

Российский физиологический журнал им. И.М. Сеченова, 2022, T. 108, № 6, стр. 712-724
Влияние лактата на митохондриальную активность в клетках эндотелия при остром токсическом действии бета-амилоида in vitro
Я. В. Горина 1, Е. Д. Хилажева 1, А. И. Мосягина 1, Е. В. Харитонова 1, *, М. Р. Капкаева 2, Е. В. Стельмашук 2, Н. К. Исаев 2, Н. А. Розанова 2, А. Б. Салмина 2
1 Красноярский государственный медицинский университет им. профессора
В.Ф. Войно-Ясенецкого Министерства здравоохранения Российской Федерации
Красноярск, Россия
2 Научный центр неврологии
Москва, Россия
* E-mail: kharitonova1988@mail.ru
Поступила в редакцию 21.02.2022
После доработки 11.05.2022
Принята к публикации 11.05.2022
- EDN: HLIYLC
- DOI: 10.31857/S0869813922060024
Аннотация
Установлено, что при остром токсическом действии бета-амилоида in vitro присутствие лактата во внеклеточном пространстве в дозозависимой манере снижает активность митохондрий в клетках эндотелия, блокада лактатных монокарбоксилатных транспортеров (МСТ) обладает таким же эффектом, но стимуляция лактатных GPR81-рецепторов на этих клетках вызывает увеличение активности митохондрий. Это позволяет предположить, что высокая концентрация лактата во внеклеточном пространстве подавляет активность митохондрий в клетках эндотелия, но это не связано с активностью GPR81-рецепторов. Вероятнее всего, эффекты GPR81 реализуются в присутствии более низких концентраций внеклеточного лактата. Поскольку развитие болезни Альцгеймера сопровождается снижением экспрессии изоформ MCT, определяющих транспорт и метаболизм лактата в нервных клетках, в комплексе с полученными нами данными дисрегуляция МСТ-транспортеров при болезни Альцгеймера способствует развитию митохондриальной дисфункции, а воспроизведение эффектов внеклеточного лактата путем активации GPR81-рецепторов частично компенсирует такое нарушение.
Болезнь Альцгеймера является одним из наиболее распространенных нейродегенеративных заболеваний у пожилых людей, эта болезнь сопровождается рядом прогрессирующих когнитивных нарушений и потерей памяти, которым как правило предшествует внеклеточное отложение в головном мозге бета-амилоида (Aβ) в виде диффузных и нерастворимых бляшек [1]. Одним из наиболее распространенных и ярко выраженных патологических признаков болезни Альцгеймера является церебральная амилоидная ангиопатия, которая развивается за счет отложения нерастворимого Aβ в артериях, артериолах и вокруг стенок капилляров [2, 3]. При этом повреждение церебральных микрососудов приводит к запуску комплекса патологических событий, включающего гипоперфузию, нейровоспаление, нарушение структурно-функциональной целостности гематоэнцефалического барьера (ГЭБ) и окислительный стресс, что в совокупности способствует прогрессированию нейродегенерации [4].
Как известно, церебральные эндотелиальные клетки, занимающие центральное место в структуре ГЭБ, контролируют постоянство химического состава внутренней среды головного мозга, регулируя транспорт ионов и молекул через ГЭБ и модулируя мозговой кровоток [5]. При этом важно отметить, что эндотелиальные клетки капилляров головного мозга содержат в 2–4 раза больше митохондрий, чем любые другие эндотелиальные клетки организма, это обусловлено высокой потребностью в активном энергетическом метаболизме и выживании [6], а также более высокой восприимчивостью к апоптозу и нарушениям в структуре белков плотных контактов в условиях гипоксии [7]. Это объясняет тот факт, что изменения структуры и функции митохондрий в церебральных эндотелиальных клетках вызывают клеточную дисфункцию, потерю целостности ГЭБ и воспаление, что в конечном итоге приводит к гибели клеток [8–10].
Примечательно, что содержание митохондрий в значительной мере снижается в мозге мышей с болезнью Альцгеймера [11–13], а поскольку митохондрии представляют собой ключевой источник энергии для клеток головного мозга, повреждение митохондрий может являться одним из самых ранних событий в развитии болезни Альцгеймера [14]. Более того, показано, что митохондрии играют решающую роль в сигнальных путях, опосредующих повреждение церебрального эндотелия при развитии болезни Альцгеймера, ассоциированной с церебральной амилоидной ангиопатией [15].
Ранее предполагалось, что лактат является конечным продуктом метаболизма, однако накопленные экспериментальные данные указывают на то, что лактат является важным источником энергии и сигнальной молекулой для нейронов [16], это позволяет предположить, что гомеостаз лактата может иметь решающее значение для функции мозга. Интересен и тот факт, что согласно недавно проведенному исследованию, циркулирующий лактат может являться основным субстратом цикла трикарбоновых кислот во всех тканях, кроме головного мозга [17]. При этом полное распределение лактата между тканями, за исключением головного мозга, указывает на то, что головной мозг имеет отличительную метаболическую микросреду, это подразумевает важную роль церебральных эндотелиальных клеток в поддержании гомеостаза лактата. Ряд исследований показал, что эндотелиальные клетки головного мозга экспрессируют монокарбоксилатный транспортер 1 (MCT1), который транспортирует лактат, пируват и кетоновые тела через клеточные мембраны, что указывает на способность эндотелиоцитов обеспечивать как транспорт лактата в мозг, так и его отток для поддержания лактатного гомеостаза [18, 19].
