

Российский физиологический журнал им. И.М. Сеченова, 2023, T. 109, № 6, стр. 703-722
Механизмы физиологического ангиогенеза
А. Н. Иванов 1, Ю. Р. Чаббаров 1, *
1 Саратовский государственный медицинский университет им. В.И. Разумовского
Саратов, Россия
* E-mail: chabbarov@bk.ru
Поступила в редакцию 12.03.2023
После доработки 05.05.2023
Принята к публикации 07.05.2023
- EDN: WHBLCG
- DOI: 10.31857/S0869813923060043
Аннотация
Ангиогенез представляет собой образование новых кровеносных сосудов из уже существующих для удовлетворения потребности ткани в перфузии. Во взрослом организме это часто реализуется в процессе роста и регенерации организма. Ангиогенез привлекает внимание исследователей с позиции его терапевтической регуляции: усиление может способствовать существенному прогрессу в терапии ишемических заболеваний, а ингибирование активно изучается для лечения опухолевых заболеваний. Регуляция ангиогенеза невозможна без точных знаний его механизмов. Ангиогенез осуществляется либо путем миграции эндотелиальных клеток существующего сосуда во внеклеточный матрикс для формирования сосуда по направлению ангиогенных стимулов, либо путем разделения сформированного сосуда на два новых в результате образования перегородки, отграничивающей два самостоятельных просвета. В данном обзоре рассматриваются основные этапы каждого из видов физиологического ангиогенеза, их механизмы и регуляция.
ВВЕДЕНИЕ
Ангиогенез – это формирование новых кровеносных сосудов из уже существующих, что отличает его от васкулогенеза, характеризующегося образованием сосудов de novo с участием ангиобластов во время эмбрионального развития или эндотелиальных клеток-предшественниц постнатально [1]. За последние два десятилетия существенно возрос и продолжает расти интерес к разработке методов лечения, нацеленных на ангиогенез. Выделяют два основных направления такого подхода: стимуляция ангиогенеза и антиангиогенная терапия. Стимуляция ангиогенеза, направленная на восстановление сниженной перфузии ткани, рассматривается как потенциальный способ лечения заболеваний сердечно-сосудистой системы и хронических ишемических процессов [2]. Главной точкой приложения антиангиогенной терапии являются злокачественные новообразования, рост которых невозможен без формирования новых сосудов [3]. Разработка направленных на ангиогенез методов лечения особенно актуальна с учетом того, что ишемические и онкологические заболевания являются причиной большинства смертей и случаев инвалидности в Российской Федерации [4]. Все методы терапевтического воздействия на ангиогенез фокусируются на определенных звеньях его регуляции, именно поэтому понимание механизмов ангиогенеза является ключом к успеху. Цель данного обзора – систематизация современной информации об этапах физиологического ангиогенеза, их механизмах и регуляции.
ИНДУКЦИЯ АНГИОГЕНЕЗА
Физиологический ангиогенез является важнейшим условием роста и развития органов и тканей, а также осуществления компенсаторно-приспособительных процессов, в особенности регенерации [1]. Основными участниками ангиогенеза являются регулирующие его молекулы и эффекторные клетки. Паттерны экспрессии и секреции ангиогенных молекул зависят от множества факторов, включая индивидуальные генетические особенности организма, эпигенетические регуляторы (особое внимание исследователи уделяют микроРНК), особенности экспрессии и строения клеточных рецепторов и функционирование сигнальных путей. Основными эффекторными клетками ангиогенеза являются эндотелиальные клетки (ЭК); также участвуют перициты, гладкомышечные клетки, фибробласты, макрофаги. Фенотип клеток может меняться под воздействием множества модифицируемых и немодифицируемых факторов внутренней и внешней среды. В зависимости от этого динамически меняется способность клеток отвечать на определенные стимулы, что также влияет на процесс ангиогенеза. Таким образом, регуляция ангиогенеза является многоуровневым процессом. Подход к изучению механизмов регуляции ангиогенеза должен включать методы геномики, эпигеномики, транскриптомики, протеомики и метаболомики (мультиомный подход). Подходы, направленные на терапевтическую модуляцию ангиогенеза, могут быть нацелены на каждый из уровней регуляции [5, 6].
Пусковым механизмом физиологического ангиогенеза является снижение тканевой концентрации кислорода, при котором усиливается синтез индуцируемых гипоксией факторов. Ключевым регулятором клеточного ответа на гипоксию является семейство транскрипционных факторов HIF, причем особенно важен HIF-1α, активирующий сигнальные пути адаптации, которые переключают метаболизм клеток для повышения их жизнеспособности в условиях гипоперфузии [7]. HIF-1α взаимодействует с энхансерами, активируя таким образом транскрипцию более 100 нижестоящих генов, которые регулируют синтез ферментов гликолиза, эритропоэтина и множества факторов роста [8]. Быстрый и эффективный переход на гликолиз является важнейшим условием адаптации ЭК к ишемии и участия в ангиогенезе. Активация гликолиза в ЭК осуществляется путем повышения экспрессии 6-фосфофрукто-2-киназы/фруктозо-2,6-бисфосфатазы-3 (PFKFB3) и активатора фосфофруктокиназы-1 [9]. Факторы роста представляют собой белковые сигнальные молекулы, стимулирующие рост, пролиферацию и дифференцировку клеток. Некоторые из них являются важнейшими регуляторами ангиогенеза и называются ангиогенные факторы роста (АФР). АФР и связанные с ними сигнальные пути играют ключевую роль в активации, регуляции и затухании ангиогенных процессов [10].
Важнейшим АФР, участвующим в регуляции почти всех этапов физиологического ангиогенеза, является семейство сосудистого эндотелиального фактора роста, состоящее из VEGF-A, VEGF-B, VEGF-C, VEGF-D, VEGF-F и PlGF и рецепторов VEGFR-1, VEGFR-2 и VEGFR-3 [8]. VEGF секретируется ЭК, перицитами, гладкомышечными клетками, лейкоцитами, клетками стромы и паренхимы органов и влияет почти на все процессы ангиогенеза [10]. VEGF-А является ключевым звеном сигнального пути VEGF/VEGFR и играет основную роль в начальных процессах ангиогенеза: повышает проницаемость сосудов, стимулирует дифференцировку, пролиферацию и миграцию ЭК [11]. Данные функции реализуются при взаимодействии VEGF с VEGFR-1 и дальнейшей активации различных сигнальных путей, включая PI3K/Akt (фосфоинозитид-3-киназа/ протеинкиназа B) и MAPK/ERK (митоген-активируемая протеинкиназа/киназа, регулируемая внеклеточным сигналом), которые запускают миграцию клеток, высвобождение протеолитических ферментов во внеклеточный матрикс (ВКМ) и высвобождение воспалительных цитокинов, многие из которых обладают проангиогенным эффектом. В результате связывания VEGF с внеклеточным доменом VEGFR-2 активируются пути PLCγ/PKC (фосфолипазы-Cγ/протеинкиназы C) и MAPK/ERK, что приводит к активации пролиферации ЭК [11]. Другим важным для ангиогенеза рецептором VEGF является VEGFR-2, запускающий высвобождение NO путем активации эндотелиальной NO-синтазы (eNOS) с участием пути PI3K/AKT [12]. Существует несколько изоформ VEGF-A, образующихся в результате альтернативного сплайсинга, который определяет количество аминокислот в составе белка, что отражается в названии изоформы [13]. В контексте ангиогенеза наибольший интерес представляет VEGF121, VEGF165 и VEGF189, хотя существуют и другие изоформы. Основное их отличие заключается в количестве гепарин-связывающих доменов, с помощью которых факторы роста закрепляются во внеклеточном матриксе посредством связывания с протеогликанами гепарансульфата внеклеточного матрикса [14]. VEGF189 имеет в своем составе два таких домена, VEGF165 – один, а изоформа VEGF-121 – ни одного, поэтому неспособна к прочному и длительному закреплению во внеклеточном матриксе [8]. Связывание VEGF и других АФР с внеклеточным матриксом необходимо, поскольку их эффект не строго специфичен к ЭК, а плюрипотентен, поэтому при системном действии митогенный эффект может проявиться и на других типах клеток [15]. Повышение концентрации АФР в нецелевой ткани может способствовать развитию патологического процесса (особенно часто это проявляется при неопластических заболеваниях). Несмотря на то что VEGF участвует во всех процессах ангиогенеза, для физиологического ангиогенеза необходимы и другие АФР [10].
