow recovery, angiogenesis, and neuronal survival after stroke. J Mol Med (Berl) 96(12): 1319–1332. https://doi.org/10.1007/s00109-018-1698-6
Zukowska-Grojec Z, Karwatowska-Prokopczuk E, Rose W, Rone J, Movafagh S, Ji H, Yeh Y, Chen WT, Kleinman HK, Grouzmann E, Grant DS (1998) Neuropeptide Y: a novel angiogenic factor from the sympathetic nerves and endothelium. Circ Res 83(2):187–195. https://doi.org/10.1161/01.res.83.2.187
Ghersi G, Chen W, Lee EW, Zukowska Z (2001) Critical role of dipeptidyl peptidase IV in neuropeptide Y-mediated endothelial cell migration in response to wounding. Peptides 22(3): 453–458. https://doi.org/10.1016/s0196-9781(01)00340-0
Duff SE, Li C, Garland JM, Kumar S (2003) CD105 is important for angiogenesis: evidence and potential applications. FASEB J 17(9): 984–992. https://doi.org/10.1096/fj.02-0634rev
Browne S, Jha AK, Ameri K, Marcus SG, Yeghiazarians Y, Healy KE (2018) TGF-beta1/CD105 signaling controls vascular network formation within growth factor sequestering hyaluronic acid hydrogels. PLoS One 13(3): e0194679. https://doi.org/10.1371/journal.pone.0194679
Bussolino F, Di Renzo MF, Ziche M, Bocchietto E, Olivero M, Naldini L, Gaudino G, Tamagnone L, Coffer A, Comoglio PM (1992) Hepatocyte growth factor is a potent angiogenic factor which stimulates endothelial cell motility and growth. J Cell Biol 119(3): 629–641. https://doi.org/10.1083/jcb.119.3.629
Xin X, Yang S, Ingle G, Zlot C, Rangell L, Kowalski J, Schwall R, Ferrara N, Gerritsen ME (2001) Hepatocyte growth factor enhances vascular endothelial growth factor-induced angiogenesis in vitro and in vivo. Am J Pathol 158(3): 1111–1120. https://doi.org/10.1016/S0002-9440(10)64058-8
Slater T, Haywood NJ, Matthews C, Cheema H, Wheatcroft SB (2019) Insulin-like growth factor binding proteins and angiogenesis: from cancer to cardiovascular disease. Cytokine Growth Factor Rev 46: 28–35. https://doi.org/10.1016/j.cytogfr.2019.03.005
Haywood NJ, Slater TA, Drozd M, Warmke N, Matthews C, Cordell PA, Smith J, Rainford J, Cheema H, Maher C, Bridge KI, Yuldasheva NY, Cubbon RM, Kearney MT, Wheatcroft SB (2019) IGFBP-1 in Cardiometabolic Pathophysiology-Insights From Loss-of-Function and Gain-of-Function Studies in Male Mice. J Endocr Soc 4(1):bvz006. https://doi.org/10.1210/jendso/bvz006
Azar WJ, Azar SH, Higgins S, Hu JF, Hoffman AR, Newgreen DF, Werther GA, Russo VC (2011) IGFBP-2 enhances VEGF gene promoter activity and consequent promotion of angiogenesis by neuroblastoma cells. Endocrinology 152(9): 3332–3342. https://doi.org/10.1210/en.2011-1121
Das SK, Bhutia SK, Azab B, Kegelman TP, Peachy L, Santhekadur PK, Dasgupta S, Dash R, Dent P, Grant S, Emdad L, Pellecchia M, Sarkar D, Fisher PB (2013) MDA-9/syntenin and IGF-BP-2 promote angiogenesis in human melanoma. Cancer Res 73(2): 844–854. https://doi.org/10.1158/0008-5472.CAN-12-1681
Granata R, Trovato L, Lupia E, Sala G, Settanni F, Camussi G, Ghidoni R, Ghigo E. Insulin-like growth factor binding protein-3 induces angiogenesis through IGF-I- and SphK1-dependent mechanisms. J Thromb Haemost (2007) 5(4): 835–845. https://doi.org/10.1111/j.1538-7836.2007.02431.x
Dallinga MG, Habani YI, Kayser RP, Van Noorden CJF, Klaassen I, Schlingemann RO (2020) IGF-binding proteins 3 and 4 are regulators of sprouting angiogenesis. Mol Biol Rep 47(4): 2561–2572. https://doi.org/10.1007/s11033-020-05339-0
Zhuge X, Murayama T, Arai H, Yamauchi R, Tanaka M, Shimaoka T, Yonehara S, Kume N, Yokode M, Kita T (2005) CXCL16 is a novel angiogenic factor for human umbilical vein endothelial cells. Biochem Biophys Res Commun 331(4): 1295–1300. https://doi.org/10.1016/j.bbrc.2005.03.200