Ранее нами было показано, что активация лактатных рецепторов GPR81 в церебральном эндотелии стимулирует митохондриальный биогенез in vitro, это дает возможноcть предположить, что метаболическое взаимодействие между астроцитами и эндотелиоцитами посредством продукции лактата в астроцитах, его транспортом за счет МСТ к церебральным эндотелиоцитам и действием через рецепторы GPR81 может играть существенную роль в регуляции структурно-функциональной целостности ГЭБ [20].
Необходимо отметить, что не до конца известны механизмы, с помощью которых Aβ в церебральном эндотелии нарушает митохондриальные метаболические процессы, такие как цикл трикарбоновых кислот, дыхательная цепь переноса электронов и окислительное фосфорилирование, а также механизмы его абберантного влияния на ферменты. При этом было высказано предположение, согласно которому наличие общих структурно-функциональных особенностей ферментов может обуславливать схожие эффекты Aβ на метаболические процессы и продукцию АТФ как в церебральном эндотелии, так и в клетках нейрональной и астроглиальной природы [21].
Более того, в настоящее время недостаточно ясно, какое влияние на функцию митохондрий в эндотелиальных клетках головного мозга при нейродегенерации альцгеймеровского типа оказывает уровень лактата.
Цель работы – изучить влияние лактата на митохондриальную активность в клетках эндотелия при остром токсическом действии бета-амилоида in vitro.
МЕТОДЫ ИССЛЕДОВАНИЯ
Получение первичной монокультуры эндотелиальных клеток микрососудов головного мозга in vitro
В работе использовались первичная культура клеток церебрального эндотелия, полученная из крыс линии Wistar. Животных содержали в клетках со свободным доступом к воде и корму при постоянной температуре 21 ± 1°С и регулярном световом цикле 12 ч день/12 ч ночь. Исследования на животных проводились с соблюдением принципов гуманности, изложенных в Директиве Европейского сообщества (2010/63/ЕС). Исследования выполняли после утверждения заявки и протокола на использование лабораторных животных для исследования на заседании биоэтической комиссии по работе с животными при локальном этическом комитете Красноярского государственного медицинского университета им. проф. В.Ф. Войно-Ясенецкого (выписка из протокола №3 от 27.10.2020 г.). Общее количество животных n = 2.
Выделение церебральных эндотелиоцитов проводилось по модифицированному протоколу Liu и соавт. [22]. Выделение мозга с удалением мозговых оболочек и крупных церебральных сосудов осуществлялось с помощью роллинга на фильтровальной бумаге. Выделяли кору головного мозга и удаляли крупные сосуды в холодном растворе Хенкса (ПанЭко, Россия). Мелконарезанную кору головного мозга центрифугировали в течение 3 мин при 150 g, после чего добавляли к осадку в двукратном объеме 25%-ный бычий сывороточный альбумин (BSA, Sigma, США), подвергали механической диссоциации пипеткой на 5 мл, полученную взвесь клеток центрифугировали в течение 10 мин при 600 g при комнатной температуре. После центрифугирования проводили забор самого нижнего слоя осадка и перенос в новую коническую пробирку на 15 мл. Этапы механической диссоциации и центрифугирования повторяли трижды, после чего проводилась ферментативная обработка осадка в 0.1%-ном растворе коллагеназы II (ПанЭко, Россия) в течение 35 мин при 37°С с периодическим перемешиванием. Далее осадок умеренно ресуспензировали и центрифугировали при 150 g в течение 5 мин. После центрифугирования осадок ресуспензировали в модифицированной среде Игла (DMEM-F12, GIBCO Invitrogen Corporation, США), содержащей 20% эмбриональной телячьей сыворотки (HyClone, Австрия), 2 мМ глутамина (glutaMAX, Gibco, Великобритания), 16.6 мМ глюкозы, и высаживали на пластиковую чашку Петри диаметром 40 мм (Медполимер, Россия), покрытую матригелем. Смену среды проводили дважды в неделю. После образования монослоя эндотелиальные клетки пересаживали в 96-луночные культуральные планшеты (Eppendorf, Германия).
Исследование влияния концентрации лактата на митохондриальную активность в эндотелиоцитах при остром токсическом действии бета-амилоида in vitro
При достижении 90%-ной конфлюентности монослоя эндотелиальных клеток производили смену среды и добавляли лактат натрия в концентрациях 2.5, 5 и 10 мМ. Также в часть экспериментальных лунок одновременно с лактатом натрия добавляли Аβ1-42 в конечной концентрации 100 нМ.
Таким образом, были сформированы следующие экспериментальные группы:
1) Контроль;
2) Контроль + 2.5 мМ лактат;
3) Контроль + 5 мМ лактат;
4) Контроль + 10 мМ лактат;
5) Aβ1-42;
6) Aβ1-42 + 2.5 мМ лактат;
7) Aβ1-42 + 5 мМ лактат;
8) Aβ1-42 + 10 мМ лактат.
