

Российский физиологический журнал им. И.М. Сеченова, 2022, T. 108, № 7, стр. 795-806
Перспективы генной терапии эпилепсии с использованием векторов на основе кальций-зависимых калиевых каналов
Е. С. Никитин 1, *, П. М. Балабан 1, А. В. Зайцев 2, 3
1 Институт высшей нервной деятельности и нейрофизиологии РАН
Москва, Россия
2 Институт эволюционной физиологии и биохимии им. И.М. Сеченова РАН
Санкт-Петербург, Россия
3 Национальный медицинский исследовательский центр им. В.А. Алмазова
Санкт-Петербург, Россия
* E-mail: nikitin@ihna.ru
Поступила в редакцию 14.04.2022
После доработки 20.06.2022
Принята к публикации 23.06.2022
- EDN: BLLTQD
- DOI: 10.31857/S0869813922070068
Аннотация
Эпилепсия является одним из распространенных неврологических заболеваний человека, при этом почти трети больных современные противосудорожные препараты не помогают полностью избавиться от эпилептических приступов. Поэтому поиск и разработка новых подходов лечения эпилепсии остается одной из актуальных проблем современной фундаментальной нейробиологии и клинической неврологии. В последние годы все большее внимание исследователей привлекает генная терапия эпилепсии. На сегодняшний день для генной терапии приоритетным направлением считается гиперэкспрессия каких-либо генов в нейронах, снижающих активность нейронных сетей в эпилептическом очаге, включая как экспрессию белков-каналов, так и тормозных нейромодуляторов. В данном обзоре мы рассматриваем возможность использования гиперэкспрессии кальций-зависимых калиевых каналов. Преимущество выбора данной подгруппы каналов для генной терапии может заключаться в том, что максимальная активация кальций-зависимых калиевых каналов и их гиперполяризующее действие реализуется при накоплении внутриклеточного кальция, что наблюдается при эпилептической активности в нейронных сетях. В клетках млекопитающих экспрессируется несколько подтипов кальций-зависимых калиевых каналов. Анализ имеющихся экспериментальных и клинических данных показывает, что каналы с промежуточной (IK-каналы) и малой проводимостью (SK-каналы) могут обладать высоким терапевтическим потенциалом для применения в генной терапии эпилепсии.
По разным оценкам от 0.4 до 1% населения страдает от эпилепсии [1, 2]. Несмотря на успехи в создании новых противоэпилептических препаратов, полного избавления от судорожных припадков не удается достичь почти у трети больных [2]. Наиболее эффективным методом лечения в таком случае является хирургическое удаление эпилептического очага [3], но и этот метод подходит не для всех пациентов из-за неприемлемых побочных последствий удаления мозговой ткани. Кроме того, есть существенный риск возникновения новых очагов после операционного вмешательства [4].
В последние годы все большее внимание исследователей привлекает генная терапия эпилепсии [5, 6]. Генная терапия традиционно определялась как способ замены дефектной копии гена его нормально работающей копией и восстановлением функции клеток [7]. Однако идиопатические формы эпилепсии, вызванные мутацией какого-то одного гена и вследствие этого нарушенной функцией канала или рецептора, встречаются относительно редко, а в большинстве случаев выявить конкретный генетический фактор не удается [8]. Кроме того, доставка генетического материала одновременно в обширные области мозга технически сложна, поэтому считается, что генная терапия имеет наибольшие перспективы для лечения фокальных форм эпилепсии [5].
Так как эпилептическая активность обусловлена нарушением баланса возбуждения и торможения в очаге, то усилия исследователей направлены в первую очередь на регуляцию возбудимости нейронов. Первоначально основные подходы были основаны на гиперэкспрессии ингибирующих пептидов, таких как галанин [9] или NPY [10], или подавлении возбудимости нейронов путем гиперэкспрессии в них калиевых каналов [11–13]. Однако эти воздействия должны быть хорошо рассчитаны и строго дозированы, так как скорректировать экспрессию в дальнейшем сложно. При недостаточной экспрессии противосудорожный эффект не достигается, а при избыточной – происходит нарушение функционирования нейронных сетей из-за избыточного торможения. Поэтому с практической точки зрения более интересны подходы, при которых эффективность торможения будет зависеть от степени возбуждения в нейронной сети, то есть будет задействован механизм обратной связи.
Одним из наиболее очевидных кандидатов для реализации такого подхода являются кальций-зависимые калиевые каналы [14]. Их активация обусловлена входом ионов кальция через ионотропные глутаматные рецепторы и потенциал-зависимые кальциевые каналы при деполяризации нейронов, наблюдаемой при преобладании процессов возбуждения в нейронной сети. Работа кальций-зависимых калиевых каналов ведет в свою очередь к гиперполяризации нейронов и ослаблению их спайковой активности. Кальций-зависимые калиевые каналы весьма разнообразны по своим характеристикам [14]. В данном обзоре мы рассмотрим, какие типы каналов могут обладать наиболее высоким терапевтическим потенциалом при генной терапии и какие экспериментальные данные получены к настоящему времени.