Yu X, Zhao R, Lin S, Bai X, Zhang L, Yuan S, Sun L (2016) CXCL16 induces angiogenesis in autocrine signaling pathway involving hypoxia-inducible factor 1alpha in human umbilical vein endothelial cells. Oncol Rep 35(3): 1557–1565. https://doi.org/10.3892/or.2015.4520
Ferrari G, Cook BD, Terushkin V, Pintucci G, Mignatti P (2009) Transforming growth factor-beta 1 (TGF-beta1) induces angiogenesis through vascular endothelial growth factor (VEGF)-mediated apoptosis. J Cell Physiol 219(2): 449–458. https://doi.org/10.1002/jcp.21706
Budi EH, Mamai O, Hoffman S, Akhurst RJ, Derynck R (2019) Enhanced TGF-beta Signaling Contributes to the Insulin-Induced Angiogenic Responses of Endothelial Cells. Science 11: 474–491. https://doi.org/10.1016/j.isci.2018.12.038
Stepanova V, Jayaraman PS, Zaitsev SV, Lebedeva T, Bdeir K, Kershaw R, Holman KR, Parfyonova YV, Semina EV, Beloglazova IB, Tkachuk VA, Cines DB (2016) Urokinase-type Plasminogen Activator (uPA) Promotes Angiogenesis by Attenuating Proline-rich Homeodomain Protein (PRH) Transcription Factor Activity and De-repressing Vascular Endothelial Growth Factor (VEGF) Receptor Expression. J Biol Chem 291(29): 15029–15045. https://doi.org/10.1074/jbc.M115.678490
van Hinsbergh VW, Engelse MA, Quax PH (2006) Pericellular proteases in angiogenesis and vasculogenesis. Arterioscler Thromb Vasc Biol 26(4): 716–728. https://doi.org/10.1161/01.ATV.0000209518.58252.17
Blackburn JS, Brinckerhoff CE (2008) Matrix metalloproteinase-1 and thrombin differentially activate gene expression in endothelial cells via PAR-1 and promote angiogenesis. Am J Pathol 173(6): 1736–1746. https://doi.org/10.2353/ajpath.2008.080512
Fan Y, Ye J, Shen F, Zhu Y, Yeghiazarians Y, Zhu W, Chen Y, Lawton MT, Young WL, Yang GY (2008) Interleukin-6 stimulates circulating blood-derived endothelial progenitor cell angiogenesis in vitro. J Cereb Blood Flow Metab 28(1): 90–98. https://doi.org/10.1038/sj.jcbfm.9600509
Kayakabe K, Kuroiwa T, Sakurai N, Ikeuchi H, Kadiombo AT, Sakairi T, Matsumoto T, Maeshima A, Hiromura K, Nojima Y (2012) Interleukin-6 promotes destabilized angiogenesis by modulating angiopoietin expression in rheumatoid arthritis. Rheumatology (Oxford) 51(9): 1571–1579. https://doi.org/10.1093/rheumatology/kes093
Heidemann J, Ogawa H, Dwinell MB, Rafiee P, Maaser C, Gockel HR, Otterson MF, Ota DM, Lugering N, Domschke W, Binion DG (2003) Angiogenic effects of interleukin 8 (CXCL8) in human intestinal microvascular endothelial cells are mediated by CXCR2. J Biol Chem 278(10): 8508–8515. https://doi.org/10.1074/jbc.M208231200
Li A, Dubey S, Varney ML, Dave BJ, Singh RK (2003) IL-8 directly enhanced endothelial cell survival, proliferation, and matrix metalloproteinases production and regulated angiogenesis. J Immunol 170(6): 3369–3376. https://doi.org/10.4049/jimmunol.170.6.3369
Hong KH, Ryu J, Han KH (2005) Monocyte chemoattractant protein-1-induced angiogenesis is mediated by vascular endothelial growth factor-A. Blood 105(4): 1405–1407. https://doi.org/10.1182/blood-2004-08-3178
Stamatovic SM, Keep RF, Mostarica-Stojkovic M, Andjelkovic AV (2006) CCL2 regulates angiogenesis via activation of Ets-1 transcription factor. J Immunol 177(4): 2651–2661. https://doi.org/10.4049/jimmunol.177.4.2651
Lobov IB, Brooks PC, Lang RA (2002) Angiopoietin-2 displays VEGF-dependent modulation of capillary structure and endothelial cell survival in vivo. Proc Natl Acad Sci U S A 99(17): 11205–11210. https://doi.org/10.1073/pnas.172161899
Felcht M, Luck R, Schering A, Seidel P, Srivastava K, Hu J, Bartol A, Kienast Y, Vettel C, Loos EK, Kutschera S, Bartels S, Appak S, Besemfelder E, Terhardt D, Chavakis E, Wieland T, Klein C, Thomas M, Uemura A, Goerdt S, Augustin HG (2012) Angiopoietin-2 differentially regulates angiogenesis through TIE2 and integrin signaling. J Clin Invest 122(6): 1991–2005. https://doi.org/10.1172/JCI58832