Через сутки культивирования клеток с модуляторами производилась оценка интенсивности флуоресценции митохондрий с использованием TMRE (Sigma, США). TMRE селективно накапливается в активных митохондриях и позволяет детектировать мембранный потенциал митохондрий в живых клетках. В лунки добавляли TMRE в рабочей концентрации 200 нМ и инкубировали в течение 15 мин при 37°C, после чего отмывали лунки модифицированным раствором Рингера–Локка 3 раза. Оценку интенсивности флуоресценции митохондрий проводили с использованием системы клеточной визуализации EVOS M7000 (Thermo Fisher Scientific, США).
Исследование влияния модуляторов GPR81 на клетки эндотелия при остром токсическом действии бета-амилоида in vitro
При достижении 90%-ной конфлюентности монослоя эндотелиальных клеток производили смену среды и добавляли химические модуляторы: Аβ1-42 в конечной концентрации 100 нМ и 3Cl-5OH-BA (агонист рецепторов лактата GPR81) в конечной концентрации 500 мкМ.
Были сформированы следующие экспериментальные группы:
1) Контроль;
2) Аβ1-42;
3) 3Cl-5OH-BA;
4) Аβ1-42 + 3Cl-5OH-BA.
Инкубацию с Аβ1-42 проводили в течение 48 ч, инкубацию с 3Cl-5OH-BA – в течение 24 ч, после чего оценивали интенсивность флуоресценции митохондрий с помощью TMRE с использованием системы клеточной визуализации EVOS M7000 (Thermo Fisher Scientific, США).
Исследование влияния модуляторов МСТ-транспортеров на клетки эндотелия при остром токсическом действии бета-амилоида in vitro
При достижении 90%-ной конфлюентности монослоя эндотелиальных клеток производили смену среды и добавляли химические модуляторы: лактат натрия в конечной концентрации 10 мМ, флоретин (блокатор монокарбоксилатных транспортеров МСТ1, МСТ4) в конечной концентрации 100 мкМ, Аβ1-42 в конечной концентрации 100 нМ.
Были сформированы следующие экспериментальные группы:
1) Лактат;
2) Лактат + Флоретин;
3) Лактат + Аβ1-42;
4) Лактат + Флоретин + Аβ1-42.
Инкубацию с Аβ1-42 проводили в течение 48 ч, инкубацию с лактатом натрия и флоретином – в течение 24 ч, после чего оценивали интенсивность флуоресценции митохондрий с помощью TMRE с использованием системы клеточной визуализации EVOS M7000 (Thermo Fisher Scientific, США).
Статистический анализ
Статистический анализ полученных результатов проводили с помощью программ GraphPad Prizm 8.0.1 (версия 8.0, США) и Stаtplus Professional (AnalystSoft Inc, США), сборка 5.9.8.5/Core v.5.9.33 и GraphPad 6.0 (США). Критерий Колмогорова–Смирнова использовали для оценки нормальности распределения. При отсутствии нормальности распределения применяли U-критерий Манна–Уитни. При сравнении более двух групп применяли непараметрический дисперсионный анализ (критерий Краскелла–Уоллиса).
Различия принимали значимыми при р ≤ 0.05. Результаты представлены в виде Ме [Q1; Q3], где Ме – медиана; [Q1; Q3] интерквартильный размах, p – уровень значимости.
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
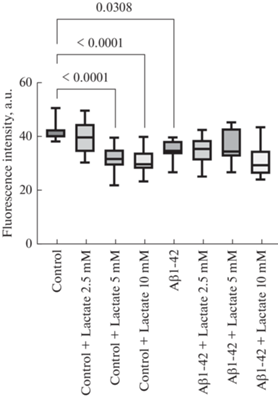
Нами было выявлено, что культивирование эндотелиальных клеток в присутствии Aβ1-42 приводило к снижению интенсивности флуоресценции митохондрий по сравнению с группой контроля (рис. 1). При изучении влияния внеклеточного лактата на активность митохондрий в клетках эндотелия найдено, что добавление лактата в концентрации 5 и 10 мМ, но не 2.5 мМ приводило к статистически значимому снижению интенсивности флуоресценции митохондрий в группе культивирования клеток в питательной среде (рис. 1). В группе Aβ1-42 присутствие лактата нивелировало влияние Aβ1-42 на митохондриальную активность (рис. 1).
Рис. 1.
Интенсивность флуоресценции митохондрий в эндотелиальных клетках микрососудов головного мозга при воздействии лактата натрия в концентрациях 2.5, 5 и 10 мМ в культуральной среде, а также в присутствии Aβ1-42. Данные представлены в виде Ме [Q1; Q3], где Ме – медиана; [Q1; Q3] интерквартильный размах, p – уровень значимости, U – критерий Манна–Уитни.
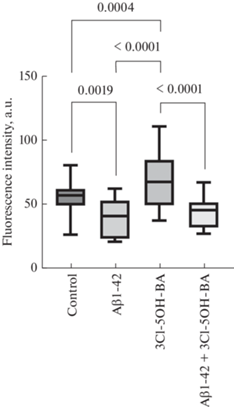
Для оценки вклада рецепторов лактата GPR81 в выявленные нами эффекты лактата было изучено действие 3Cl-5OH-BA, агониста рецепторов лактата GPR81, на активность митохондрий в клетках эндотелия.