ОБЩАЯ ХАРАКТЕРИСТИКА КАЛЬЦИЙ-ЗАВИСИМЫХ КАЛИЕВЫХ КАНАЛОВ
В геноме млекопитающих ~80 генов кодируют α-субъединицы калиевых каналов, формирующие проводящую пору, и еще ~10 генов – вспомогательные регуляторные β-субъединицы [15]. Кроме того, посттранскрипционные модификации увеличивают разнообразие, поскольку варианты альтернативного сплайсинга одного гена могут сосуществовать в одном нейроне [16, 17].
Кальций-зависимые калиевые каналы являются одним из четырех основных семейств калиевых каналов, включающих в себя: (1) каналы утечки (tandem pore domain, TWIK/TRAAK/TREK/TASK), (2) каналы входящего выпрямляющего тока (Kir1–6, обычно активируются G-белками), (3) потенциал-активируемые каналы (Kv1–12, основные быстрые каналы, реполяризующие нейрон после потенциала действия) и (4) кальций-зависимые каналы, подразделяемые на основе проводимости на 3 группы: BK (large conductance или big potassium, KCa1.1) – каналы большой проводимости от 100 до 300 пСм; SK (small potassium, KCa2.1–2.3) – каналы малой проводимости от 5 до 25 пСм; IK (intermediate potassium, KCa3.1) – каналы промежуточной проводимости от 25 до 100 пСм. Особенностью кальций-зависимых калиевых каналов является их активация или модуляция внутриклеточным кальцием. Современная номенклатура этих каналов приведена в табл. 1.
Таблица 1.
Международные номенклатурные названия кальций-зависимых калиевых каналов человека и кодирующих их генов
| IUPHAR | HGNC | Другие часто используемые аббревиатуры | Полные названия |
|---|---|---|---|
| KCa1.1 | KCNMA1 | Slo, Slo1, BK, Maxi-K | Big potassium; large conductance calcium-activated potassium channels |
| KCa2.1 | KCNN1 | SKCa1, SK1 | Small potassium; small conductance calcium-activated potassium channels |
| KCa2.2 | KCNN2 | SKCa2, SK2 | |
| KCa2.3 | KCNN3 | SKCa3, SK3 | |
| KCa3.1 | KCNN4 | IKCa1, IK | Intermediate potassium; intermediate conductance calcium-activated potassium channels |
| KCa4.1 | KCNT1 | Slack, Slo2.2 | Potassium channel subfamily T, member 1 |
| KCa4.2 | KCNT2 | Slick, Slo2.1 | Potassium channel subfamily T, member 2 |
| KCa5.1 | KCNU1 | Slo3 | Potassium channel, subfamily U, member 1 |
Кальций-зависимый канал представляет собой тетрамер из 4 α-субъединиц и 4 β-субъединиц. Альфа-субъединица состоит из шести (SK) или семи (BK) трансмембранных единиц и большой внутриклеточной области. Именно эта субъединица образует пору, сенсор напряжения и кальций-чувствительную область (рис. 1). Сенсором напряжения служит трансмембранная область S4, содержащая несколько остатков аргинина, аналогично другим потенциал-зависимым калиевым каналам. Линкер между областями S5 и S6 служит для формирования поры канала, селективного для ионов калия. Внутри клетки основной частью является “кальциевая чаша”. Считается, что эта “чаша” является местом связывания кальция в BK-каналах [18].
Рис. 1.
Молекулярная структура BK-канала. Схожую структуру имеют и другие подтипы этого семейства каналов (из https://en.wikipedia.org/wiki/Calcium-activated_potassium_channel).
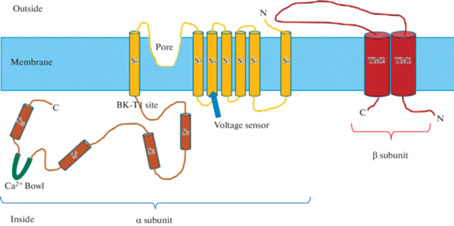
Бета-субъединица канала является регуляторной субъединицей канала. Регуляторная β-субъединица состоит из двух предполагаемых трансмембранных (ТМ) единиц, соединенных внеклеточной петлей (рис. 1). Для BK-каналов известно четыре различных вида β-субъединиц [18]. Каналы, содержащие β1- или β4-субъединицы, редко инактивируются [19, 20], тогда как экспрессия β2-субъединицы, наоборот, приводит к инактивации BK-тока, что наблюдается в хромаффинных клетках и гиппокампальных нейронах [19]. К аналогичному тормозному эффекту приводит экспрессия β3-субъединицы [21].
Кальций-зависимые каналы открываются при повышении уровня внутриклеточного кальция или модулируются им, что позволяет этим каналам отставлено реагировать на значительные изменения в уровне внутриклеточного кальция, к которым обычно приводит интенсивная электрическая активность нейронов. Однако BK-каналы также активируются при деполяризации мембраны и обеспечивают быструю следовую гиперполяризацию после потенциала действия. Считается, что эти различные режимы активации BK-каналов не зависят друг от друга.