Nör JE, Mitra RS, Sutorik MM, Mooney DJ, Castle VP, Polverini PJ (2000) Thrombospondin-1 induces endothelial cell apoptosis and inhibits angiogenesis by activating the caspase death pathway. J Vasc Res 37(3): 209–218. https://doi.org/10.1159/000025733
Kaur S, Martin-Manso G, Pendrak ML, Garfield SH, Isenberg JS, Roberts DD (2010) Thrombospondin-1 inhibits VEGF receptor-2 signaling by disrupting its association with CD47. J Biol Chem 285(50): 38923–38932. https://doi.org/10.1074/jbc.M110.172304
Xu X, Mao W, Chen Q, Zhuang Q, Wang L, Dai J, Wang H, Huang Z (2014) Endostar, a modified recombinant human endostatin, suppresses angiogenesis through inhibition of Wnt/beta-catenin signaling pathway. PLoS One 9(9): e107463. https://doi.org/10.1371/journal.pone.0107463
Ergün S, Kilic N, Wurmbach JH, Ebrahimnejad A, Fernando M, Sevinc S, Kilic E, Chalajour F, Fiedler W, Lauke H, Lamszus K, Hammerer P, Weil J, Herbst H, Folkman J (2001) Endostatin inhibits angiogenesis by stabilization of newly formed endothelial tubes. Angiogenesis 4(3): 193–206. https://doi.org/10.1023/a:1014027218980
Ikenaka Y, Yoshiji H, Kuriyama S, Yoshii J, Noguchi R, Tsujinoue H, Yanase K, Namisaki T, Imazu H, Masaki T, Fukui H (2003) Tissue inhibitor of metalloproteinases-1 (TIMP-1) inhibits tumor growth and angiogenesis in the TIMP-1 transgenic mouse model. Int J Cancer 105(3): 340–346. https://doi.org/10.1002/ijc.11094
Reed MJ, Koike T, Sadoun E, Sage EH, Puolakkainen P (2003) Inhibition of TIMP1 enhances angiogenesis in vivo and cell migration in vitro. Microvasc Res 65(1): 9–17. https://doi.org/10.1016/s0026-2862(02)00026-2
Seo DW, Li H, Guedez L, Wingfield PT, Diaz T, Salloum R, Wei BY, Stetler-Stevenson WG (2003) TIMP-2 mediated inhibition of angiogenesis: an MMP-independent mechanism. Cell 114(2): 171–180. https://doi.org/10.1016/s0092-8674(03)00551-8
Lahat N, Bitterman H, Engelmayer-Goren M, Rosenzweig D, Weiss-Cerem L, Rahat MA (2011) Reduced TIMP-2 in hypoxia enhances angiogenesis. Am J Physiol Cell Physiol 300(3): C557–C566. https://doi.org/10.1152/ajpcell.00177.2010
Wu J, Strawn TL, Luo M, Wang L, Li R, Ren M, Xia J, Zhang Z, Ma W, Luo T, Lawrence DA, Fay WP (2015) Plasminogen activator inhibitor-1 inhibits angiogenic signaling by uncoupling vascular endothelial growth factor receptor-2-alphaVbeta3 integrin cross talk. Arterioscler Thromb Vasc Biol 35(1): 111–120. https://doi.org/10.1161/ATVBAHA.114.304554
Bajou K, Herkenne S, Thijssen VL, D’Amico S, Nguyen NQ, Bouché A, Tabruyn S, Srahna M, Carabin JY, Nivelles O, Paques C, Cornelissen I, Lion M, Noel A, Gils A, Vinckier S, Declerck PJ, Griffioen AW, Dewerchin M, Martial JA, Carmeliet P, Struman I (2014) PAI-1 mediates the antiangiogenic and profibrinolytic effects of 16K prolactin. Nat Med 20(7): 741–747. https://doi.org/10.1038/nm.3552
Kutikhin AG, Velikanova EA, Mukhamadiyarov RA, Glushkova TV, Borisov VV, Matveeva VG, Antonova LV, Filip’ev DE, Golovkin AS, Shishkova DK, Burago AY, Frolov AV, Dolgov VY, Efimova OS, Popova AN, Malysheva VY, Vladimirov AA, Sozinov SA, Ismagilov ZR, Russakov DM, Lomzov AA, Pyshnyi DV, Gutakovsky AK, Zhivodkov YA, Demidov EA, Peltek SE, Dolganyuk VF, Babich OO, Grigoriev EV, Brusina EB, Barbarash OL, Yuzhalin AE (2016) Apoptosis-mediated endothelial toxicity but not direct calcification or functional changes in anti-calcification proteins defines pathogenic effects of calcium phosphate bions. Sci Rep 6: 27255. https://doi.org/10.1038/srep27255
Shishkova D, Velikanova E, Sinitsky M, Tsepokina A, Gruzdeva O, Bogdanov L, Kutikhin A (2019) Calcium Phosphate Bions Cause Intimal Hyperplasia in Intact Aortas of Normolipidemic Rats through Endothelial Injury. Int J Mol Sci 20(22): 5728. https://doi.org/10.3390/ijms20225728