Активация рецепторов лактата GPR81 в группе 3Cl-5OH-BA приводила к статистически значимому увеличению интенсивности флуоресценции митохондрий в эндотелиальных клетках по сравнению с группой контроля (рис. 2). Присутствие в среде культивирования Aβ1-42 вызывало статистически значимое снижение интенсивности флуоресценции митохондрий в эндотелиальных клетках по сравнению с группой контроля. Также была отмечена тенденция, свидетельствующая о том, что активация рецепторов лактата GPR81 нивелирует токсическое воздействие Aβ1-42 на митохондриальную активность, однако при межгрупповом сравнении статистически значимых различий не выявлено (рис. 2).
Рис. 2.
Интенсивность флуоресценции митохондрий в эндотелиальных клетках микрососудов головного мозга при воздействии Аβ1-42 и агониста рецепторов лактата GPR81 – 3Cl-5OH-BA. Данные представлены в виде Ме [Q1; Q3], где Ме – медиана; [Q1; Q3] интерквартильный размах, p – уровень значимости, U-критерий Манна–Уитни.
В ходе исследования влияния модуляторов МСТ-транспортеров лактата на активность митохондрий в клетках эндотелия нами установлено, что присутствие Аβ1-42 и блокирование MCT-транспортеров флоретином приводило к статистически значимому снижению интенсивности флуоресценции митохондрий в эндотелиальных клетках, однако подавляющее действие флоретина на митохондриальный биогенез являлось менее выраженным по сравнению с Аβ1-42 (рис. 3). Совместное воздействие модуляторов вызывало выраженное снижение интенсивности флуоресценции митохондрий (рис. 3).
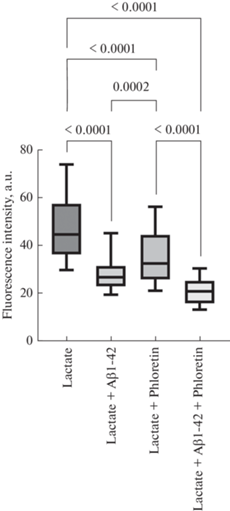
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Известно, что метаболические изменения в нервных клетках головного мозга пациентов с болезнью Альцгеймера могут предшествовать таким ключевым нейропатологическим событиям, как отложение амилоидных бляшек и формирование нейрофибриллярных белков [23]. Все большее количество экспериментальных данных свидетельствует о том, что снижение интенсивности гликолиза и нарушение функций митохондрий, особенно в отношении регуляции окислительного фосфорилирования, вероятно, являются значимыми факторами в развитии и прогрессировании болезни Альцгеймера [24, 25].
Согласно гипотезе митохондриального каскада, мутантные митохондриальные гены, которые предрасполагают к снижению скорости митохондриального дыхания, могут выступать в качестве триггеров развития абберантного функционирования митохондрий при болезни Альцгеймера [14]. Так, у животных с экспериментальной моделью болезни Альцгеймера патологические изменения функций митохондрий наблюдаются еще до отложения Aβ [26]. Аналогичная ситуация установлена и в моделях in vitro, а именно выраженные нарушения структурно-функциональной целостности митохондрий и окислительный стресс в условиях отсутствия Aβ [27], что дает дополнительные доказательства ключевой роли митохондриальной дисфункции в этиологии болезни Альцгеймера.
Важно отметить и то, что нарушение гликолиза также наблюдается на ранних стадиях у пациентов с болезнью Альцгеймера, в частности, ярко выраженное снижение аэробного гликолиза в тех областях головного мозга, которые в наибольшей степени подвержены повреждающему действию Aβ [28], а также в тех областях, где наблюдаются высокие уровни накопления гиперфосфорилированного тау-белка [29].
Примечательным является тот факт, что отобранные линии нервных клеток, устойчивые к токсическому эффекту Aβ, демонстрируют гиперэкспрессию киназы пируватдегидрогеназы – фермента, подавляющего митохондриальное дыхание и переводящего клетку в режим аэробного гликолиза, на фоне повышения активности лактатдегидрогеназы A и продукции лактата [30], что коррелирует со снижением уровня активных форм кислорода. При этом химическое или генетическое ингибирование лактатдегидрогеназы A в значительной мере повышает чувствительность клеток к токсическому действию Aβ. Такое переключение метаболизма с митохондриального дыхания на аэробный гликолиз, известного как эффект Варбурга [31], частично обусловлено действием HIF-1α (hypoxia-inducible factor 1-α, фактор, индуцируемый гипоксией 1-α), который приводит к снижению митохондриального дыхания и связанной с этим продукции активных форм кислорода, что в свою очередь увеличивает устойчивость нервных клеток к апоптозу [32, 33]. Более того, сверхэкспрессия киназы пируватдегидрогеназы или лактатдегидрогеназы A в культуре нервных клеток, выделенных из ткани головного мозга крыс, в значительной мере увеличивает устойчивость к Aβ и другим нейротоксинам за счет снижения потенциала митохондриальной мембраны и продукции активных форм кислорода. При этом стоит отметить, что клетки потребляют меньший объем кислорода, сохраняя уровень АТФ как в контрольных условиях культивирования, так и после добавления Aβ [30].
Интересно, что добавление субстратов окислительной энергии пирувата и 3-β-гидроксибутирата в условиях in vitro и in vivo уменьшало развитие Aβ-индуцированной дисфункции нейронов за счет активации дыхательной цепи переноса электронов в митохондриях в условиях развития болезни Альцгеймера [34].