Калиевые каналы SK [22] и IK [23] подтипов являются потенциал-независимыми и активируются внутриклеточным кальцием. Эти каналы отвечают за следовую гиперполяризацию после пачечной активности нейронов с достаточно медленной кинетикой (десятки и даже сотни миллисекунд). Ген IK-каналов (SK4/KCNN4) демонстрирует гомологию ~40% с генами каналов SK типа 1–3 [24], что может повлечь за собой образование химерных гетеромерных каналов в нейронах, одновременно экспрессирующих KCa3.1 и один из подтипов SK-каналов [25].
В качестве сенсора внутриклеточного кальция эти каналы используют универсальный цитоплазматический кальциевый кофактор кальмодулин (CaM), аллостерический комплекс которого с кальцием вызывает открытие этих каналов [26]. Одним из наиболее ярких проявлений активации KCa3.1 каналов в нейроне является продолжительная (медленная) следовая гиперполяризация, отставленная от быстрой следовой гиперполяризации, которую вызывают потенциал-зависимые каналы, и сохраняющаяся сотни миллисекунд [23, 27]. Как следствие, KCa3.1-зависимая медленная следовая гиперполяризация улучшает способность нейрона к быстрой адаптации своего электрического спайкового разряда, увеличивая интервалы между последующими потенциалами действия [27].
В целом, открытие калиевых каналов после генерации разрядов снижает возбудимость нейрона и возвращает потенциал на мембране к значению потенциала покоя или даже ниже, вызывая следовую гиперполяризацию (состояние с пониженной вероятностью генерации электрических разрядов). Кроме того, калиевые каналы регулируют потенциал покоя, ограничивают частоту и длительность отдельных потенциалов действия, а также вносят вклад в рефрактерный период нейрона [26]. Важные функциональные особенности калиевых каналов определяют не только их вспомогательную роль в поддержании физиологических параметров электрической активности, но также их участие в жизненно-важных функциях нейрона, без которых невозможен устойчивый баланс возбуждения и торможения в ЦНС, который нарушается при развитии эпилепсии.
КАНАЛОПАТИИ КАЛИЕВЫХ КАНАЛОВ И ЭПИЛЕПСИЯ
Предполагается, что определенные каналопатии могут быть причиной развития судорожной активности в ЦНС и развития эпилепсии. Генетический скрининг пациентов с различными формами эпилепсии выявил многочисленные корреляции между проявлением заболевания и мутациями потери/усиления функции или нарушения проводимости различных калиевых каналов [28]. По крайней мере несколькими независимыми исследователями в каждом случае, у пациентов были подтверждены эпилептогенные мутации (нелетальные, но сопровождающиеся эпилептическим фенотипом) представителей группы кальций-зависимых каналов (BK и KCa4.1), группы выпрямляющих каналов (Kir4 и Kir6), и множество потенциал-активируемых каналов (Kv1, Kv2, Kv3, Kv4, Kv7.2/3, Kv8.2, Kv10, Kv11.1). Из всех патологий каналов значительно выделяется нейроспецифический канал Kv7.2/3, мутации которого встречались примерно в половине случаев. Обычно мутации Kv7.2/3 вызывают формы эпилепсии, проявляющиеся в раннем детстве и варьирующие по степени тяжести от легкого неонатального синдрома, до тяжелых форм фармакорезистентной эпилепсии и ранней энцефалопатии, сопровождающейся интеллектуальной инвалидностью [29]. Для гена Kv7.3 (KCNQ3) были также определены позиции мутаций, наиболее сильно влияющие на прогноз тяжести заболевания [28]. В одном из вариантов семейной аутосомно-доминантной ночной лобной эпилепсии (ADNFLE) была выявлена мутация в гене KCNT1 (KCa4.1) [30].
KCNMA1-каналопатии (BK-каналы) часто сопровождаются наличием двигательных дисфункций и развитием эпилепсии (у 35 из 69 человек с KCNMA1-каналопатией диагностирована эпилепсия) [31], однако к настоящему времени не выявлено связи между увеличением (gain of function) или ослаблением (loss of function) функциональной активности BK-каналов и клиническими проявлениями эпилепсии. Любые нарушения функциональной активности BK-каналов могут сопровождаться судорогами [31, 32].
В нейронах нижних бугров четверохолмия крыс с генетической предрасположенностью к эпилепсии (линия GEPR) показан сниженный уровень экспрессии белков SK1- и SK3-каналов, в то время как экспрессия SK2-каналов повышена [33]. Интересно, что активация SK-каналов с помощью 1-этил-2-бензимидазолинона (1-EBIO) позволяла эффективно подавлять аудиогенные судороги, характерные для этой линии крыс [34].