Большой вклад в регуляцию ангиогенеза вносит сигнальный путь фактора роста фибробластов (FGF), основные представители которого – FGF-1, FGF-2 и рецепторы FGFR-1–FRFR-4. FGF-1 (aFGF или кислый фактор роста фибробластов) способствует миграции ЭК, морфогенезу сосудов и участвует в активации сигнальных путей других факторов роста. FGF-2 (bFGF или основный фактор роста фибробластов) стимулирует миграцию, пролиферацию и дифференцировку ЭК; активирует ремоделирование внеклеточного матрикса в процессе ангиогенеза [10, 16, 17]. Среди сигнальных путей, участвующих в реализации эффектов FGF, есть совпадения с путями VEGF: MAPK/ERK, PI3K/AKT и PLCγ/PKC. Сигнальные пути, задействованные в ангиогенезе, имеют множество точек взаимодействия, в которых реализуются механизмы обратной связи, конкуренции, избыточности, синергизма и антагонизма. Именно поэтому в контексте регуляции ангиогенеза недостаточно рассмотреть изолированные эффекты отдельных молекул, а нужно оценивать их взаимодействие в динамике [18].
Фактор роста гепатоцитов (HGF), также известный как фактор рассеивания (SF), участвует в эмбриогенезе, регенерации и ангиогенезе и реализует эффекты путем связывания с рецептором c-Met и расположенным на ЭК [19]. HGF продуцируется мезенхимальными клетками (фибробластами, гладкомышечными клетками сосудов). Проангиогенное действие HGF проявляется в стимулировании миграции, пролиферации и инвазии ЭК, активации тубулярного морфогенеза (выстраивание ЭК в вытянутые тяжи, которые предшествуют формированию сосуда) и увеличении экспрессии VEGF и MMP-1 [19, 20].
Для образования зрелых функциональных сосудов важно не только наличие всех необходимых медиаторов ангиогенеза, но и соответствие их концентраций и продолжительности действия довольно узкому физиологическому диапазону. Этот аспект хорошо изучен применительно к VEGF: терапевтический диапазон довольно узок, а выход за его пределы приводит к формированию патологических сосудов. При сверхэкспрессии VEGF образуются гемангиомы, негерметичные сосуды с аномальной морфологией и неравномерно расширенным просветом, а слишком низкая концентрация VEGF приводит к формированию недостаточного для восстановления перфузии количества сосудов, причем эти сосуды имеют уменьшенный просвет, не успевают созреть и быстро регрессируют [21].
Важно отметить, что существует некоторый базальный уровень секреции АФР, который существенно ниже, чем при ишемии. В условиях нормоксии не происходит ангиогенеза, поскольку эффект проангиогенных молекул уравновешен антиангиогенными. Вкратце рассмотрим важнейшие из них, начиная с вазогибина-1 (VASH-1), который продуцируется ЭК в ответ на повышение концентрации VEGF, FGF-2, HIF-1α. Экспрессия VASH-1 увеличивается в терминальной зоне ангиогенеза для ограничения дальнейшего прорастания сосудов [22]. Пигментный фактор эпителиального происхождения (PEDF) является ингибитором индуцированного ишемией ангиогенеза и вызывает апоптоз в активированных ЭК; он также играет важную роль в регуляции формирования сосудов глаза [23]. Хондромодулин-1 (ChM-1) – специфичный для хряща фактор, который проявляет антиангиогенную активность в различных тканях [24]. Ангиостатин оказывает антиангиогенный эффект посредством нескольких механизмов: ингибирует интегрин αvβ3, способствуя уменьшению адгезии клеток; блокирует АТФ-синтазу ЭК, лишая их необходимой для участия в ангиогенезе энергии; ингибирует ангиомотин, который является вырабатываемые тромбоцитами стимулятором миграции ЭК [25–27]. Тромбоспондины (TSP-1 и TSP-2), вырабатываемые тромбоцитами и мегакариоцитами, уменьшают интенсивность миграции и пролиферации ЭК, а также могут запускать апоптоз ЭК и модулировать активность некоторых регуляторов ангиогенеза [25–27]. Снижают интенсивность образования сосудов ингибиторы многочисленных ферментов, участвующих в ангиогенезе (TIMP, ингибитор эластазы и др.) [25].
Переход сосудистой сети от состояния покоя к ангиогенезу называется ангиогенным переключением. Оно происходит, когда равновесие между ангиогенными и антиангиогенными факторами смещается в пользу первых за счет их высвобождения из депо, повышения биодоступности и усиленной продукции в результате приобретения клетками проангиогенного фенотипа. Ангиогенному переключению также способствует снижение продукции и биодоступности антиангиогенных молекул, однако их концентрация более стабильна [12]. Важнейшими молекулами, участвующими в ангиогенном переключении, являются VEGF-A, FGF-2, Ang-2, TGF-β, PDGF, NO и IL-8 [12, 28]. Чаще всего ангиогенное переключение рассматривается в контексте онкогенеза, поскольку оно является обязательным условием роста опухоли, перфузионные потребности которой очень высоки и не могут быть удовлетворены без формирования дополнительных сосудов. Поскольку опухоль может расти только в том случае, если формируется достаточная для ее кровоснабжения сосудистая сеть, в данном контексте ангиогенное переключение рассматривается как одно из ключевых звеньев патогенеза опухолей. Помимо этого, формируемая в результате опухолевого ангиогенеза сосудистая сеть является важным механизмом распространения метастазов, а в дальнейшем – их активации и роста. На этом основано перспективное направление лечения в онкологии – антиангиогенная терапия [29]. Опухолевый ангиогенез является патологическим, поскольку приводит к формированию сосудов с нарушенной морфологией и функциями, что во многом связано с отклонением соотношения между регуляторами ангиогенеза от физиологического. Во многом этому способствует сверхэкспрессия VEGF-A, которая приводит к чрезмерной пролиферации ЭК, формирования множества сосудов, которые не успевают созревать и имеют повышенную проницаемость [29, 30]. Патологический ангиогенез при каждом отдельном заболевании имеет свои особенности, которые связаны с паттернами высвобождения проангиогенных и антиангиогенных факторов и их соотношением.
Таким образом, процесс ангиогенеза запускается сложным каскадом реакций, который включает в себя изменение транскрипции генов, индуцируемых локальными метаболическими изменениями тканей, приводящими к выделению комплекса АФР, среди которых наибольшее значение имеют факторы семейства VEGF. АФР обеспечивают комплекс межклеточных взаимодействий между ЭК, перицитами, гладкомышечными клетками, лейкоцитами, клетками стромы и паренхимы органов. Межклеточные регуляторные реакции, сопряженные с изменением продукции АФР, вызывают активацию многочисленных внутриклеточных сигнальных механизмов ЭК, что приводит к формированию их ангиогенного фенотипа. Важно отметить, что в указанном регуляторном каскаде большое значение имеют множественные отрицательные обратные связи. В условиях нормоксии тканей продукция АФР физиологически уравновешивается выделением антиангиогенных факторов (VASH-1, ангиостатин и др.). При индукции ангиогенеза происходит изменение баланса продукции АФР и антиангиогенных факторов, что определяется как ангиогенное переключение – сигнальный механизм, адекватное функционирование которого определяет сано- или патогенетический характер ремоделирования сосудистого русла.