Таким образом, в ходе наших исследований установлено, что в физиологических условиях возрастание концентраций внеклеточного лактата привело к торможению митохондриальной активности. Однако в условиях токсического действия бета-амилоида in vitro присутствие внеклеточного лактата повысило резистентность митохондрий к токсическому действию Aβ1-42, что может быть связано со способностью лактата вызывать торможение окислительного фосфорилирования и образования активных форм кислорода, тем самым способствуя повышению устойчивости клеток эндотелия к апоптозу при развитии болезни Альцгеймера.
Наличие лактат-чувствительного рецептора GPR81 в головном мозге [35] указывает на то, что лактат является не только метаболитом и энергетическим субстратом, но и сигнальной молекулой, которая может модулировать активность нейронов посредством различных механизмов. По данным одного из исследований [36], церебральный GPR81 экспрессируется преимущественно в постсинаптических мембранах синапсов возбуждающих нейронов, а также, хотя и в меньшей степени, в перисинаптических астроглиальных отростках и в гематоэнцефалическом барьере, в частности, в эндотелиальных клетках и периваскулярных астроцитарных отростках. Дальнейшие исследования продемонстрировали присутствие GPR81 в мозжечке, гиппокампе и коре головного мозга мыши [36], а также в изолированных кортикальных астроцитах крыс и мышей [37], что указывают на способность лактата реализовывать сигнальные эффекты посредством активации рецепторов в различных областях ЦНС. Следовательно, рецепторы GPR81 могут опосредовать эффекты лактата в контексте реализации синаптической функции, энергетического метаболизма и мозгового кровотока [35, 38].
Согласно недавно проведенному исследованию [37], стимуляция внеклеточным L-лактатом и селективным агонистом GPR81 – 3-хлор-5-гидроксибензойной кислотой (3Cl-5OH-BA) подобно стимуляции адренергических рецепторов, повышает внутриклеточный уровень цАМФ и L-лактата в астроцитах за счет активации аденилатциклазы. Интересно, что 3Cl-5OH-BA также повышал содержание цитозольного цАМФ и в астроцитах, выделенных из ткани головного мозга мышей, нокаутных по L-лактат-специфическому рецептору GPR81, это указывает на существование нового L-лактат-рецепторноподобного механизма, не зависящего от активности рецептора GPR81, посредством которого происходит активация аэробного гликолиза и продукции L-лактата по механизму положительной обратной связи.
Известно, что митохондриальный цАМФ влияет на физиологию митохондрий, а именно митохондриальные микродомены цАМФ могут интегрировать вне- и внутримитохондриальные стимулы для регуляции большинства митохондриальных функций [39]. Структурно-функциональные нарушения митохондрий, развивающиеся при болезни Альцгеймера [40], сопровождаются аберрантной передачей сигналов цАМФ [41], что в свою очередь приводит к реализации ряда эффектов, а именно стимуляции ионных каналов, которые блокируют активность нейронной сети и вызывают стимуляцию экспрессии белка-предшественника амилоида и амилоидогенного процессинга [42].
Таким образом, в ходе нашего исследования установлено, что активация лактатных рецепторов вызывала повышение интенсивности митохондриального дыхания в клетках эндотелия, в то время как воздействие Aβ1-42 приводило к митохондриальной дисфункции. Это дает возможность предположить, что активация GPR81-рецепторов лактата при амилоидопатии может быть эффективна для коррекции энергетического дефицита, вызванного действием Aβ на митохондрии.
Как известно, монокарбоксилатные транспортеры, в частности MCT1, в ЦНС впервые были функционально идентифицированы на эндотелиальных клетках ГЭБ [43]. Значительная экспрессия MCT1 в сосудах головного мозга во взрослом возрасте предполагает их способность проходить через ГЭБ и выступать в роли ключевых энергетических субстратов для мозга, что особенно важно в условиях дисметаболизма глюкозы при развитии болезни Альцгеймера [44, 45].
Примечательно, что MCT2 также обнаружен на внутриклеточной мембране митохондрий нейронов [46, 47], что в комплексе с присутствием изоформы лактатдегидрогеназы B указывает на непосредственное окисление лактата в митохондриях [48], это в свою очередь дает возможность рассматривать монокарбоксилатные транспортеры как часть митохондриального комплекса, который способствует проникновению и использованию окислительных субстратов.
Более того, поглощение L-лактата через МСТ сопровождается котранспортом протонов, вызывая внутриклеточное закисление [49], что может модулировать энергетический метаболизм головного мозга путем ингибирования фосфофруктокиназы – гликолитического фермента, чувствительного к небольшим изменениям рН [50].
Интересными являются результаты недавно проведенного исследования [51], согласно которому активация MCT2 in vivo улучшала функцию митохондрий и в значительной мере снижала прогрессирование когнитивных нарушений за счет усиления AMPK-опосредованного митохондриального биогенеза.
В целом можно заключить, что блокирование транспорта лактата MCT-транспортерами оказывает угнетающее действие на митохондриальную активность в церебральном эндотелии, приводя к энергетическому дефициту в митохондриях. Наблюдаемая при этом митохондриальная дисфункция еще более усугубляется при действии Aβ, обладающего токсическим эффектом в отношении митохондриальной активности.