НАРУШЕНИЯ ЭКСПРЕССИИ КАЛИЕВЫХ КАНАЛОВ ПРИ ЭПИЛЕПСИИ
Не только каналопатии могут быть причиной развития эпилепсии, но и эпилептическая активность может повлиять на экспрессию и активность различных каналов. Это становится очевидным при изучении моделей эпилепсии на животных, когда изменения функций каналов наблюдаются после повреждающего воздействия на мозг. Например, экспрессия Kv4.2-канала временно повышается в областях CA1 и CA3 гиппокампа крыс в латентную фазу литий-пилокарпиновой модели эпилепсии, тогда как в хронической фазе экспрессия снижается [35].
В гиппокампе хронических крыс-эпилептиков часто наблюдается снижение функций кальций-зависимых BK-каналов [36], однако может наблюдаться и обратная картина. В модели эпилепсии, вызванной применением пикротоксина, токи через BK-каналы усиливаются у ювенильных крыс [37]. Неоднозначное действие BK-каналов на судорожную активность показало и применение фармакологических агентов. Так, некоторый эффект на ослабление судорог вызывали как активаторы, так и блокаторы BK-каналов [38].
Долговременное снижение экспрессии SK3-каналов как на уровне белка, так и на уровне мРНК, было показано в гиппокампе крыс после эпилептического статуса, вызванного пилокарпином [39]. Также, недавние исследования [40] показали, что в пилокарпиновой модели височной эпилепсии в гиппокампе крыс происходит значительное долговременное снижение экспрессии KCa3.1-каналов, что сопровождается снижением медленной кальций-зависимой следовой гиперполяризации. Снижение уровня экспрессии KCa3.1-каналов (IK-каналов) опосредовано сигнальным путем, связанным с цАМФ-зависимой протеинкиназой А (PKA). Интересно, что применение ингибиторов PKA приводит к восстановлению функции KCa3.1-каналов и нормализации возбудимости нейронов [40]. Эти данные свидетельствуют о вовлеченности кальций-зависимых калиевых каналов в развитие эпилепсии, в то время как их аллели могут определять предрасположенность к этому заболеванию.
ГЕННАЯ ТЕРАПИЯ ЭПИЛЕПСИИ С ИСПОЛЬЗОВАНИЕМ ВЕКТОРОВ НА ОСНОВЕ КАЛИЕВЫХ КАНАЛОВ
Традиционно, генно-терапевтические подходы разделяют на четыре основные категории: (1) замена гена при моногенных заболеваниях, (2) добавление гена при сложных патологиях, (3) изменение экспрессии генов и таргетинг РНК и (4) редактирование генома для внесения изменений [7]. При эпилепсии поражаются соматические нервные клетки, подвергнувшиеся полной дифференцировке и объединенные в сложные сети, организация и все функции которых остаются не до конца изученными. Поэтому на сегодняшний день для генной терапии эпилепсии используют подход № 2 (gain of function), добавляя новый ген для снижения активности нейрона-мишени. В качестве мишени обычно выбираются глутаматергические пирамидные нейроны кортикальных структур (неокортекса и гиппокампа).
Первые попытки генной терапии, возникшие на волне прорывных методов оптогенетики [11, 12], не увенчались значительными успехами, а исследователи, в свою очередь, ставили себе целью больше демонстрацию возможности управляемого снижения активности нейронной сети, а не разработку реального терапевтического метода. Кроме того, сложные оптогенетические методы требуют имплантации вглубь головного мозга передающих световодов, соединенных с внешним источником для фотоактивации светочувствительных каналов, а также удаления части ткани на пути световода, поэтому методы оптогенетической стимуляции вряд ли когда-либо будут применяться в клинике в таком виде. Поэтому в современных разработках предпочтение отдается каналам, активация которых происходит без необходимости использования внешних сигналов, а в ответ на какие-то внутренние изменения в нервных клетках по системе петли обратной связи.
В экспериментальных моделях генной терапии эпилепсии используют экспрессию дополнительных калиевых каналов или другие опосредованные способы модификации калиевой проводимости, которая представляется наиболее перспективным средством достижения терапевтических целей из-за важнейшей способности калиевых каналов к глубокой гиперполяризации нейрона. На первый взгляд, как альтернативу калиевым каналам можно было бы использовать анионные (хлорные) ионные каналы, также гиперполяризующие нейрон при их активации. Наиболее известный пример внутрисетевого торможения в ЦНС – это торможение с помощью ГАМК, медиатора интернейронов, активирующего ГАМК-чувствительные хлорные каналы у пирамидных нейронов. Однако хлорный потенциал реверсии сильно варьирует от нейрона к нейрону, а также различен в нейрональных компартментах одного нейрона.
По литературным данным, в гиперполяризованых нейронах открытие хлорных ГАМК-чувствительных каналов может вызвать серии потенциалов действия [41], в то время как оптогенетическая активация светочувствительных хлорных каналов может вызывать как подавление, так и усиление электрической спайковой активности [42, 43]. В недавней работе [44] было продемонстрировано, что проиктогенный эффект активации ГАМКергических интернейронов в определенных условиях усиливает судороги, однако при дополнительной оверэкспрессии калий-хлорного транспортера КСС2 в постсинаптических пирамидных нейронах активация ГАМКергических интернейронов уже подавляет судорожную активность. Известно, что значение потенциала реверсии хлорного тока расположено близко от потенциала покоя нейрона, поэтому даже небольшие сдвиги концентраций хлора во внутриклеточном и внеклеточном пространстве, происходящие при судорогах, способны его инвертировать, и тогда открытие хлорных каналов начинает уже возбуждать нейрон вместо торможения. Ситуацию изменяет усиление функции калий-хлорных транспортеров, которые быстро возвращают ионы хлора к исходным концентрациям, используя как движущую силу более стабильный и негативный градиент ионов калия.