ПРОРАСТАНИЕ СОСУДОВ КАК МЕХАНИЗМ АНГИОГЕНЕЗА
Механизм процесса ангиогенеза может быть реализован двумя способами: прорастанием – SA (от англ. – Sprouting angiogenesis) и расщеплением – IA (от англ. –Intussusceptive angiogenesis) [31]. Суть механизма SA заключается в миграции ЭК сформированного сосуда во внеклеточный матрикс и росте сосуда в направлении ангиогенных стимулов [31]. Инициация SA происходит за счет увеличения под действием VEGF проницаемости стенки сосуда и опосредованного VEGF, NO и другими вазодилататорами локального снижения тонуса гладкомышечных клеток стенки сосуда, что в итоге способствует проникновению белков плазмы (фибриноген, витронектин и фибронектин) во внеклеточный матрикс [1, 10]. VEGF взаимодействует с расположенными на ЭК рецепторами VEGFR-2, что приводит к формированию верхушечного (или концевого) фенотипа ЭК. Верхушечные ЭК секретируют Delta-like ligand 4 (Dll4), который взаимодействует с интегральными белками-рецепторами Notch соседних ЭК, что способствует снижению экспрессии VERGFR-2 и увеличению экспрессии VEGFR-1. Активация VEGFR-1 приводит к образованию ЭК стеблевого фенотипа [32]. VEGF стимулирует секрецию Dll4 всеми ЭК, однако верхушечный фенотип приобретают те из них, которые начинают ее раньше и осуществляют активнее под действием более высокой концентрации VEGF [17]. Передача сигналов по пути Dll4/Notch также способствует усилению ангиогенеза за счет повышения интенсивности секреции FGF-2, VEGF-A и HGF [33]. Помимо Dll4, у рецепторов Notch есть и другой лиганд: белок Jagged1 (Jag1), вырабатываемый стеблевыми ЭК и подавляющий передачу сигнала Dll4–Notch, что обеспечивает возможность взаимодействия VEGF с VEGFR-2 ЭК для образования новых верхушечных ЭК [32, 34]. В поддержании оптимального соотношения концентраций Dll4 и Jag1, регулирующих переход ЭК из одного фенотипа в другой, также участвуют: сигнальный путь Wnt/β-катенин, ответственный за активацию секреции Dll4 [35], цитокин TNF-α, усиливающий эффект Jag1 и снижающий активность Dll4 [34]. Поскольку отличительной чертой SA является инвазия ЭК во внеклеточный матрикс, сигнальный путь Notch является одним из ключевых для данного способа ангиогенеза.
Функция верхушечных ЭК заключается в определении направления роста сосуда за счет миграции по градиенту проангиогенных стимулов. Верхушечные ЭК секретируют плазмин, матриксные металлопротеиназы и другие протеолитические ферменты для разрушения базальной мембраны сосуда, в результате чего становится возможной инвазия ЭК во внеклеточный матрикс [1, 36]. Плазмин запускает лизис белков внеклеточного матрикса и активирует MMP-1, MMP-2, MMP-3 MMP-7 и MMP-9 [37]. Базальные мембраны играют регуляторную роль в ангиогенезе, поскольку их компоненты, такие как коллаген и ламинин, могут оказывать стимулирующее действие, а такие как эндостатин, тумостатин и другие – ингибирующее [38, 39]. Важная функция коллагена I типа заключается в запуске упорядочивания ЭК в тяжи, приобретения ими веретенообразной формы и морфогенеза сосуда [38, 39]. Для миграции ЭК в направлении ангиогенного стимула необходима деградация и ремоделирование внеклеточного матрикса, что также осуществляется с помощью секретируемых верхушечными ЭК протеолитических ферментов [37, 40]. Внеклеточный матрикс служит резервуаром АФР, которые, как было сказано выше, связываются с белками посредством гепарин-связывающих доменов. В состоянии покоя данные АФР имеют очень низкую биодоступность, но при высвобождении в результате протеолиза внеклеточного матрикса способны к мощному стимулированию и поддержанию ангиогенеза [38]. В процессе ангиогенеза повышается концентрация не только стимуляторов ангиогенеза, но и его ингибиторов. Эндостатин образуется в результате протеолиза коллагена XVIII типа, тумстатин-коллагена IV типа, тетрастатин, пентастатин и гексастатин-коллагена IV типа. Данные молекулы ингибируют пролиферацию и миграцию ЭК и запускают их апоптоз. Таким образом постоянно происходит коррекция соотношения между про- и антиангиогенными молекулами для предотвращения гиперактивации ангиогенеза [37].
Движение ЭК в направлении проангиогенных стимулов реализуется в основном за счет хемотаксиса по градиенту VEGF. На филоподиях верхушечных ЭК активно экспрессируется VEGFR-2, который регистрирует градиент концентраций VEGF, от которого зависит запуск перестройки актиновых волокон цитоскелета для формирования филоподий и ламеллоподий, которые удлиняются в направлении максимального сосредоточения VEGF [1, 36]. На поверхности активированных VEGF ЭК экспрессируются интегрины, посредством которых осуществляется адгезия ЭК к коллагену и фибрину внеклеточного матрикса. Это способствует активации сигнального пути MAPK, результатом чего является повышение жизнеспособности ЭК. Внеклеточный матрикс служит каркасом, к которому с помощью интегринов адгезируются ЭК, что необходимо для нормального протекания ангиогенеза, поскольку при низкой адгезии запускается апоптоз ЭК, при средней – становится возможной их дифференцировка, а при высокой – пролиферация и правильная пространственная ориентация. После того как филоподии верхушечных ЭК зафиксируются на субстрате, происходит сокращение актиновых филаментов для перемещения всей массы клетки по градиенту VEGF [10, 17]. Отличительной чертой верхушечных ЭК является сверхэкспрессия гликолитического регулятора PFKFB3, которая необходима для интенсивного гликолиза, обеспечивающего высокие энергетические потребности клеток в условиях гипоксии [9]. Гликолитические ферменты связываются с F-актином и компартментализируются в ламеллиподии и филоподии, где быстрое и локализованное производство АТФ посредством гликолиза делает возможным ремоделирование цитоскелета во время миграции клеток [9].
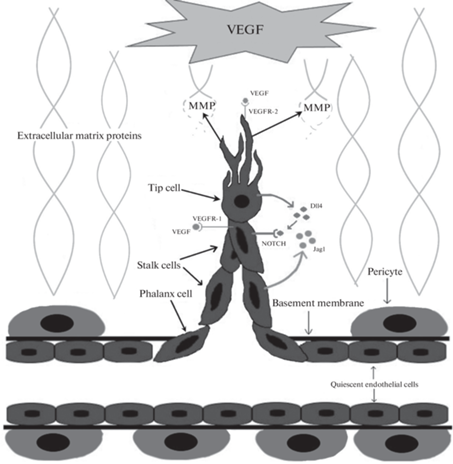
Ключевая особенность стеблевых ЭК сводится к высокой пролиферативной активности, благодаря которой они, следуя за верхушечными клетками, формируют ствол будущего сосуда. Стеблевые ЭК также являются основными участницами процесса формирования просвета сосуда и синтеза компонентов базальной мембраны [36]. Есть и третий фенотип ЭК – фаланговый – представленный покоящимися ЭК. Они обладают меньшей пролиферативной активностью, чем стеблевые клетки и движутся в направлении проангиогенного стимула с минимальной скоростью. Фаланговые ЭК образуют гладкий монослой из плотно прилегающих друг к другу клеток и участвуют в созревании и стабилизации сосуда за счет укрепления межклеточных контактов, построения базальной мембраны, обеспечения взаимодействия ЭК с перицитами. Рис. 1 иллюстрирует описанные выше процессы, происходящие в начале SA.
Рис. 1.
Начальные процессы SA. MMP – матриксные металлопротеиназы, Extracellular matrix proteins – белки внеклеточного матрикса, Tip cell – верхушечная ЭК, Stalk cells – стеблевые ЭК, Phalanx cell – фаланговая ЭК, Basement membrane – базальная мембрана, Pericyte – перицит, Quiescent endothelial cells – покоящиеся ЭК, которые не участвуют в ангиогенезе.