Shishkova D, Markova V, Sinitsky M, Tsepokina A, Velikanova E, Bogdanov L, Glushkova T, Kutikhin A (2020) Calciprotein Particles Cause Endothelial Dysfunction under Flow. Int J Mol Sci 21(22): 8802. https://doi.org/10.3390/ijms21228802
Shishkova DK, Velikanova EA, Bogdanov LA, Sinitsky MY, Kostyunin AE, Tsepokina AV, Gruzdeva OV, Mironov AV, Mukhamadiyarov RA, Glushkova TV, Krivkina EO, Matveeva VG, Hryachkova ON, Markova VE, Dyleva YA, Belik EV, Frolov AV, Shabaev AR, Efimova OS, Popova AN, Malysheva VY, Kolmykov RP, Sevostyanov OG, Russakov DM, Dolganyuk VF, Gutakovsky AK, Zhivodkov YA, Kozhukhov AS, Brusina EB, Ismagilov ZR, Barbarash OL, Yuzhalin AE, Kutikhin AG (2021) Calciprotein Particles Link Disturbed Mineral Homeostasis with Cardiovascular Disease by Causing Endothelial Dysfunction and Vascular Inflammation. Int J Mol Sci 22(22): 12458. https://doi.org/10.3390/ijms222212458
Kutikhin AG, Feenstra L, Kostyunin AE, Yuzhalin AE, Hillebrands JL, Krenning G (2021) Calciprotein Particles: Balancing Mineral Homeostasis and Vascular Pathology. Arterioscler Thromb Vasc Biol 41(5): 1607–1624. https://doi.org/10.1161/ATVBAHA.120.315697
Shay-Salit A, Shushy M, Wolfovitz E, Yahav H, Breviario F, Dejana E, Resnick N (2002) VEGF receptor 2 and the adherens junction as a mechanical transducer in vascular endothelial cells. Proc Natl Acad Sci U S A 99(14): 9462–9467. https://doi.org/10.1073/pnas.142224299
Jin ZG, Ueba H, Tanimoto T, Lungu AO, Frame MD, Berk BC (2003) Ligand-independent activation of vascular endothelial growth factor receptor 2 by fluid shear stress regulates activation of endothelial nitric oxide synthase. Circ Res 93(4): 354–363. https://doi.org/10.1161/01.RES.0000089257.94002.96.34
Lee HJ, Koh GY (2003) Shear stress activates Tie2 receptor tyrosine kinase in human endothelial cells. Biochem Biophys Res Commun 304(2): 399–404. https://doi.org/10.1016/S0006-291X(03)00592-8
Osawa M, Masuda M, Kusano K, Fujiwara K (2002) Evidence for a role of platelet endothelial cell adhesion molecule-1 in endothelial cellmechanosignal transduction: is it a mechanoresponsive molecule? J Cell Biol 158(4): 773–785. https://doi.org/10.1083/jcb.200205049
Tzima E, Irani-Tehrani M, Kiosses WB, Dejana E, Schultz DA, Engelhardt B, Cao G, DeLisser H, Schwartz MA (2005) A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature 437(7057): 426–431. https://doi.org/10.1038/nature03952
Ishida T, Peterson TE, Kovach NL, Berk BC (1996) MAP kinase activation by flow in endothelial cells. Role of beta 1 integrins and tyrosine kinases. Circ Res 79(2): 310–316. https://doi.org/10.1161/01.RES.79.2.310
Li S, Kim M, Hu YL, Jalali S, Schlaepfer DD, Hunter T, Chien S, Shyy JY (1997) Fluid shear stress activation of focal adhesion kinase. Linking to mitogen-activated protein kinases. J Biol Chem 272(48): 30455–30462. https://doi.org/10.1074/jbc.272.48.30455
Jalali S, del Pozo MA, Chen K, Miao H, Li Y, Schwartz MA, Shyy JY, Chien S (2001) Integrin-mediated mechanotransduction requires its dynamic interaction with specific extracellular matrix (ECM) ligands. Proc Natl Acad Sci U S A 98(3): 1042–1046. https://doi.org/10.1073/pnas.031562998
Tzima E, del Pozo MA, Shattil SJ, Chien S, Schwartz MA (2001) Activation of integrins in endothelial cells by fluid shear stress mediates Rho-dependent cytoskeletal alignment. EMBO J 20(17): 4639–4647. https://doi.org/10.1093/emboj/20.17.4639
Pang Z, Antonetti DA, Tarbell JM (2005) Shear stress regulates HUVEC hydraulic conductivity by occludin phosphorylation. Ann Biomed Eng 33(11): 1536–1545. https://doi.org/10.1007/s10439-005-7786-0
Brakemeier S, Eichler I, Hopp H, Köhler R, Hoyer J (2002) Up-regulation of endothelial stretch-activated cation channels by fluid shear stress. Cardiovasc Res 53(1): 209–218. https://doi.org/10.1016/S0008-6363(01)00476-X