Поэтому гиперполяризация нейронов в эпилептическом очаге с помощью дополнительной калиевой проводимости с теоретической точки зрения представляется на сегодняшний день наиболее эффективным подходом. Увеличить калиевую проводимость можно, используя калиевые каналы, которые будут быстро активироваться при гипервозбуждении в нейронной сети, обладать достаточно высокой проводимостью и относительно медленной инактивацией, чтобы успешно подавлять эпилептическую активность. Таким набором свойств потенциально обладают некоторые подтипы потенциал-активируемых калиевых каналов и кальций-зависимых калиевых каналов. Среди кальций-зависимых калиевых каналов оптимальным сочетанием свойств, вероятно, обладают IK и в меньшей степени, из-за малой проводимости, SK-каналы.
К настоящему времени нет опубликованных работ по генной терапии эпилепсии с использованием какого-либо подтипа кальций-зависимых калиевых каналов. Среди моделей генной терапии эпилепсии с использованием калиевой проводимости наиболее глубоко была разработана модель вектора на основе потенциал-активируемых калиевых каналов Kv1.1 (рис. 2). Группа исследователей из UCL (Университетский колледж Лондона) впоследствии дополнительно модифицировала этот канал генно-инженерными методами для уменьшения его инактивации и увеличения длительности открытого состояния с целью улучшения противосудорожного эффекта [13]. При возникновении судорог канал активировался многочисленными потенциалами действия в эпилептическом очаге и обеспечивал дополнительное обратное торможение сверхактивных нейронов.
Рис. 2.
Генная терапия эпилепсии модифицированными каналами Kv1.1 (ген KCNA1) в модели заболевания, вызванного токсином столбняка, введенным в кору больших полушарий грызуна. (а) – Репрезентативные записи судорог в теменной коре через две недели после инъекции столбнячного токсина (TeNT) в первичную зрительную кору (также приведены увеличенные участки 1–4). (b) – Схема эксперимента с последовательностью экспериментальных воздействий и измерений. (c) – Внедрение в нейроны вектора Lenti-CMV-KCNA1 ограничивалось небольшой областью ~500–1000 мкм вблизи места инъекции. (d) – Число судорог у животных, получивших инъекцию Kv1 (KCNA1), достоверно снижается по сравнению с контрольными животными (GFP), что говорит об ослаблении модельной эпилепсии при терапии калиевыми каналами. (e) – Суммарная гистограмма показывает достоверное снижение судорожной активности у животных, получивших инъекцию Kv1 (KCNA1) (синяя кривая) по сравнению с контрольными животными (красная кривая). С незначительными модификациями из [13].
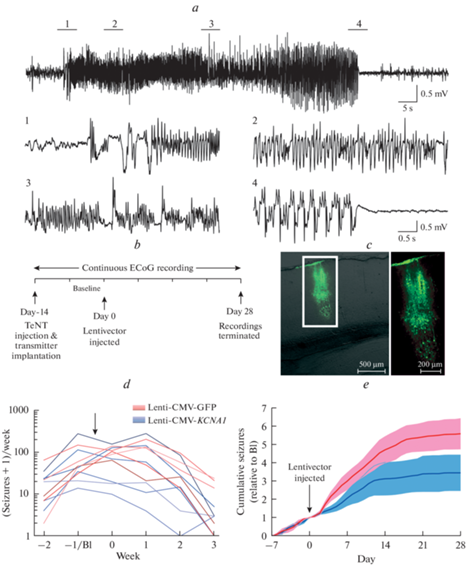
Лентивирусный вектор с модифицированным каналом Kv1.1 протестировали на модели фокальной эпилепсии, вызванной инъекцией в мозг животных столбнячного токсина, необратимо повреждающего нейроны и вызывающего локальную патологическую активность. Инъекция вектора с Kv1.1 в эпилептический очаг вызывала достоверное снижение интенсивности эпилептических разрядов и стабильную компенсацию судорожной активности [13]. По результатам работы исследователи из UCL даже запатентовали последовательность генетически-модифицированного канала Kv1.1 как средство от эпилепсии, рассчитывая на скорое внедрение в клинику этого способа лечения.
Также стоит отметить другое важное исследование по генной терапии с использованием вирусного вектора для оверэкспрессии пре-динорфина [45]. Когда нейроны подвергаются судорожной активации, большее количество созревшего динорфина выбрасывается наружу в межнейронное соединение и активирует опиоидные рецепторы, которые, в свою очередь, открывают тормозные калиевые Kir-каналы. Это обеспечивает механизм обратной связи и позволяет нейронам самим реагировать на свою гиперактивность, выбрасывая дополнительный динорфин в случае необходимости.