Для ангиогенеза недостаточно приобретших ангиогенный фенотип ЭК, входящих в состав расположенных в области гипоперфузии сосудов, поэтому необходимо привлечение дополнительных ЭК и эндотелиальных клеток-предшественниц в зону ангиогенеза. Термин “эндотелиальные клетки-предшественницы” имеет множество определений, но ключевой смысл заключается в том, что это клетки, способные дифференцироваться в зрелые ЭК [18]. Эндотелиальные клетки-предшественницы формируются в красном костном мозге, откуда при появлении таких стимулов, как гипоксия, АФР (особенно VEGF) и цитокины (особенно ГМ-КСФ), рекрутируются в ишемизированную ткань; также эндотелиальные клетки-предшественницы формируются локально в стенке сосудов [41].
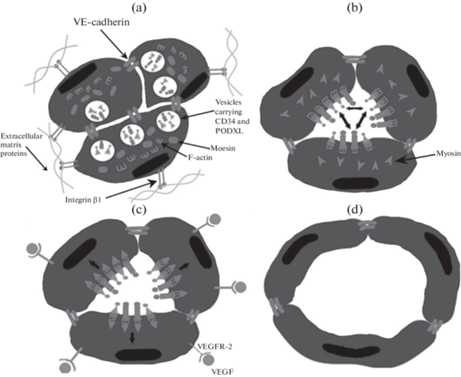
Следующим этапом ангиогенеза является образование в тяже ЭК просвета (люменогенез). Данный процесс может осуществляться по внеклеточному и внутриклеточному механизму, и в реализации их обоих важную роль играет состав внеклеточного матрикса и взаимодействие с ним ЭК [42]. Внеклеточный люменогенез начинается с поляризации ЭК, которая запускается интегрин-опосредованным взаимодействием ЭК и внеклеточного матрикса и поддерживается VE-кадгерином (рис. 2a). Оформление люминальной поверхности, выстилающей просвет сосуда, и базальной, которая обращена к внеклеточному матриксу, начинается со смещения межклеточных контактов в направлении от центра тяжа к периферии с соответствующей реорганизацией цитоскелета [43]. Параллельно происходит опосредованный VE-кадгерином и белком-регулятором клеточного цикла Cdc42 экзоцитоз расположенных в везикулах ЭК гликопротеинов CD34 и PODXL (подокаликсин) на люминальную поверхность (рис. 2b). Люминальные гликопротеины имеют отрицательный заряд, что способствует взаимному отталкиванию ЭК с постепенным формированием просвета (рис. 2b). Люминальные гликопротеины соединяются с F-актином цитоскелета (рис. 2b) при участии белков ERM (эзрин, радиксин и моэзин), из которых моэзин играет основную роль. Далее VEGF связывается с VEGFR-2, что приводит к активации протеинкиназы ROCK, отвечающей за образование актино-миозинового комплекса, который за счет сократительной активности расширяет просвет сосуда (рис. 2c) до необходимого диаметра (рис. 2d) [43].
Рис. 2.
Люменогенез путем межклеточного отталкивания. (a) – Интегрин-опосредованное взаимодействие ЭК и внеклеточного матрикса для оформления люминальной и базальной поверхностей ЭК. (b) – Смещение VE-кадгерина на периферию; экзоцитоз люминальных гликопротеинов и взаимное отталкивание ЭК; связывание люминальных гликопротеинов с F-актином. (c) – Формирование актино-миозинового комплекса и расширение просвета сосуда в результате его сократительной активности. (d) – Просвет сформирован. VE-cadherin – VE-кадгерин, Vesicles carrying CD34 and PODXL – везикулы, несущие люминальные гликопротеины CD34 и PODXL (подокалексин), Moesin – моэзин, F-actin – F-актин, Integrin β1 – интегрин β1, Extracellular matrix proteins – белки внеклеточного матрикса, Myosin – миозин.
Процесс нормального люменогенеза невозможен без еще одного – внутриклеточного механизма, суть которого заключается в образовании посредством пиноцитоза крупных вакуолей и мелких пузырьков, которые в дальнейшем сливаются для формирования центрального просвета, который соединяется с аналогичными просветами соседних клеток (рис. 3a, b) [44]. Для инициации данного процесса необходимо интегрин-опосредованное взаимодействие ЭК с внеклеточным матриксом, причем состав внеклеточного матрикса определяет активность внутриклеточного люменогенеза. Активирующим действием обладают коллаген I типа и фибрин [42]. Важную роль в стимулировании такого способа образования просвета играет MT1-MMP [44]. В большинстве сосудов для формирования стабильного просвета необходимо сочетание обоих способов люменогенеза. Паттерн формирования просвета зависит от измеряемой в количестве клеток толщины сосуда. Клеточное отталкивание реализуется в сосудах с бо́льшим диаметром и начинается на периферии сосуда, в то время как в ЭК в центре клеточного тяжа преобладает внутриклеточный механизм [45].
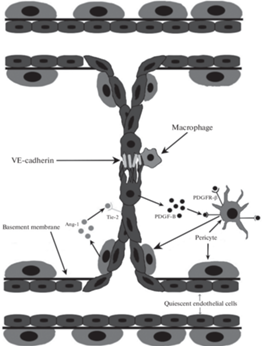
Следующим этапом SA является формирование одного непрерывного сосуда путем слияния в области максимальной концентрации VEGF двух предшественников, проросших с разных сторон (рис. 4) [46]. Между верхушечными ЭК устанавливается контакт, который далее стабилизируется макрофагами и VE-кадгерином [10, 17]. VE-кадгерин модулирует экспрессию VEGFR-2, что способствует снижению миграционной активности ЭК для предотвращения формирования избыточных контактов [47]. Макрофаги участвуют в образовании анастомоза между предшественниками сосудов за счет продукции VEGF-C, который активирует VEGFR-3 верхушечных ЭК, что способствует активации NOTCH для перехода верхушечных ЭК в стеблевые для прекращения миграции и стабилизации контакта между ЭК [48]. Для полноценного функционирования сосуда он должен быть стабилизирован путем рекрутирования перицитов и формирования прочных межклеточных контактов между ЭК. Наличие полноценного этапа созревания является важнейшей отличительной чертой физиологического ангиогенеза по сравнению с патологическим. В этом процессе ключевая роль принадлежит сигнальным путям PDGF и Ang/Tie (ангиопоэтин/рецепторная тирозинкиназа) [49, 50]. Хотя сигнальный путь PDGF участвует в регуляции начальных стадий ангиогенеза, его наиболее важная и специфическая функция реализуется именно на данном этапе. PDGF экспрессируется ЭК и запускают миграцию, пролиферацию и встраивание в состав сосуда клеток, экспрессирующих PDGFR-β, в число которых входят перициты и гладкомышечные клетки [50]. Рекрутирование перицитов необходимо для формирования непрерывного матрикса базальной мембраны капилляра путем отложения ламинина, фибронектина и коллагена IV типа [50]. Перициты способствуют стабилизации сосуда путем реорганизации внеклеточного матрикса, упорядочивания ЭК, снижения пролиферативной активности ЭК и экспрессии TGF-β1 – стимулятора дифференцировки клеток мезенхимального происхождения в перициты [49]. Из секретируемых перицитами проангиогенных молекул особо важна роль ангиопоэтина-1 (Ang-1), который является агонистом локализованных на ЭК рецепторов Tie2. Передача сигналов Ang-1/Tie2 за счет активации путей eNOS и PI3K/AKT оказывает антиапоптотическое действие; способствует стабилизации и повышению жизнеспособности ЭК; усиливает взаимодействие между ЭК и перицитами; индуцирует сужение межклеточных щелей и увеличение прочности соединений между ЭК; стабилизирует VE-кадгерин; запускает перестройку цитоскелета ЭК и способствует уменьшению проницаемости сосуда. Ang-1 также снижает чувствительность ЭК к проангиогенным стимулам, что ведет к затуханию процессов ангиогенеза и переходу сосудистой сети к состоянию покоя, которое окончательно произойдет после восстановления перфузии ткани [51, 52].
Рис. 4.
Образование непрерывного сосуда из двух предшественников. Macrophage – макрофаг, Pericyte – перицит, Quiescent endothelial cells – покоящиеся ЭК, Basement membrane – базальная мембрана.