Ranade SS, Qiu Z, Woo SH, Hur SS, Murthy SE, Cahalan SM, Xu J, Mathur J, Bandell M, Coste B, Li YS, Chien S, Patapoutian A (2014) Piezo1, a mechanically activated ion channel, is required for vascular development in mice. Proc Natl Acad Sci U S A 111(28): 10347–10352. https://doi.org/10.1073/pnas.1409233111
Baeyens N, Mulligan-Kehoe MJ, Corti F, Simon DD, Ross TD, Rhodes JM, Wang TZ, Mejean CO, Simons M, Humphrey J, Schwartz MA (2014) Syndecan 4 is required for endothelial alignment in flow and atheroprotective signaling. Proc Natl Acad Sci U S A 111(48): 17308–17313. https://doi.org/10.1073/pnas.1413725111
Chachisvilis M, Zhang YL, Frangos JA (2006) G protein-coupled receptors sense fluid shear stress in endothelial cells. Proc Natl Acad Sci U S A 103(42): 15463–15468. https://doi.org/10.1073/pnas.0607224103
Gudi S, Nolan JP, Frangos JA (1998) Modulation of GTPase activity of G proteins by fluid shear stress and phospholipid composition. Proc Natl Acad Sci U S A 95(5): 2515–2519.https://doi.org/10.1073/pnas.95.5.251
Gudi S, Huvar I, White CR, McKnight NL, Dusserre N, Boss GR, Frangos JA (2003) Rapid activation of Ras by fluid flow is mediated by Galpha(q) and Gbetagamma subunits of heterotrimeric G proteins in human endothelial cells. Arterioscler Thromb Vasc Biol 23(6): 994–1000. https://doi.org/10.1161/01.ATV.0000073314.51987.84
Florian JA, Kosky JR, Ainslie K, Pang Z, Dull RO, Tarbell JM (2003) Heparan sulfate proteoglycan is a mechanosensor on endothelial cells. Circ Res 93(10): e136–142. https://doi.org/10.1161/01.RES.0000101744.47866.D5
Lopez-Quintero SV, Cancel LM, Pierides A, Antonetti D, Spray DC, Tarbell JM (2013) High glucose attenuates shear-induced changes in endothelial hydraulic conductivity by degrading the glycocalyx. PLoS One 8(11): e78954. https://doi.org/10.1371/journal.pone.0078954
Isshiki M, Ando J, Korenaga R, Kogo H, Fujimoto T, Fujita T, Kamiya A (1998) Endothelial Ca2+ waves preferentially originate at specific loci in caveolin-rich cell edges. Proc Natl Acad Sci U S A 95(9): 5009–5014. https://doi.org/10.1073/pnas.95.9.5009
Yu J, Bergaya S, Murata T, Alp IF, Bauer MP, Lin MI, Drab M, Kurzchalia TV, Stan RV, Sessa WC (2006) Direct evidence for the role of caveolin-1 and caveolae in mechanotransduction and remodeling of blood vessels. J Clin Invest 116(5): 1284–1291. https://doi.org/10.1172/JCI27100
Iomini C, Tejada K, Mo W, Vaananen H, Piperno G (2004) Primary cilia of human endothelial cells disassemble under laminar shear stress. J Cell Biol 164(6): 811–817. https://doi.org/10.1083/jcb.200312133
AbouAlaiwi WA, Takahashi M, Mell BR, Jones TJ, Ratnam S, Kolb RJ, Nauli SM (2009) Ciliary polycystin-2 is a mechanosensitive calcium channel involved in nitric oxide signaling cascades. Circ Res 104(7): 860–869. https://doi.org/10.1161/CIRCRESAHA.108.192765
Helmke BP, Goldman RD, Davies PF (2000) Rapid displacement of vimentin intermediate filaments in living endothelial cells exposed to flow. Circ Res 86(7): 745–752. https://doi.org/10.1161/01.RES.86.7.745
Helmke BP, Thakker DB, Goldman RD, Davies PF (2001) Spatiotemporal analysis of flow-induced intermediate filament displacement in living endothelial cells. Biophys J 80(1): 184–194. https://doi.org/10.1016/S0006-3495(01)76006-7
Malek AM, Izumo S (1996) Mechanism of endothelial cell shape change and cytoskeletal remodeling in response to fluid shear stress. J Cell Sci 109 (Pt 4): 713–726. https://doi.org/10.1242/jcs.109.4.713
Satcher R, Dewey CF Jr, Hartwig JH (1997) Mechanical remodeling of the endothelial surface and actin cytoskeleton induced by fluid flow. Microcirculation 4(4): 439–453. https://doi.org/10.3109/10739689709146808