ЗАКЛЮЧЕНИЕ
Фокальная эпилепсия часто бывает резистентной к лекарствам, а хирургическая операция по удалению эпилептогенного очага не всегда выполнима или эффективна. Поэтому разработка пространственно-ограниченной, специфической генной терапии, которая избирательно изменяет активность нейронов в очаге припадка, может стать важным шагом в лечении эпилепсии. В случае успеха эта стратегия также поможет свести к минимуму побочные эффекты, наблюдаемые при использовании фармакологических подходов.
Поскольку калиевые каналы чрезвычайно важны для внутреннего ограничения собственной активности и возбудимости нейрона, то воздействие на калиевую проводимость методом генной терапии имеет громадный потенциал для лечения эпилептических расстройств. С теоретической точки зрения гиперэкспрессия кальций-зависимых калиевых каналов IK подтипа, обладающих достаточно высокой проводимостью и медленной кинетикой инактивации, представляется одним из наиболее перспективных вариантов для подавления гипервозбудимости нейронов в эпилептическом очаге. Однако такой вариант генной терапии еще не реализован и необходима его экспериментальная проверка. Тем не менее, наличие достаточно большого спектра методов генной терапии, уже доказавших свою эффективность в доклинических исследованиях, позволяет предположить, что клиническое испытание некоторых из этих методов для терапии эпилепсии начнется уже в ближайшие годы.
Список литературы
Banerjee PN, Filippi D, Hauser AW (2009) The descriptive epidemiology of epilepsy—A review. Epilepsy Res 85: 31–45.https://doi.org/10.1016/j.eplepsyres.2009.03.003
Janmohamed M, Brodie MJ, Kwan P (2020) Pharmacoresistance – Epidemiology, mechanisms, and impact on epilepsy treatment. Neuropharmacology 168: 107790.https://doi.org/10.1016/j.neuropharm.2019.107790
Engel J (2018) The current place of epilepsy surgery. Curr Opin Neurol 31: 192–197.https://doi.org/10.1097/WCO.0000000000000528
Schramm J (2008) Temporal lobe epilepsy surgery and the quest for optimal extent of resection: a review. Epilepsia 49: 1296–307.https://doi.org/10.1111/j.1528-1167.2008.01604.x
Walker MC, Kullmann DM (2020) Optogenetic and chemogenetic therapies for epilepsy. Neuropharmacology 168: 107751.https://doi.org/10.1016/j.neuropharm.2019.107751
Simonato M (2014) Gene therapy for epilepsy. Epilepsy Behav 38: 125–130.https://doi.org/10.1016/j.yebeh.2013.09.013
Wang D, Gao G (2014) State-of-the-art human gene therapy: part II. Gene therapy strategies and clinical applications. Discov Med 18: 151–161.
Thomas RH, Berkovic SF (2014) The hidden genetics of epilepsy—a clinically important new paradigm. Nat Rev Neurol 10: 283–292.https://doi.org/10.1038/nrneurol.2014.62
McCown TJ (2006) Adeno-associated Virus-Mediated Expression and Constitutive Secretion of Galanin Suppresses Limbic Seizure Activity in Vivo. Mol Ther 14: 63–68.https://doi.org/10.1016/J.YMTHE.2006.04.004
Noè F, Pool AH, Nissinen J, Gobbi M, Bland R, Rizzi M, Balducci C, Ferraguti F, Sperk G, During MJ, Pitkänen A, Vezzani A (2008) Neuropeptide Y gene therapy decreases chronic spontaneous seizures in a rat model of temporal lobe epilepsy. Brain 131: 1506–1515.https://doi.org/10.1093/BRAIN/AWN079
Bernard C (2012) Treating Epilepsy with a Light Potassium Diet. Sci Transl Med 4.https://doi.org/10.1126/SCITRANSLMED.3005297
Wykes RC, Heeroma JH, Mantoan L, Zheng K, MacDonald DC, Deisseroth K, Hashemi KS, Walker MC, Schorge S, Kullmann DM (2012) Optogenetic and potassium channel gene therapy in a rodent model of focal neocortical epilepsy. Sci Transl Med 4: 161ra152.https://doi.org/10.1126/scitranslmed.3004190
Snowball A, Chabrol E, Wykes RC, Shekh-Ahmad T, Cornford JH, Lieb A, Hughes MP, Massaro G, Rahim AA, Hashemi KS, Kullmann DM, Walker MC, Schorge S (2019) Epilepsy Gene Therapy Using an Engineered Potassium Channel. J Neurosci 39: 3159–3169.https://doi.org/10.1523/JNEUROSCI.1143-18.2019
Nikitin ES, Balaban PM (2021) Diversity and Functional Features of Calcium-Dependent Potassium Channels as Determinants of Their Role in the Plasticity of Cerebral Neurons. Neurosci Behav Physiol 519 (51): 1239–1243.https://doi.org/10.1007/S11055-021-01186-Z