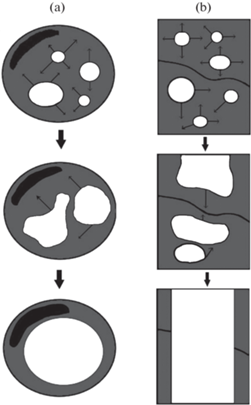
Ангиогенез завершается реорганизацией вновь сформированных сосудов из гомогенной паутины в дифференцированную иерархичную сосудистую сеть, которая способна обеспечить необходимую перфузию ткани с максимальной эффективностью (рис. 5a, b). В процессе реорганизации происходит элиминирование избыточных, незрелых и неправильно сформированных сосудов, отростков и анастомозов путем апоптоза ЭК или их миграции и встраивания в состав соседних сосудов (механизмы варьируются в зависимости от ткани). Функциональные сосуды претерпевают изменения толщины стенки, диаметра просвета и формы для оптимизации их гемодинамической функции [46]. Основным регулятором этого процесса является напряжение сдвига, которое тем больше, чем активнее кровоток в сосуде. В сосудах с интенсивным кровотоком высокое напряжение сдвига способствует активации Akt, которая посредством Kruppel-подобного фактора 2 (KLF2) повышает экспрессию эндотелиальной синтазы оксида азота (eNOS) и супероксиддисмутазы (SOD), что приводит к повышению жизнеспособности ЭК и расширению просвета сосудов [53]. В клетках сосудов с минимальной активностью кровотока аналогичной активации сигнальных путей повышения жизнеспособности не происходит, поэтому эти сосуды подвергаются разрушению [54]. Проангиогенные факторы повышают жизнеспособность ЭК, поэтому снижение их концентрации при достижении достаточного уровня перфузии ткани приводит к гибели сосудов, не успевших созреть и стабилизироваться [55]. Уменьшение уровня проангиогенных молекул способствует смещению равновесия в сторону антиангиогенных, играющих важную роль в запуске апоптоза. В инициации регрессии сосудов важная функция отведена Ang-2, который в условиях сниженной концентрации VEGF, характерной для завершающей стадии ангиогенеза, нарушает передачу сигналов PI3K/AKT, запускает инволюцию нефункционирующих и незрелых сосудов за счет апоптоза ЭК и дальнейшей регрессии капилляров. Это не единственная функция Ang-2: в условиях высокой концентрации VEGF он выступает в роли синергиста Ang-1/Tie2, способствуя тем самым созреванию сосудов в условиях гипоперфузии. Такая зависимость при физиологическом ангиогенезе обуславливает своевременный переход от фазы образования предшественника сосуда к его созреванию и является звеном динамической регуляции активности ангиогенеза с учетом потребности ткани [54].
Рис. 5.
Ремоделирование сосудистой сети. (a) – Сосудистая сеть до ремоделирования. Стрелками отмечены области с активным кровотоком, а перечеркнутыми стрелками – области с минимальным кровотоком. (b) – Сосудистая сеть после ремоделирования.
После того как по образованным сосудам устанавливается кровоток и возникает состояние нормоксии, кислород связывается с HIF-1α, в результате чего снижается его экспрессия и начинается деградация. Уменьшение концентрации HIF-1α приводит к снижению продукции проангиогенных факторов, и процессы ангиогенеза стихают [16].
Таким образом, SA протекает в несколько этапов:
1. Появление стимула к активации ангиогенеза (медиаторы-индукторы ангиогенеза);
2. Миграция ЭК, имеющих верхушечный фенотип;
3. Образование тяжа за счет пролиферации стеблевых ЭК;
4. Формирование просвета;
5. Слияние двух предшественников сосуда в месте повышенной концентрации VEGF с образованием единого непрерывного сосуда;
6. Созревание сосуда за счет рекрутирования перицитов, укрепления межклеточных контактов и формирования базальной мембраны;
7. Образование зрелой сосудистой сети благодаря удалению избыточных и патологических сосудов и функциональной дифференцировки перфузируемых;
8. Восстановление кровотока, снижение концентрации проангиогенных факторов, стихание ангиогенеза.
РАСЩЕПЛЕНИЕ СОСУДОВ КАК МЕХАНИЗМ АНГИОГЕНЕЗА
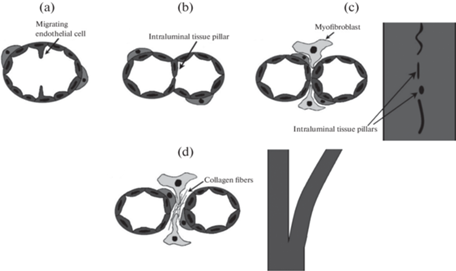
IA – это процесс образования новых сосудов за счет разделения имеющегося на две части путем формирования внутри него перегородки, отграничивающей два самостоятельных просвета. Данный механизм ангиогенеза реализуется в капиллярах, мелких артериях и венах [56]. IA существенно быстрее и энергетически выгоднее SA, потому что не требует разрушения базальной мембраны и внеклеточного матрикса, миграции ЭК в направлении локализованного во внеклеточном матриксе проангиогенного стимула и активной клеточной пролиферации [31]. Формирование тканевого столба в процессе IA осуществляется в несколько этапов. Вначале под действием Ang-1 и Ang-2 происходит выпячивание ЭК в просвет в двух противоположных друг другу точках окружности сосуда (рис. 6a) с формированием из них листков эндотелия, которые впоследствии сливаются с образованием внутрипросветного тканевого столба (рис. 6b) [31, 56, 57]. Далее тканевый столб стабилизируется за счет упрочнения межклеточных контактов, после чего в его центре формируется канал, который инвазируется цитоплазматическими отростками миофибробластов (рис. 6c). Затем под действием PDGF происходит рекрутирование перицитов (рис. 6c) в латеральную часть тканевого столба для стабилизации межклеточных взаимодействий между ЭК. Формирование столба завершается синтезом перицитами и миофибробластами коллагена (рис. 6d) и увеличением толщины столба (рис. 6d) [1, 31]. Несколько столбов увеличиваются в размерах и сливаются друг с другом, в результате чего сосуд окончательно разделяется на два (рис. 6d).
Рис. 6.
Этапы IA. (a) – Выпячивание ЭК. (b) – Образование внутрипросветного тканевого столба. (c) – Перфорация тканевого столба в центре и инвазия отростками миофибробластов; рекрутирование перицитов. Слева изображен поперечный срез сосуда, справа – вид сверху. (d) – Синтез коллагена перицитами и миофибробластами; разрастание тканевых столбов и окончательное разделение на два самостоятельных сосуда. Слева изображен поперечный срез сосуда, справа – вид сверху. Migrating endothelial cell – мигрирующая (выпячивающаяся) в просвет ЭК, Intraluminal tissue pillar – внутрипросветный тканевый столб, Myofibroblast – миофибробласт, Collagen fibrils – волокна коллагена.
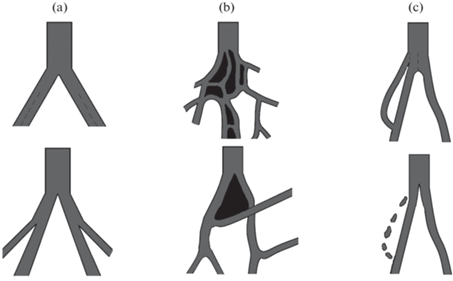
Существует три вида IA: рост микрососудов (рис. 7a), их разветвление (рис. 7b) и ремоделирование ветвления (рис. 7c). Рост микрососудов осуществляется за счет развития тканевых столбов в центре сосуда на протяженном участке, что, в свою очередь, приводит к формированию многочисленных капилляров одинакового размера. Разветвление микрососудов осуществляется путем формирования тканевых столбов на протяжении всего существующего сосуда. Данный вид IA способствует переходу от гомогенной капиллярной сети к иерархичному сосудистому руслу с дифференцировкой на артериолы, капилляры и венулы. При ремоделировании ветвления тканевые столбы образуются вблизи бифуркации, и их дальнейший рост приводит либо ее к сужению и смещению в проксимальном направлении, либо к перекрыванию просвета гемодинамически невыгодной ветви сосуда. То есть ремоделирование ветвления представляет собой процесс обрезки лишних сосудов или изменение угла отхождения ветви сосуда для оптимизации кровоснабжения ткани [57].