Osborn EA, Rabodzey A, Dewey CF Jr, Hartwig JH (2006) Endothelial actin cytoskeleton remodeling during mechanostimulation with fluid shear stress. Am J Physiol Cell Physiol 290(2): C444–C452. https://doi.org/10.1152/ajpcell.00218.2005
Dekker RJ, van Thienen JV, Rohlena J, de Jager SC, Elderkamp YW, Seppen J, de Vries CJ, Biessen EA, van Berkel TJ, Pannekoek H, Horrevoets AJ (2005) Endothelial KLF2 links local arterial shear stress levels to the expression of vascular tone regulating genes. Am J Pathol 167(2): 609–618. https://doi.org/10.1016/S0002-9440(10)63002-7
Dekker RJ, Boon RA, Rondaij MG, Kragt A, Volger OL, Elderkamp YW, Meijers JC, Voorberg J, Pannekoek H, Horrevoets AJ (2006) KLF2 provokes a gene expression pattern that establishes functional quiescent differentiation of the endothelium. Blood 107(11): 4354–4363. https://doi.org/10.1182/blood-2005-08-3465
Parmar KM, Larman HB, Dai G, Zhang Y, Wang ET, Moorthy SN, Kratz JR, Lin Z, Jain MK, Gimbrone MA Jr, García-Cardeña G (2006) Integration of flow-dependent endothelial phenotypes by Kruppel-like factor 2. J Clin Invest 116(1): 49–58. https://doi.org/10.1172/JCI24787
Villarreal G Jr, Zhang Y, Larman HB, Gracia-Sancho J, Koo A, García-Cardeña G (2010) Defining the regulation of KLF4 expression and its downstream transcriptional targets in vascular endothelial cells. Biochem Biophys Res Commun 391(1): 984–989. https://doi.org/10.1016/j.bbrc.2009.12.002
Zhou G, Hamik A, Nayak L, Tian H, Shi H, Lu Y, Sharma N, Liao X, Hale A, Boerboom L, Feaver RE, Gao H, Desai A, Schmaier A, Gerson SL, Wang Y, Atkins GB, Blackman BR, Simon DI, Jain MK (2012) Endothelial Kruppel-like factor 4 protects against atherothrombosis in mice. J Clin Invest 122(12): 4727–4731. https://doi.org/10.1172/JCI66056
Jiang YZ, Jiménez JM, Ou K, McCormick ME, Zhang LD, Davies PF (2014) Hemodynamic disturbed flow induces differential DNA methylation of endothelial Kruppel-Like Factor 4 promoter in vitro and in vivo. Circ Res 115(1): 32–43. https://doi.org/10.1161/CIRCRESAHA.115.303883
Dai G, Vaughn S, Zhang Y, Wang ET, Garcia-Cardena G, Gimbrone MA Jr (2007) Biomechanical forces in atherosclerosis-resistant vascular regions regulate endothelial redox balance via phosphoinositol 3-kinase/Akt-dependent activation of Nrf2. Circ Res 101(7): 723–733. https://doi.org/10.1161/CIRCRESAHA.107.152942
Zakkar M, Van der Heiden K, Luong le A, Chaudhury H, Cuhlmann S, Hamdulay SS, Krams R, Edirisinghe I, Rahman I, Carlsen H, Haskard DO, Mason JC, Evans PC (2009) Activation of Nrf2 in endothelial cells protects arteries from exhibiting a proinflammatory state. Arterioscler Thromb Vasc Biol 29(11): 1851–1857. https://doi.org/10.1161/ATVBAHA.109.193375
Wang KC, Yeh YT, Nguyen P, Limqueco E, Lopez J, Thorossian S, Guan KL, Li YJ, Chien S (2016) Flow-dependent YAP/TAZ activities regulate endothelial phenotypes and atherosclerosis. Proc Natl Acad Sci U S A 113(41): 11525–11530. https://doi.org/10.1073/pnas.1613121113
Wang L, Luo JY, Li B, Tian XY, Chen LJ, Huang Y, Liu J, Deng D, Lau CW, Wan S, Ai D, Mak KK, Tong KK, Kwan KM, Wang N, Chiu JJ, Zhu Y, Huang Y (2016) Integrin-YAP/TAZ-JNK cascade mediates atheroprotective effect of unidirectional shear flow. Nature 540(7634): 579–582. https://doi.org/10.1038/nature20602
SenBanerjee S, Lin Z, Atkins GB, Greif DM, Rao RM, Kumar A, Feinberg MW, Chen Z, Simon DI, Luscinskas FW, Michel TM, Gimbrone MA Jr, García-Cardeña G, Jain MK (2004) KLF2 Is a novel transcriptional regulator of endothelial proinflammatory activation. J Exp Med 199(10): 1305–1315. https://doi.org/10.1084/jem.20031132
Lin Z, Kumar A, SenBanerjee S, Staniszewski K, Parmar K, Vaughan DE, Gimbrone MA Jr, Balasubramanian V, García-Cardeña G, Jain MK (2005) Kruppel-like factor 2 (KLF2) regulates endothelial thrombotic function. Circ Res 96(5): e48–е57. https://doi.org/10.1161/01.RES.0000159707.05637.a1