Trimmer JS (2015) Subcellular Localization of K+ Channels in Mammalian Brain Neurons: Remarkable Precision in the Midst of Extraordinary Complexity. Neuron 85: 238–256.https://doi.org/10.1016/j.neuron.2014.12.042
Bell TJ, Miyashiro KY, Sul J-Y, Buckley PT, Lee MT, McCullough R, Jochems J, Kim J, Cantor CR, Parsons TD, Eberwine JH (2010) Intron retention facilitates splice variant diversity in calcium-activated big potassium channel populations. Proc Natl Acad Sci U S A 107: 21152–21157. https://doi.org/10.1073/pnas.1015264107
Tian Y, Liao IH, Zhan X, Gunther JR, Ander BP, Liu D, Lit L, Jickling GC, Corbett BA, Bos-Veneman NGP, Hoekstra PJ, Sharp FR (2011) Exon expression and alternatively spliced genes in tourette syndrome. Am J Med Genet Part B Neuropsychiatr Genet 156: 72–78.https://doi.org/10.1002/ajmg.b.31140
Ghatta S, Nimmagadda D, Xu X, O’Rourke ST (2006) Large-conductance, calcium-activated potassium channels: structural and functional implications. Pharmacol Ther 110: 103–116. https://doi.org/10.1016/j.pharmthera.2005.10.007
Wallner M, Meera P, Toro L (1999) Molecular basis of fast inactivation in voltage and Ca2+-activated K+ channels: A transmembrane β-subunit homolog. Proc Natl Acad Sci U S A 96: 4137–4142.https://doi.org/10.1073/pnas.96.7.4137
Meera P, Wallner M, Toro L (2000) A neuronal β subunit (KCNMB4) makes the large conductance, voltage- and Ca2+-activated K+ channel resistant to charybdotoxin and iberiotoxin. Proc Natl Acad SciU S A 97: 5562–5567.https://doi.org/10.1073/pnas.100118597
Xia X-M, Ding JP, Lingle CJ (1999) Molecular Basis for the Inactivation of Ca2+- and Voltage-Dependent BK Channels in Adrenal Chromaffin Cells and Rat Insulinoma Tumor Cells. J Neurosci 19: 5255–5264.https://doi.org/10.1523/JNEUROSCI.19-13-05255.1999
Köhler M, Hirschberg B, Bond CT, Kinzie JM, Marrion N V., Maylie J, Adelman JP (1996) Small-Conductance, Calcium-Activated Potassium Channels from Mammalian Brain. Science (80) 273: 1709–1714.https://doi.org/10.1126/science.273.5282.1709
King B, Rizwan AP, Asmara H, Heath NC, Engbers JDT, Dykstra S, Bartoletti TM, Hameed S, Zamponi GW, Turner RW (2015) IKCa Channels Are a Critical Determinant of the Slow AHP in CA1 Pyramidal Neurons. Cell Rep 11: 175–182.https://doi.org/10.1016/j.celrep.2015.03.026
Joiner WJ, Wang L-Y, Tang MD, Kaczmarek LK (1997) hSK4, a member of a novel subfamily of calcium-activated potassium channels. Proc Natl Acad Sci U S A 94: 11013–11018.https://doi.org/10.1073/pnas.94.20.11013
Higham J, Sahu G, Wazen R-M, Colarusso P, Gregorie A, Harvey BSJ, Goudswaard L, Varley G, Sheppard DN, Turner RW, Marrion N V (2019) Preferred Formation of Heteromeric Channels between Coexpressed SK1 and IKCa Channel Subunits Provides a Unique Pharmacological Profile of Ca2+-Activated Potassium Channels. Mol Pharmacol 96: 115–126.https://doi.org/10.1124/mol.118.115634
Bean BP (2007) The action potential in mammalian central neurons. Nat Rev Neurosci 8: 451–465.https://doi.org/10.1038/nrn2148
Roshchin MV, Ierusalimsky VN, Balaban PM, Nikitin ES (2020) Ca2+-activated KCa3.1 potassium channels contribute to the slow afterhyperpolarization in L5 neocortical pyramidal neurons. Sci Rep 10: 14484.https://doi.org/10.1038/s41598-020-71415-x
Nikitin E, Vinogradova L (2021) Potassium channels as prominent targets and tools for the treatment of epilepsy. Expert Opin Ther Targets 25: 223–235.https://doi.org/10.1080/14728222.2021.1908263
Miceli F, Soldovieri MV, Ambrosino P, Barrese V, Migliore M, Cilio MR, Taglialatela M (2013) Genotype–phenotype correlations in neonatal epilepsies caused by mutations in the voltage sensor of K v 7.2 potassium channel subunits. Proc Natl Acad Sci U S A 110: 4386–4391.https://doi.org/10.1073/pnas.1216867110
Heron SE, Smith KR, Bahlo M, Nobili L, Kahana E, Licchetta L, Oliver KL, Mazarib A, Afawi Z, Korczyn A, Plazzi G, Petrou S, Berkovic SF, Scheffer IE, Dibbens LM (2012) Missense mutations in the sodium-gated potassium channel gene KCNT1 cause severe autosomal dominant nocturnal frontal lobe epilepsy. Nat Genet 44: 1188–1190.https://doi.org/10.1038/ng.2440