Список литературы
Scheltens P, Blennow K, Breteler MB, Strooper B, Frisoni GB, Salloway S, Flier WMV (2016) Alzheimer’s disease. Lancet 388(10043): 505–517. https://doi.org/10.1016/S0140-6736(15)01124-1
Charidimou A, Boulouis G, Gurol ME, Ayata C, Bacska BJ, Frosch MP, Viswanathan A, Greenberg SM (2017) Emerging concepts in sporadic cerebral amyloid angiopathy. Brain 140(7): 1829–1850. https://doi.org/10.1093/brain/awx047
Kim SH, Ahn JH, Yang H, Lee P, Koh GY, Jeong Y (2020) Cerebral amyloid angiopathy aggravates perivascular clearance impairment in an Alzheimer’s disease mouse model. Acta Neuropathol Commun 8(1): 181. https://doi.org/10.1186/s40478-020-01042-0
Parodi-Rullán R, Ghiso J, Cabrera E, Rostagno A, Fossati S (2020) Alzheimer’s amyloid β heterogeneous species differentially affect brain endothelial cell viability, blood-brain barrier integrity, and angiogenesis. Aging Cell 19(11): e13258. https://doi.org/10.1111/acel.13258
Iadecola C (2017) The Neurovascular Unit Coming of Age: A Journey through Neurovascular Coupling in Health and Disease. Neuron 96(1): 17–42. https://doi.org/10.1016/j.neuron.2017.07.030
Oldendorf WH, Cornford ME, Brown WJ (1977) The large apparent work capability of the blood-brain barrier: a study of the mitochondrial content of capillary endothelial cells in brain and other tissues of the rat. Ann Neurol 1(5): 409–417. https://doi.org/10.1002/ana.410010502
Zille M, Ikhsan M, Jiang Y, Lampe J, Wenzel J, Schwaninger M (2019) The impact of endothelial cell death in the brain and its role after stroke: A systematic review. Cell Stress 3(11): 330–347. https://doi.org/10.15698/cst2019.11.203
Solesio ME, Peixoto PM, Debure L, Madamba SM, Leon MJ, Wisniewski T, Pavlov EV, Fossati S (2018) Carbonic anhydrase inhibition selectively prevents amyloid β neurovascular mitochondrial toxicity. Aging Cell 17(4): e12787. https://doi.org/10.1111/acel.12787
Doll DN, Hu XH, Sun J, Lewis SE, Simpkins JW, Ren X (2015) Mitochondrial crisis in cerebrovascular endothelial cells opens the blood–brain barrier. Stroke 46(6): 1681–1689. https://doi.org/10.1161/STROKEAHA.115.009099
Aliev G, Smith MA, Torre JC, Perry G (2004) Mitochondria as a primary target for vascular hypoperfusion and oxidative stress in Alzheimer’s disease. Mitochondrion 4(5–6): 649–463. https://doi.org/10.1016/j.mito.2004.07.018
Balietti M, Giorgetti B, Casoli T, Solazzi M, Tamagnini F, Burattini C, Aicardi G, Fattoretti P (2013) Early selective vulnerability of synapses and synaptic mitochondria in the hippocampal CA1 region of the Tg2576 mouse model of Alzheimer’s disease. J Alzheimers Dis 34: 887–896. https://doi.org/10.3233/JAD-121711
Du H, Guo L, Yan S, Sosunov AA, McKhann GM, Yan SS (2010) Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model. Proc Natl Acad Sci USA 107: 18670–18675. https://doi.org/10.1073/pnas.1006586107
Yao J, Irwin RW, Zhao L, Nilsen J, Hamilton RT, Brinton RD (2009) Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA 106: 14670–14675. https://doi.org/10.1073/pnas.0903563106
Swerdlow RH, Khan SM (2004) A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med Hypotheses 63(1): 8–20. https://doi.org/10.1016/j.mehy.2003.12.045
Solé M, Miñano-Molina AJ, Unzeta M (2015) Cross-talk between Aβ and endothelial SSAO/VAP-1 accelerates vascular damage and Aβ aggregation related to CAA-AD. Neurobiol Aging 36(2): 762–775. https://doi.org/10.1016/j.neurobiolaging.2014.09.030
Magistretti PJ, Allaman I (2018) Lactate in the brain: from metabolic end-product to signalling molecule. Nat Rev Neurosci 19: 235–249. https://doi.org/10.1038/nrn.2018.19
Hui S, Ghergurovich JM, Morscher RJ, Jang C, Teng X, Lu W, Esparza LA, Reya T, Zhan L, Guo JY (2017) Glucose feeds the TCA cycle via circulating lactate. Nature 551: 115–118. https://doi.org/10.1038/nature24057
Gerhart DZ, Enerson BE, Zhdankina OY, Leino RL, Drewes LR (1997) Expression of monocarboxylate transporter MCT1 by brain endothelium and glia in adult and suckling rats. Am J Physiol 273(1 Pt 1): E207–E213. https://doi.org/10.1152/ajpendo.1997.273.1.E207
Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, Deng S, Liddelow SA, Zhang C, Daneman R, Maniatis T, Barres B, Wu JQ (2014) An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 34(36): 11929–11947.
Хилажева ЕД, Писарева НВ, Моргун АВ, Бойцова ЕБ, Таранушенко ТЕ, Фролова ОВ, Салмина АБ (2017) Активация лактатных рецепторов GPR81 стимулирует митохондриальный биогенез в клетках эндотелия церебральных микрососудов. Анналы клиническ эксперим неврол 11(1): 34–39. [Khilazheva ED, Pisareva NV, Morgun AV, Boytsova EB, Taranushenko TE, Frolova OV, Salmina AB (2017) Activation of lactate receptors GPR81 stimulates mitochondrial biogenesis in endothelial cells of cerebral microvessels. Ann Clin Exper Neurol 11(1): 34–39. (In Russ)].