van Thienen JV, Fledderus JO, Dekker RJ, Rohlena J, van Ijzendoorn GA, Kootstra NA, Pannekoek H, Horrevoets AJ (2006) Shear stress sustains atheroprotective endothelial KLF2 expression more potently than statins through mRNA stabilization. Cardiovasc Res 72(2): 231–240. https://doi.org/10.1016/j.cardiores.2006.07.008
Davies PF, Civelek M, Fang Y, Fleming I (2013) The atherosusceptible endothelium: endothelial phenotypes in complex haemodynamic shear stress regions in vivo. Cardiovasc Res 99(2): 315–327. https://doi.org/10.1093/cvr/cvt101
Chen XL, Varner SE, Rao AS, Grey JY, Thomas S, Cook CK, Wasserman MA, Medford RM, Jaiswal AK, Kunsch C (2003) Laminar flow induction of antioxidant response element-mediated genes in endothelial cells. A novel anti-inflammatory mechanism. J Biol Chem 278(2): 703–711. https://doi.org/10.1074/jbc.M203161200
Hosoya T, Maruyama A, Kang MI, Kawatani Y, Shibata T, Uchida K, Warabi E, Noguchi N, Itoh K, Yamamoto M (2005) Differential responses of the Nrf2-Keap1 system to laminar and oscillatory shear stresses in endothelial cells. J Biol Chem 280(29): 27244–27250. https://doi.org/10.1074/jbc.M502551200
Chen XL, Dodd G, Thomas S, Zhang X, Wasserman MA, Rovin BH, Kunsch C (2006) Activation of Nrf2/ARE pathway protects endothelial cells from oxidant injury and inhibits inflammatory gene expression. Am J Physiol Heart Circ Physiol 290(5): H1862–Н1870. https://doi.org/10.1152/ajpheart.00651.2005
Fledderus JO, Boon RA, Volger OL, Hurttila H, Ylä-Herttuala S, Pannekoek H, Levonen AL, Horrevoets AJ (2008) KLF2 primes the antioxidant transcription factor Nrf2 for activation in endothelial cells. Arterioscler Thromb Vasc Biol 28(7): 1339–1346. https://doi.org/10.1161/ATVBAHA.108.165811
Lazaro I, Lopez-Sanz L, Bernal S, Oguiza A, Recio C, Melgar A, Jimenez-Castilla L, Egido J, Madrigal-Matute J, Gomez-Guerrero C (2018) Nrf2 Activation Provides Atheroprotection in Diabetic Mice Through Concerted Upregulation of Antioxidant, Anti-inflammatory, and Autophagy Mechanisms. Front Pharmacol 9: 819. https://doi.org/10.3389/fphar.2018.00819
Nakajima H, Mochizuki N (2017) Flow pattern-dependent endothelial cell responses through transcriptional regulation. Cell Cycle 16(20): 1893–1901. https://doi.org/10.1080/15384101.2017.1364324
Panciera T, Azzolin L, Cordenonsi M, Piccolo S (2017) Mechanobiology of YAP and TAZ in physiology and disease. Nat Rev Mol Cell Biol 18(12): 758–770. https://doi.org/10.1038/nrm.2017.87
Yu Y, Su X, Qin Q, Hou Y, Zhang X, Zhang H, Jia M, Chen Y (2020) Yes-associated protein and transcriptional coactivator with PDZ-binding motif as new targets in cardiovascular diseases. Pharmacol Res 159: 105009. https://doi.org/10.1016/j.phrs.2020.105009
Yuan P, Hu Q, He X, Long Y, Song X, Wu F, He Y, Zhou X (2020) Laminar flow inhibits the Hippo/YAP pathway via autophagy and SIRT1-mediated deacetylation against atherosclerosis. Cell Death Dis 11(2): 141. https://doi.org/10.1038/s41419-020-2343-1
Moonen JR, Lee ES, Schmidt M, Maleszewska M, Koerts JA, Brouwer LA, van Kooten TG, van Luyn MJ, Zeebregts CJ, Krenning G, Harmsen MC (2015) Endothelial-to-mesenchymal transition contributes to fibro-proliferative vascular disease and is modulated by fluid shear stress. Cardiovasc Res 108(3): 377–386. https://doi.org/10.1093/cvr/cvv175
Chen PY, Qin L, Baeyens N, Li G, Afolabi T, Budatha M, Tellides G, Schwartz MA, Simons M (2015) Endothelial-to-mesenchymal transition drives atherosclerosis progression. J Clin Invest 125(12): 4514–4528. https://doi.org/10.1172/JCI82719