Рис. 7.
Виды IA. (а) – Рост микрососудов. (b) – Разветвление микрососудов. (c) – Ремоделирование ветвления.
Главным пусковым фактором IA, как и при SA, является гипоксия. Отличие в регуляции данных механизмов ангиогенеза заключается в том, что при IA бо́льшее значение имеют механические факторы. Паттерн формирования столбов зависит от локальных особенностей гемодинамики, обуславливающих разные потребности в кровоснабжении ткани. Пространственное распределение градиентов силы сдвига определяет направление развития столбов: они развиваются в областях с низким напряжением сдвига и растут вдоль них [31, 57]. Возникновение градиента силы сдвига может быть обусловлено либо постоянным усилением кровотока, либо чередованием воздействия кровотока высокой и низкой интенсивности (в мышцах при чередовании физической работы и покоя). Рост столбов вдоль оси сосуда приводит к удвоению сосуда, а развитие столбов в направлении, не совпадающем с осью, отвечает за ремоделирование угла ветвления и обрезку неэффективных сосудов. Насчет биохимической регуляции IA мнения исследователей менее согласованы, чем при SA, хотя спектры молекул, участвующих в регуляции IA и SA схожи (VEGF, FGF-1, FGF-2, HGF и другие) [56, 57]. Для IA характерна инициирующая роль Ang-1, более равномерное и непрерывное распределение VEGF, чем при SA [31].
Совсем недавно была выдвинута гипотеза о существовании еще одного вида ангиогенеза – коалеcцентного (Coalescent angiogenesis). Сутью данного механизма является слияние многочисленных микрососудов в один крупный, который обладает более высокой гемодинамической эффективностью. Коалесцентный ангиогенез запускается в гомогенной сосудистой сети, которая не способна обеспечить достаточную перфузию. Инициация коалесцентного ангиогенеза, предположительно, обусловлена локальными особенностями силы сдвига. Формирование крупного сосуда способствует элиминации неэффективных микрососудов с перенаправлением тока крови по “предпочтительному пути”, то есть гемодинамически наиболее выгодному направлению [58].
В процессе коалесцентного ангиогенеза происходит элиминация тканевых островков, которые разделяют микрососуды изначальной капиллярной сети, поэтому при одномоментной визуализации сосудистого русла коалесцентный ангиогенез практически неотличим от IA. Для выявления различий и установления механизма ангиогенеза необходима длительная видеозапись, которая позволит определить, сливаются сосуды или расщепляются [58, 59].
Данный механизм формирования сосудов был обнаружен только при эмбриональном развитии [58, 59], поэтому необходимы дальнейшие исследования для определения правильности его отнесения к ангиогенезу, а не васкулогенезу. Осуществимость коалесцентного ангиогенеза во взрослом организме, а также особенности его регуляции еще только предстоит выяснить. Необходимо определить, является ли данный механизм самостоятельным видом ангиогенеза или должен рассматриваться как один из этапов созревания сосудистой сети.
ЗАКЛЮЧЕНИЕ
Совокупность представленных данных свидетельствует, что процесс ангиогенеза, запускаемый сложным регуляторным каскадом, включающим комплекс реакций метаболома, транскриптома, протеома, может реализовываться двумя механизмами: SA и IA. Основная функция IA заключается в формировании органоспецифической ангиоархитектоники и ремоделировании сосудов для удовлетворения местной потребности ткани в кислороде в ходе адаптации или при развитии организма. Функция SA заключается в первичном увеличении количества сосудов. Эти два механизма ангиогенеза с рядом общих регуляторных путей дополняют друг друга и совместно участвуют в повышении плотности сосудистой сети. Каждый из механизмов представляет собой сложный многостадийный процесс, регулируемый динамически изменяющимися комбинациями биологически активных веществ.
Ключевыми стадиями SA следует считать: появление стимула к инициации ангиогенеза, миграция верхушечных ЭК, пролиферация стеблевых ЭК с образованием тяжа, формирование просвета, слияние двух предшественников сосуда, созревание сосуда, ремоделирование сосудистой сети. Критическими моментами IA, которые также могут быть выделены в качестве стадий, являются: выпячивание ЭК, образование внутрипросветного тканевого столба, перфорация тканевого столба и инвазия отростками миофибробластов, рекрутирование перицитов, рост и окончательное созревание тканевых столбов, завершающееся разделением изначального сосуда на два новых.
На настоящий момент точные регуляторные реакции, обеспечивающие переключения между указанными механизмами, не выявлены. Вместе с тем накопленные данные позволяют предположить, что реализация IA находится в большей зависимости от гемодинамических условий, таких как напряжение сдвига. Сложная регуляция ангиогенеза обеспечивает точное приспособление формирующегося сосудистого русла к потребностям тканей. Сложность, взаимосвязанность и в ряде случаев недостаточная изученность регуляторных реакций на настоящем этапе затрудняют разработку эффективных и безопасных стратегий управления ангиогенным процессом, которые могут быть использованы в клинической практике. Детализация регуляции стадий и механизмов ангиогенеза открывает новые перспективы для развития технологий лечения широкого спектра заболеваний и патологических состояний.
Список литературы
Oliveira de Oliveira LB, Faccin Bampi V, Ferreira Gomes C, Braga da Silva JL, Encarnação Fiala Rechsteiner SM (2014) Morphological characterization of sprouting and intussusceptive angiogenesis by SEM in oral squamous cell carcinoma: Sprouting and intussusceptive angiogenesis in oral cancer. Scanning 36: 293–300. https://doi.org/10.1002/sca.21104
Deev R, Plaksa I, Bozo I, Isaev A (2017) Results of an International Postmarketing Surveillance Study of pl-VEGF165 Safety and Efficacy in 210 Patients with Peripheral Arterial Disease. Am J Cardiovasc Drugs 17: 235–242. https://doi.org/10.1007/s40256-016-0210-3
Lheureux S, Oaknin A, Garg S, Bruce JP, Madariaga A, Dhani NC, Bowering V, White J, Accardi S, Tan Q, Braunstein M, Karakasis K, Cirlan I, Pedersen S, Li T, Fariñas-Madrid L, Lee YC, Liu ZA, Pugh TJ, Oza AM (2020) EVOLVE: A Multicenter Open-Label Single-Arm Clinical and Translational Phase II Trial of Cediranib Plus Olaparib for Ovarian Cancer after PARP Inhibition Progression. Clin Cancer Res 26: 4206–4215. https://doi.org/10.1158/1078-0432.CCR-19-4121
Kosolapov VP, Yarmonova MV (2021) The analysis of high cardiovascular morbidity and mortality in the adult population as a medical and social problem and the search for ways to solve it. Jour 20: 58–64. https://doi.org/10.52420/2071-5943-2021-20-1-58-64
Hasin Y, Seldin M, Lusis A (2017) Multi-omics approaches to disease. Genome Biol 18: 83. https://doi.org/10.1186/s13059-017-1215-1
Zhang Y, Wang H, Oliveira RHM, Zhao C, Popel AS (2022) Systems biology of angiogenesis signaling: Computational models and omics. WIREs Mech Dis 14(4): e1550. https://doi.org/10.1002/wsbm.1550
Wang J-C, Li G-Y, Li P-P, Sun X, Li W-M, Li Y, Lu S-Y, Liu P-J (2017) Suppression of hypoxia-induced excessive angiogenesis by metformin via elevating tumor blood perfusion. Oncotarget 8: 73892–73904. https://doi.org/10.18632/oncotarget.18029
Apte RS, Chen DS, Ferrara N (2019) VEGF in Signaling and Disease: Beyond Discovery and Development. Cell 176: 1248–1264. https://doi.org/10.1016/j.cell.2019.01.021
Zhang Y, Zhong Y, Liu W, Zheng F, Zhao Y, Zou L, Liu X (2022) PFKFB3-mediated glycometabolism reprogramming modulates endothelial differentiation and angiogenic capacity of placenta-derived mesenchymal stem cells. Stem Cell Res Ther 13: 391. https://doi.org/10.1186/s13287-022-03089-3
Melincovici CS, Boşca AB, Şuşman S, Mărginean M, Mihu C, Istrate M, Moldovan IM, Roman AL, Mihu CM (2018) Vascular endothelial growth factor (VEGF) – key factor in normal and pathological angiogenesis. Rom J Morphol Embryol 59: 455–467.