Parodi-Rullán R, Sone JY, Fossat S (2019) Endothelial Mitochondrial Dysfunction in Cerebral Amyloid Angiopathy and Alzheimer’s Disease. J Alzheimers Dis 72(4): 1019–1039. https://doi.org/10.3233/JAD-190357
Liu JP, Song M, Horton RM, Hu Y (2013) Reducing spread in climate model projections of a September ice-free Arctic. Proc Natl Acad Sci USA 110: 12571–12576. https://doi.org/10.1073/pnas.1219716110
Morgen K, Frölich L (2015) The metabolism hypothesis of Alzheimer’s disease: from the concept of central insulin resistance and associated consequences to insulin therapy. J Neural Transm 122(4): 499–504. https://doi.org/10.1007/s00702-015-1377-5
Yao J, Irwin RW, Zhao L, Nilsen J, Hamilton RT, Brinton RD (2009) Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA 106(34): 14670–14675. https://doi.org/10.1073/pnas.0903563106
Dragicevic N, Mamcarz M, Zhu Y, Buzzeo R, Tan J, Arendash GW, Bradshaw PC (2010) Mitochondrial amyloid-beta levels are associated with the extent of mitochondrial dysfunction in different brain regions and the degree of cognitive impairment in Alzheimer’s transgenic mice. J Alzheimers Dis 20 Suppl 2: S535–S550. https://doi.org/10.3233/JAD-2010-100342
Hartl D, Schuldt V, Forler S, Zabel C, Klose J, Rohe MJ (2012) Presymptomatic alterations in energy metabolism and oxidative stress in the APP23 mouse model of Alzheimer disease. Proteom Res 11(6): 3295–3304. https://doi.org/10.1021/pr300021e
Gan X, Huang S, Wu L, Wang Y, Hu G, Li G, Zhang H, Yu H, Swerdlow RH, Chen JX, Yan SS (2014) Inhibition of ERK-DLP1 signaling and mitochondrial division alleviates mitochondrial dysfunction in Alzheimer’s disease cybrid cell. Biochim Biophys Acta 1842(2): 220–231. https://doi.org/10.1016/j.bbadis.2013.11.009
Vlassenko AG, Vaishnavi SN, Couture L, Sacco D, Shannon BJ, Mach RH, Morris JC, Raichle ME, Mintun MA (2010) Spatial correlation between brain aerobic glycolysis and amyloid-β (Aβ) deposition. Proc Natl Acad Sci USA 107(41): 17763–17767. https://doi.org/10.1073/pnas.1010461107
Vlassenko AG, Gordon BA, Goyal MS, Su Y, Blazey TM, Durbin TJ, Couture LE, Christensen JJ, Jafri H, Morris JC, Raichle ME, Benzinger TL (2018) Aerobic glycolysis and tau deposition in preclinical Alzheimer’s disease. Neurobiol Aging 67: 95–98. https://doi.org/10.1016/j.neurobiolaging.2018.03.014
Newington JT, Rappon T, Albers S, Wong DY, Rylett RJ, Cumming RC (2012) Overexpression of pyruvate dehydrogenase kinase 1 and lactate dehydrogenase A in nerve cells confers resistance to amyloid β and other toxins by decreasing mitochondrial respiration and reactive oxygen species production. J Biol Chem 287(44): 37245–37258. https://doi.org/10.1074/jbc.M112.366195
Garcia-Heredia JM, Carnero A (2015) Decoding Warburg’s hypothesis: tumor-related mutations in the mitochondrial respiratory chain. Oncotarget 6(39): 41582–41599. https://doi.org/10.18632/oncotarget.6057
Kim J, Tchernyshyov I, Semenza GL, Dang CV (2006) HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab 3(3): 177–185. https://doi.org/10.1016/j.cmet.2006.02.002
Semenza GL, Jiang BH, Leung SW, Passantino R, Concordet JP, Maire P, Giallongo A (1996) Hypoxia response elements in the aldolase A, enolase, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J Biol Chem 271(51): 32 529–32 537. https://doi.org/10.1074/jbc.271.51.32529
Zilberter M, Ivanov A, Ziyatdinova S, Mukhtarov M, Malkov A, Alpár A, Tortoriello G, Botting CH, Fülöp L, Osypov AA, Pitkänen A, Tanila H, Harkany T, Zilberter Y (2013) Dietary energy substrates reverse early neuronal hyperactivity in a mouse model of Alzheimer’s disease. J Neurochem 125(1): 157–171. https://doi.org/10.1111/jnc.12127
Morland C, Lauritzen KH, Puchades M, Holm-Hansen S, Andersson K, Gjedde A, Attramadal H, Storm-Mathisen J, Hildegard Bergersen L (2015) The lactate receptor, G-protein-coupled receptor 81/hydroxycarboxylic acid receptor 1: Expression and action in brain. J Neurosci Res 93(7): 1045–1055. https://doi.org/10.1002/jnr.23593
Lauritzen KH, Morland C, Puchades M, Holm-Hansen S, Hagelin EM, Lauritzen F, Attramadal H, Storm-Mathisen J, Gjedde A, Bergersen LH (2014) Lactate receptor sites link neurotransmission, neurovascular coupling, and brain energy metabolism. Cereb Cortex 24(10): 2784–2795. https://doi.org/10.1093/cercor/bht136
Vardjan N, Chowdhury HH, Horvat A, Velebit J, Malnar M, Muhič M, Kreft M, Krivec ŠG, Bobnar ST, Miš K, Pirkmajer S, Offermanns S, Henriksen G, Storm-Mathisen J, Bergersen LH, Zorec R (2018) Enhancement of Astroglial Aerobic Glycolysis by Extracellular Lactate-Mediated Increase in cAMP. Front Mol Neurosci 11: 148. https://doi.org/10.3389/fnmol.2018.00148
Бойцова ЕБ, Моргун АВ, Мартынова ГП, Тохидпур A, Писарева НВ, Рузаева ВА, Салмина АБ (2016) GPR81 рецепторы лактата в регуляции функциональной активности клеток. Сибирск мед обозр 5: 17–27. [Boytsova EB, Morgun AV, Martynova GP, Tohidpur A, Pisareva NV, Ruzaeva VA, Salmina AB (2016) GPR81 lactate receptors in the regulation of cell functional activity. Siberian Med Rev 5: 17–27. (In Russ)].