Mahmoud MM, Serbanovic-Canic J, Feng S, Souilhol C, Xing R, Hsiao S, Mammoto A, Chen J, Ariaans M, Francis SE, Van der Heiden K, Ridger V, Evans PC (2017) Shear stress induces endothelial-to-mesenchymal transition via the transcription factor Snail. Sci Rep 7(1): 3375. https://doi.org/10.1038/s41598-017-03532-z
Evrard SM, Lecce L, Michelis KC, Nomura-Kitabayashi A, Pandey G, Purushothaman KR, d’Escamard V, Li JR, Hadri L, Fujitani K, Moreno PR, Benard L, Rimmele P, Cohain A, Mecham B, Randolph GJ, Nabel EG, Hajjar R, Fuster V, Boehm M, Kovacic JC (2016) Endothelial to mesenchymal transition is common in atherosclerotic lesions and is associated with plaque instability. Nat Commun 7: 11853. https://doi.org/10.1038/ncomms11853
Souilhol C, Harmsen MC, Evans PC, Krenning G (2018) Endothelial-mesenchymal transition in atherosclerosis. Cardiovasc Res 114(4): 565–577. https://doi.org/10.1093/cvr/cvx253
Dejana E, Hirschi KK, Simons M (2017) The molecular basis of endothelial cell plasticity. Nat Commun 8: 14361. https://doi.org/10.1038/ncomms14361
Jackson AO, Zhang J, Jiang Z, Yin K (2017) Endothelial-to-mesenchymal transition: A novel therapeutic target for cardiovascular diseases. Trends Cardiovasc Med 27(6): 383–393. https://doi.org/10.1016/j.tcm.2017.03.003
Cho JG, Lee A, Chang W, Lee MS, Kim J (2018) Endothelial to Mesenchymal Transition Represents a Key Link in the Interaction between Inflammation and Endothelial Dysfunction. Front Immunol 9: 294. https://doi.org/10.3389/fimmu.2018.0029
Schwartz MA, Vestweber D, Simons M (2018) A unifying concept in vascular health and disease. Science 360(6386): 270–271. https://doi.org/10.1126/science.aat3470
Chen PY, Schwartz MA, Simons M (2020) Endothelial-to-Mesenchymal Transition, Vascular Inflammation, and Atherosclerosis. Front Cardiovasc Med 7: 53. https://doi.org/10.3389/fcvm.2020.00053
Sánchez-Duffhues G, García de Vinuesa A, van de Pol V, Geerts ME, de Vries MR, Janson SG, van Dam H, Lindeman JH, Goumans MJ, Ten Dijke P (2019) Inflammation induces endothelial-to-mesenchymal transition and promotes vascular calcification through downregulation of BMPR2. J Pathol 247(3): 333–346. https://doi.org/10.1002/path.5193
Mehta V, Pang KL, Givens CS, Chen Z, Huang J, Sweet DT, Jo H, Reader JS, Tzima E (2021) Mechanical forces regulate endothelial-to-mesenchymal transition and atherosclerosis via an Alk5-Shc mechanotransduction pathway. Sci Adv 7(28):eabg5060. https://doi.org/10.1126/sciadv.abg5060
Egorova AD, Khedoe PP, Goumans MJ, Yoder BK, Nauli SM, ten Dijke P, Poelmann RE, Hierck BP (2011) Lack of primary cilia primes shear-induced endothelial-to-mesenchymal transition. Circ Res 108(9): 1093–1101. https://doi.org/10.1161/CIRCRESAHA.110.231860
Libby P, Lüscher T (2020) COVID-19 is, in the end, an endothelial disease. Eur Heart J 41(32): 3038–3044. https://doi.org/10.1093/eurheartj/ehaa623
Fogarty H, Townsend L, Morrin H, Ahmad A, Comerford C, Karampini E, Englert H, Byrne M, Bergin C, O’Sullivan JM, Martin-Loeches I, Nadarajan P, Bannan C, Mallon PW, Curley GF, Preston RJS, Rehill AM, McGonagle D, Ni Cheallaigh C, Baker RI, Renné T, Ward SE, O’Donnell JS; Irish COVID-19 Vasculopathy Study (iCVS) investigators (2021) Persistent endotheliopathy in the pathogenesis of long COVID syndrome. J Thromb Haemost 19(10): 2546–2553. https://doi.org/10.1111/jth.15490
Gorog DA, Storey RF, Gurbel PA, Tantry US, Berger JS, Chan MY, Duerschmied D, Smyth SS, Parker WAE, Ajjan RA, Vilahur G, Badimon L, Berg JMT, Cate HT, Peyvandi F, Wang TT, Becker RC (2022) Current and novel biomarkers of thrombotic risk in COVID-19: a Consensus Statement from the International COVID-19 Thrombosis Biomarkers Colloquium. Nat Rev Cardiol 1–21. https://doi.org/10.1038/s41569-021-00665-7
Дополнительные материалы отсутствуют.
Инструменты
Российский физиологический журнал им. И.М. Сеченова