Noren DP, Chou WH, Lee SH, Qutub AA, Warmflash A, Wagner DS, Popel AS, Levchenko A (2016) Endothelial cells decode VEGF-mediated Ca2+ signaling patterns to produce distinct functional responses. Sci Signal 9: ra20. https://doi.org/10.1126/scisignal.aad3188
Balberova OV, Bykov EV, Shnayder NA, Petrova MM, Gavrilyuk OA, Kaskaeva DS, Soloveva IA, Petrov KV, Mozheyko EY, Medvedev GV, Nasyrova RF (2021) The “Angiogenic Switch” and Functional Resources in Cyclic Sports Athletes. Int J Mol Sci 22: 6496. https://doi.org/10.3390/ijms22126496
Mamer SB, Wittenkeller A, Imoukhuede PI (2020) VEGF-A splice variants bind VEGFRs with differential affinities. Sci Rep 10: 14413. https://doi.org/10.1038/s41598-020-71484-y
Teran M, Nugent MA (2015) Synergistic Binding of Vascular Endothelial Growth Factor-A and Its Receptors to Heparin Selectively Modulates Complex Affinity. J Biol Chem 290: 16451–16462. https://doi.org/10.1074/jbc.M114.627372
Unterleuthner D, Neuhold P, Schwarz K, Janker L, Neuditschko B, Nivarthi H, Crncec I, Kramer N, Unger C, Hengstschläger M, Eferl R, Moriggl R, Sommergruber W, Gerner C, Dolznig H (2020) Cancer-associated fibroblast-derived WNT2 increases tumor angiogenesis in colon cancer. Angiogenesis 23: 159–177. https://doi.org/10.1007/s10456-019-09688-8
Zhao C, Popel AS (2015) Computational Model of MicroRNA Control of HIF-VEGF Pathway: Insights into the Pathophysiology of Ischemic Vascular Disease and Cancer. PLoS Comput Biol 11: e1004612. https://doi.org/10.1371/journal.pcbi.1004612
Naik MU, Chatterjee S, Naik UP (2020) Fibroblast Growth Factor-2-Induced Endothelial Cell Migration Is Regulated by Junctional Adhesion Molecule-A Through Its Tyrosine Phosphorylation and Interaction With Afadin. The FASEB J 34: 1–1. https://doi.org/10.1096/fasebj.2020.34.s1.06430
Weinstein N, Mendoza L, Gitler I, Klapp J (2017) A Network Model to Explore the Effect of the Micro-environment on Endothelial Cell Behavior during Angiogenesis. Front Physiol 8: 960. https://doi.org/10.3389/fphys.2017.00960
Vasuri F, Fittipaldi S, Abualhin M, Degiovanni A, Gargiulo M, Stella A, Pasquinelli G (2014) Biochemical and immunomorphological evaluation of hepatocyte growth factor and c-Met pathway in patients with critical limb ischemia. Eur J Vasc Endovasc Surg 48: 430–437. https://doi.org/10.1016/j.ejvs.2014.05.002
Koudstaal S, Bastings MMC, Feyen DAM, Waring CD, van Slochteren FJ, Dankers PYW, Torella D, Sluijter JPG, Nadal-Ginard B, Doevendans PA, Ellison GM, Chamuleau SAJ (2014) Sustained delivery of insulin-like growth factor-1/hepatocyte growth factor stimulates endogenous cardiac repair in the chronic infarcted pig heart. J Cardiovasc Transl Res 7: 232–241. https://doi.org/10.1007/s12265-013-9518-4
Salabarria A-C, Braun G, Heykants M, Koch M, Reuten R, Mahabir E, Cursiefen C, Bock F (2019) Local VEGF-A blockade modulates the microenvironment of the corneal graft bed. Am J Transplant 19: 2446–2456. https://doi.org/10.1111/ajt.15331
Yazdani S, Kasajima A, Tamaki K, Nakamura Y, Fujishima F, Ohtsuka H, Motoi F, Unno M, Watanabe M, Sato Y, Sasano H (2014) Angiogenesis and vascular maturation in neuroendocrine tumors. Human Pathol 45: 866–874. https://doi.org/10.1016/j.humpath.2013.09.024
Michalczyk ER, Chen L, Fine D, Zhao Y, Mascarinas E, Grippo PJ, DiPietro LA (2018) Pigment Epithelium-Derived Factor (PEDF) as a Regulator of Wound Angiogenesis. Sci Rep 8: 11142. https://doi.org/10.1038/s41598-018-29465-9
Nagai T, Sato M, Kobayashi M, Yokoyama M, Tani Y, Mochida J (2014) Bevacizumab, an anti-vascular endothelial growth factor antibody, inhibits osteoarthritis. Arthritis Res Ther 16: 427. https://doi.org/10.1186/s13075-014-0427-y
Yadav L, Puri N, Rastogi V, Satpute P, Sharma V (2015) Tumour Angiogenesis and Angiogenic Inhibitors: A Review. J Clin Diagn Res 9: XE01–XE05. https://doi.org/10.7860/JCDR/2015/12016.6135
El-Kenawi AE, El-Remessy AB (2013) Angiogenesis inhibitors in cancer therapy: mechanistic perspective on classification and treatment rationales. Br J Pharmacol 170: 712–729. https://doi.org/10.1111/bph.12344
Zhang X, Crawford R, Xiao Y (2014) Anti-angiogenic factors are essential regulators in cartilage homeostasis and osteoarthritis. Osteoarthritis and Cartilage 22: S132. https://doi.org/10.1016/j.joca.2014.02.243
Granstam E, Aurell S, Sjövall K, Paul A (2021) Switching anti-VEGF agent for wet AMD: evaluation of impact on visual acuity, treatment frequency and retinal morphology in a real-world clinical setting. Graefes Arch Clin Exp Ophthalmol 259: 2085–2093. https://doi.org/10.1007/s00417-020-05059-y
Mabeta P, Hull R, Dlamini Z (2022) LncRNAs and the Angiogenic Switch in Cancer: Clinical Significance and Therapeutic Opportunities. Genes (Basel) 13: 152. https://doi.org/10.3390/genes13010152
Luo H, Shen Y, Liao W, Li Q, Wu N, Zhong J, Xiao C, Gan J, Yang Y, Dong E, Zhang G, Liu B, Yue X, Xu L, Liu Y, Zhao C, Zhong Q, Yang H (2022) The inhibition of protein translation promotes tumor angiogenic switch. Mol Biomed 3: 18. https://doi.org/10.1186/s43556-022-00081-4
Mentzer SJ, Konerding MA (2014) Intussusceptive angiogenesis: expansion and remodeling of microvascular networks. Angiogenesis 17: 499–509. https://doi.org/10.1007/s10456-014-9428-3
Kangsamaksin T, Murtomaki A, Kofler NM, Cuervo H, Chaudhri RA, Tattersall IW, Rosenstiel PE, Shawber CJ, Kitajewski J (2015) NOTCH decoys that selectively block DLL/NOTCH or JAG/NOTCH disrupt angiogenesis by unique mechanisms to inhibit tumor growth. Cancer Discov 5: 182–197. https://doi.org/10.1158/2159-8290.CD-14-0650
Zalpoor H, Aziziyan F, Liaghat M, Bakhtiyari M, Akbari A, Nabi-Afjadi M, Forghaniesfidvajani R, Rezaei N (2022) The roles of metabolic profiles and intracellular signaling pathways of tumor microenvironment cells in angiogenesis of solid tumors. Cell Commun Signal 20: 186. https://doi.org/10.1186/s12964-022-00951-y
Travisano SI, Oliveira VL, Prados B, Grego-Bessa J, Piñeiro-Sabarís R, Bou V, Gómez MJ, Sánchez-Cabo F, MacGrogan D, de la Pompa JL (2019) Coronary arterial development is regulated by a Dll4-Jag1-EphrinB2 signaling cascade. eLife 8: e49977. https://doi.org/10.7554/eLife.49977