Miller JP, Moldenhauer HJ, Keros S, Meredith AL (2021) An emerging spectrum of variants and clinical features in KCNMA1-linked channelopathy. Channels 15: 447–464.https://doi.org/10.1080/19336950.2021.1938852
N’Gouemo P (2014) BK Ca channel dysfunction in neurological diseases. Front Physiol 5: 373.https://doi.org/10.3389/fphys.2014.00373
N’Gouemo P, Yasuda RP, Faingold CL (2009) Protein expression of small conductance calcium-activated potassium channels is altered in inferior colliculus neurons of the genetically epilepsy-prone rat. Brain Res 1270: 107–111.https://doi.org/10.1016/j.brainres.2009.02.034
Khandai P, Forcelli PA, N’Gouemo P (2020) Activation of small conductance calcium-activated potassium channels suppresses seizure susceptibility in the genetically epilepsy-prone rats. Neuropharmacology 163: 107865.https://doi.org/10.1016/j.neuropharm.2019.107865
Su T, Cong WD, Long YS, Luo AH, Sun WW, Deng WY, Liao WP (2008) Altered expression of voltage-gated potassium channel 4.2 and voltage-gated potassium channel 4-interacting protein, and changes in intracellular calcium levels following lithium-pilocarpine-induced status epilepticus. Neuroscience 157: 566–576.https://doi.org/10.1016/j.neuroscience.2008.09.027
Pacheco Otalora LF, Hernandez EF, Arshadmansab MF, Francisco S, Willis M, Ermolinsky B, Zarei M, Knaus H-G, Garrido-Sanabria ER (2008) Down-regulation of BK channel expression in the pilocarpine model of temporal lobe epilepsy. Brain Res 1200: 116–131.https://doi.org/10.1016/j.brainres.2008.01.017
Shruti S, Clem RL, Barth AL (2008) A seizure-induced gain-of-function in BK channels is associated with elevated firing activity in neocortical pyramidal neurons. Neurobiol Dis 30: 323–330.https://doi.org/10.1016/j.nbd.2008.02.002
Leo A, Citraro R, Constanti A, De Sarro G, Russo E (2015) Are big potassium-type Ca2+-activated potassium channels a viable target for the treatment of epilepsy? Expert Opin Ther Targets 19: 911–926.https://doi.org/10.1517/14728222.2015.1026258
Oliveira MS, Skinner F, Arshadmansab MF, Garcia I, Mello CF, Knaus H-G, Ermolinsky BS, Otalora LFP, Garrido-Sanabria ER (2010) Altered expression and function of small-conductance (SK) Ca2+-activated K+ channels in pilocarpine-treated epileptic rats. Brain Res 1348: 187–199.https://doi.org/10.1016/j.brainres.2010.05.095
Tiwari MN, Mohan S, Biala Y, Yaari Y (2019) Protein Kinase A-Mediated Suppression of the Slow Afterhyperpolarizing KCa3.1 Current in Temporal Lobe Epilepsy. J Neurosci 39: 9914–9926.https://doi.org/10.1523/JNEUROSCI.1603-19.2019
Chavas J, Marty A (2003) Coexistence of Excitatory and Inhibitory GABA Synapses in the Cerebellar Interneuron Network. J Neurosci 23: 2019–2031.https://doi.org/10.1523/JNEUROSCI.23-06-02019.2003
Malyshev AY, Roshchin MV, Smirnova GR, Dolgikh DA, Balaban PM, Ostrovsky MA (2017) Chloride conducting light activated channel GtACR2 can produce both cessation of firing and generation of action potentials in cortical neurons in response to light. Neurosci Lett 640: 76–80.https://doi.org/10.1016/j.neulet.2017.01.026
Messier JE, Chen H, Cai Z-L, Xue M (2018) Targeting light-gated chloride channels to neuronal somatodendritic domain reduces their excitatory effect in the axon. Elife 7.https://doi.org/10.7554/eLife.38506
Magloire V, Cornford J, Lieb A, Kullmann DM, Pavlov I (2019) KCC2 overexpression prevents the paradoxical seizure-promoting action of somatic inhibition. Nat Commun 10: 1225.https://doi.org/10.1038/s41467-019-08933-4
Agostinho AS, Mietzsch M, Zangrandi L, Kmiec I, Mutti A, Kraus L, Fidzinski P, Schneider UC, Holtkamp M, Heilbronn R, Schwarzer C (2019) Dynorphin-based “release on demand” gene therapy for drug-resistant temporal lobe epilepsy. EMBO Mol Med 11: e9963.https://doi.org/10.15252/emmm.201809963
Дополнительные материалы отсутствуют.
Инструменты
Российский физиологический журнал им. И.М. Сеченова