Amer YO, Hebert-Chatelain E (2018) Mitochondrial cAMP-PKA signaling: What do we really know? Biochim Biophys Acta Bioenerg 1859(9): 868–877. https://doi.org/10.1016/j.bbabio.2018.04.005
Dixit S, Fessel JP, Harrison FE (2017) Mitochondrial dysfunction in the APP/PSEN1 mouse model of Alzheimer’s disease and a novel protective role for ascorbate. Free Radic Biol Med 112: 515–523. https://doi.org/10.1016/j.freeradbiomed.2017.08.021
Iijima-Ando K, Hearn SA, Shenton C, Gatt A, Zhao L, Iijima K (2009) Mitochondrial mislocalization underlies Abeta42-induced neuronal dysfunction in a Drosophila model of Alzheimer’s disease. PLoS One 4(12): e8310. https://doi.org/10.1371/journal.pone.0008310
Canepa E, Domenicotti C, Marengo B, Passalacqua M, Marinari UM, Pronzato MA, Fedele E, Ricciarelli R (2013) Cyclic adenosine monophosphate as an endogenous modulator of the amyloid-β precursor protein metabolism. IUBMB Life 65(2): 127–133. https://doi.org/10.1002/iub.1109
Terasaki T, Takakuwa S, Moritani S, Tsuji A (1991) Transport of monocarboxylic acids at the blood-brain barrier: studies with monolayers of primary cultured bovine brain capillary endothelial cells. J Pharmacol Exp Ther 258(3): 932–937.
An Y, Varma VR, Varma S, Casanova R, Dammer E, Pletnikova O, Chia CW, Egan JM, Ferrucci L, Troncoso J, Levey AI, Lah J, Seyfried NT, Legido-Quigley C, O’Brien R, Thambisetty M (2018) Evidence for brain glucose dysregulation in Alzheimer’s disease. Alzheimers Dement 14(3): 318–329. https://doi.org/10.1016/j.jalz.2017.09.011
Lu W, Huang J, Sun S, Huang S, Gan S, Xu J, Yang M, Xu S, Jiang X (2015) Changes in lactate content and monocarboxylate transporter 2 expression in Aβ25−35-treated rat model of Alzheimer’s disease. Neurol Sci 36(6): 871–876. https://doi.org/10.1007/s10072-015-2087-3
Hashimoto T, Hussien R, Cho HS, Kaufer D, Brooks GA (2008) Evidence for the mitochondrial lactate oxidation complex in rat neurons: demonstration of an essential component of brain lactate shuttles. PLoS One 3(8): e2915. https://doi.org/10.1371/journal.pone.0002915
Tescaroll F, Covolan L, Pellerin L (2014) Glutamate reduces glucose utilization while concomitantly enhancing AQP9 and MCT2 expression in cultured rat hippocampal neurons. Front Neurosci 8: 246. https://doi.org/10.3389/fnins.2014.00246
Hashimoto T, Brooks GA (2008) Mitochondrial lactate oxidation complex and an adaptive role for lactate production. Med Sci Sports Exerc 40(3): 486–494. https://doi.org/10.1249/MSS.0b013e31815fcb04
Nedergaar M, Goldman SA (1993) Carrier-mediated transport of lactic acid in cultured neurons and astrocytes. Am J Phys 265: R282–R289. https://doi.org/10.1152/ajpregu.1993.265.2.R282
Dienel GA (2012) Brain lactate metabolism: the discoveries and the controversies. J Cereb Blood Flow Metab 32: 1107–1138. https://doi.org/10.1038/jcbfm.2011.175
Yu X, Zhang R, Wei C, Gao Y, Yu Y, Wang L, Jiang J, Zhang X, Li J, Chen X (2021) MCT2 overexpression promotes recovery of cognitive function by increasing mitochondrial biogenesis in a rat model of stroke. Anim Cells Syst (Seoul) 25(2): 93–101. https://doi.org/10.1080/19768354.2021.1915379
Дополнительные материалы отсутствуют.
Инструменты
Российский физиологический журнал им. И.М. Сеченова