Kasprzak A (2020) Angiogenesis-Related Functions of Wnt Signaling in Colorectal Carcinogenesis. Cancers (Basel) 12: E3601. https://doi.org/10.3390/cancers12123601
Santos-Oliveira P, Correia A, Rodrigues T, Ribeiro-Rodrigues TM, Matafome P, Rodríguez-Manzaneque JC, Seiça R, Girão H, Travasso RDM (2015) The Force at the Tip–Modelling Tension and Proliferation in Sprouting Angiogenesis. PLoS Comput Biol 11: e1004436. https://doi.org/10.1371/journal.pcbi.1004436
Neve A, Cantatore FP, Maruotti N, Corrado A, Ribatti D (2014) Extracellular matrix modulates angiogenesis in physiological and pathological conditions. Biomed Res Int 2014: 756078. https://doi.org/10.1155/2014/756078
Ruehle MA, Eastburn EA, LaBelle SA, Krishnan L, Weiss JA, Boerckel JD, Wood LB, Guldberg RE, Willett NJ (2020) Extracellular matrix compression temporally regulates microvascular angiogenesis. Sci Adv 6: eabb6351. https://doi.org/10.1126/sciadv.abb6351
Conway RE, Joiner K, Patterson A, Bourgeois D, Rampp R, Hannah BC, McReynolds S, Elder JM, Gilfilen H, Shapiro LH (2013) Prostate specific membrane antigen produces pro-angiogenic laminin peptides downstream of matrix metalloprotease-2. Angiogenesis 16: 847–860. https://doi.org/10.1007/s10456-013-9360-y
Lai K-C, Lu C-C, Tang Y-J, Chiang J-H, Kuo D-H, Chen F-A, Chen I-L, Yang J-S (2014) Allyl isothiocyanate inhibits cell metastasis through suppression of the MAPK pathways in epidermal growth factor‑stimulated HT29 human colorectal adenocarcinoma cells. Oncol Rep 31: 189–196. https://doi.org/10.3892/or.2013.2865
Chambers SEJ, Pathak V, Pedrini E, Soret L, Gendron N, Guerin CL, Stitt AW, Smadja DM, Medina RJ (2021) Current concepts on endothelial stem cells definition, location, and markers. Stem Cells Transl Med 10 Suppl 2: S54–S61. https://doi.org/10.1002/sctm.21-0022
Charpentier MS, Conlon FL (2014) Cellular and molecular mechanisms underlying blood vessel lumen formation. Bioessays 36: 251–259. https://doi.org/10.1002/bies.201300133
Charpentier MS, Tandon P, Trincot CE, Koutleva EK, Conlon FL (2015) A Distinct Mechanism of Vascular Lumen Formation in Xenopus Requires EGFL7. PLoS One 10: e0116086. https://doi.org/10.1371/journal.pone.0116086
Davis GE, Stratman AN, Sacharidou A, Koh W (2011) Molecular basis for endothelial lumen formation and tubulogenesis during vasculogenesis and angiogenic sprouting. Int Rev Cell Mol Biol 288: 101–165. https://doi.org/10.1016/B978-0-12-386041-5.00003-0
Boas SEM, Merks RMH (2014) Synergy of cell-cell repulsion and vacuolation in a computational model of lumen formation. J R Soc Interface 11: 20131049. https://doi.org/10.1098/rsif.2013.1049
Lenard A, Daetwyler S, Betz C, Ellertsdottir E, Belting H-G, Huisken J, Affolter M (2015) Endothelial Cell Self-fusion during Vascular Pruning. PLoS Biol 13: e1002126. https://doi.org/10.1371/journal.pbio.1002126
Lenard A, Ellertsdottir E, Herwig L, Krudewig A, Sauteur L, Belting H-G, Affolter M (2013) In vivo analysis reveals a highly stereotypic morphogenetic pathway of vascular anastomosis. Dev Cell 25: 492–506. https://doi.org/10.1016/j.devcel.2013.05.010
Zarkada G, Heinolainen K, Makinen T, Kubota Y, Alitalo K (2015) VEGFR3 does not sustain retinal angiogenesis without VEGFR2. Proc Natl Acad Sci U S A 112: 761–766. https://doi.org/10.1073/pnas.1423278112
Zhang Z, Warner KA, Mantesso A, Nör JE (2022) PDGF-BB signaling via PDGFR-β regulates the maturation of blood vessels generated upon vasculogenic differentiation of dental pulp stem cells. Front Cell Dev Biol 10: 977725. https://doi.org/10.3389/fcell.2022.977725
Gianni-Barrera R, Bartolomeo M, Vollmar B, Djonov V, Banfi A (2014) Split for the cure: VEGF, PDGF-BB and intussusception in therapeutic angiogenesis. Biochem Soc Trans 42: 1637–1642. https://doi.org/10.1042/BST20140234
Khan M, Aziz AA, Shafi NA, Abbas T, Khanani AM (2020) Targeting Angiopoietin in Retinal Vascular Diseases: A Literature Review and Summary of Clinical Trials Involving Faricimab. Cells 9: E1869. https://doi.org/10.3390/cells9081869
Chu H, Sun Y, Gao Y, Guan X, Yan H, Cui X, Zhang X, Li X, Li H, Cheng M (2019) Function of Kruppel‑like factor 2 in the shear stress‑induced cell differentiation of endothelial progenitor cells to endothelial cells. Mol Med Report 19(3): 1739–1746.https://doi.org/10.3892/mmr.2019.9819
Dekker RJ, van Thienen JV, Rohlena J, de Jager SC, Elderkamp YW, Seppen J, de Vries CJM, Biessen EAL, van Berkel TJC, Pannekoek H, Horrevoets AJG (2005) Endothelial KLF2 Links Local Arterial Shear Stress Levels to the Expression of Vascular Tone-Regulating Genes. Am J Pathol 167: 609–618. https://doi.org/10.1016/S0002-9440(10)63002-7
Korn C, Augustin HG (2015) Mechanisms of Vessel Pruning and Regression. Dev Cell 34: 5–17. https://doi.org/10.1016/j.devcel.2015.06.004
Shahik SMD, Salauddin A, Hossain MDS, Noyon SH, Moin AT, Mizan S, Raza MDT (2021) Screening of novel alkaloid inhibitors for vascular endothelial growth factor in cancer cells: an integrated computational approach. Genomics Inform 19: e6. https://doi.org/10.5808/gi.20068
Díaz-Flores L, Gutiérrez R, García MP, Gayoso S, Carrasco JL, Díaz-Flores Lucio, González-Gómez M, Madrid JF (2020) Intussusceptive Angiogenesis and Peg–Socket Junctions between Endothelial Cells and Smooth Muscle Cells in Early Arterial Intimal Thickening. IJMS 21: 8049. https://doi.org/10.3390/ijms21218049
Ackermann M, Tsuda A, Secomb TW, Mentzer SJ, Konerding MA (2013) Intussusceptive remodeling of vascular branch angles in chemically-induced murine colitis. Microvasc Res 87: 75–82. https://doi.org/10.1016/j.mvr.2013.02.002
Nitzsche B, Rong WW, Goede A, Hoffmann B, Scarpa F, Kuebler WM, Secomb TW, Pries AR (2022) Coalescent angiogenesis-evidence for a novel concept of vascular network maturation. Angiogenesis 25: 35–45. https://doi.org/10.1007/s10456-021-09824-3
Gifre-Renom L, Jones EAV (2021) Vessel Enlargement in Development and Pathophysiology. Front Physiol 12: 639645. https://doi.org/10.3389/fphys.2021.639645
Дополнительные материалы отсутствуют.
Инструменты
Российский физиологический журнал им. И.М. Сеченова