

Российский физиологический журнал им. И.М. Сеченова, 2023, T. 109, № 8, стр. 1028-1044
Роль метаболитов триптофанового обмена и короткоцепочечных жирных кислот в патогенезе аутоиммунных заболеваний
О. П. Шатова 1, 2, 4, *, Е. М. Ягодкина 1, С. С. Кайдошко 1, А. А. Заболотнева 1, 4, А. В. Шестопалов 1, 3, 4
1 Российский национальный исследовательский медицинский университет им. Н.И. Пирогова
Москва, Россия
2 Российский университет дружбы народов им. Патриса Лумумбы
Москва, Россия
3 Национальный медицинский исследовательский центр детской гематологии,
онкологии и иммунологии им. Дмитрия Рогачева
Москва, Россия
4 Национальный медицинский исследовательский центр эндокринологии
Москва, Россия
* E-mail: shatova.op@gmail.com
Поступила в редакцию 03.05.2023
После доработки 12.07.2023
Принята к публикации 13.07.2023
- EDN: NKMVBY
- DOI: 10.31857/S0869813923080095
Аннотация
С каждым годом распространенность аутоиммунных заболеваний в мире неуклонно растет. Этиология и патогенез аутоиммунных заболеваний крайне сложные и во многом остаются неясными. Тем не менее, все больше данных исследований последних лет указывает на критическую роль микроорганизмов в формировании нормального иммунного ответа и аутоиммунных реакций в организме хозяина. При этом одна из ведущих ролей отводится кишечной микробиоте, представленной триллионами микробов, образующих широкий спектр сигнальных и иммуннорегуляторных метаболитов. Формируя сложную взаимозависимую систему “хозяин–микробиота”, симбиотические бактерии во многом определяют развитие и функционирование иммунных клеток человека. В настоящем обзоре мы рассматриваем роль кишечной микробиоты и ее ключевых метаболитов (а именно короткоцепочечных жирных кислот и метаболитов триптофана) в патогенезе аутоиммунных заболеваний и обсуждаем возможные механизмы влияния обозначенных сигнальных молекул на иммунные клетки хозяина.
ВВЕДЕНИЕ
Аутореактивность или аутоиммунитет – это комплексный патологический процесс, в основе которого лежит нарушение толерантности и, как следствие, патологический иммунный ответ в отношении компонентов собственных тканей (аутоантигенов), приводящий к развитию широкого спектра аутоиммунных заболеваний [1]. В свою очередь аутоиммунные заболевания разделяют на органоспецифические (например, сахарный диабет 1-го типа (СД1Т), болезнь Крона и аутоиммунный тиреоидит) и системные (например, ревматоидный артрит и системная красная волчанка) [2]. С ростом распространенности аутоиммунных заболеваний и связанной с этим интенсификацией их исследования становится все более очевидной вовлеченность кишечной микробиоты в реакции аутоиммунного ответа. Ранее считалось, что развитие аутоиммунных заболеваний обусловлено преимущественно наследственной предрасположенностью, однако в настоящее время все большая роль отводится влиянию факторов окружающей среды, в том числе микробному воздействию. Этот факт связан с появлением “гигиенической гипотезы”, отмечающей протективные эффекты патогенов (возбудителей инфекций и паразитов) и комменсальной кишечной микробиоты против развития аутоиммунных заболеваний у хозяина [3]. В процессе эволюции микробиота стала неотъемлемым компонентом человеческого организма. Так, микроорганизмы, заселяющие кишечник, участвуют в пищеварении, обмене веществ, развитии органов и регуляции дифференцировки иммунных клеток и нейронов [4–6]. Большая часть микробиоты обитает во внешнем слое кишечной слизи и способствует развитию и функционированию иммунной системы хозяина посредством продукции своих метаболитов, молекулярных паттернов, ассоциированных с микроорганизмами, и антигенов. Нарушение баланса образующихся метаболитов или нарушение регуляции любого из процессов, сопутствующих их образованию, будет способствовать развитию воспаления и иммуноопосредованным заболеваниям. Тремя наиболее изученными в настоящее время категориями метаболитов, участвующих во взаимодействиях “хозяин–микробиота”, являются (1) короткоцепочечные жирные кислоты (КЦЖК), продуцируемые бактериями в основном в результате ферментации неперевариваемых растительных волокон; (2) вторичные желчные кислоты, предшественники которых образуются в печени, а затем трансформируются микробиотой кишечника и (3) микробные метаболиты триптофана (Trp).
В настоящем обзоре мы рассматриваем роль кишечной микробиоты и ее метаболитов (а именно КЦЖК и метаболитов Trp) в патогенезе аутоиммунных заболеваний и обсуждаем возможные механизмы влияния обозначенных сигнальных молекул на иммунные клетки хозяина.
1. ДИСБИОЗ КИШЕЧНИКА КАК ФАКТОР РАЗВИТИЯ АУТОРЕАКТИВНОСТИ
Механизмы влияния дисбиоза кишечной микробиоты на развитие аутоиммунных заболеваний до сих пор точно не определены. Предположительно, они могут включать в себя молекулярную мимикрию, нарушение функции кишечного барьера и развитие так называемого “синдрома протекающей кишки”, аутофагию эпителиальных клеток кишечника, формирование мембранных везикул и влияние микроРНК [7]. Более того, было показано, что системный аутоиммунитет очень сильно зависит от разнообразия и состава кишечного микробиома [8]. Несмотря на то, что в определении “аутоиммунного заболевания” отсутствует какая-либо информация о вовлеченности кишечной микробиоты в его патогенез, целый пласт научных исследований посвящен изучению роли таксономической и функциональной представленности микробиоты в механизмах развития аутоиммунных заболеваний [2, 9–10]. Аберрантный кишечный микробиом способствует развитию оксидативного стресса в кишечнике, его барьерной дисфункции, воспалению и аутореактивности [8]. Более того, дисбиоз кишечника связан с хроническим вялотекущим воспалением и с активацией аутореактивных CD4+ и CD8+ Т-клеток, запускающих спонтанные аутоиммунные реакции в органах-мишенях [10–12] (рис. 1).
Рис. 1.
Развитие дисбиоза кишечника способствует интенсификации образования реактивных форм кислорода (РФК) и окислительной модификации белков и липидов, что оказывает влияние на целостность кишечного барьера. IL – интерлейкин.
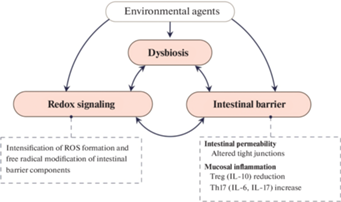
У людей с различными аутоиммунными заболеваниями прослеживается тенденция к снижению биомассы и таксономического разнообразия микроорганизмов кишечника, метаболиты которых обладают иммуномодулирующими свойствами как, например, КЦЖК [13, 14] или метаболиты Trp [15, 16]. В противовес данному изменению обнаруживается увеличение количества сигнальных молекул, усиливающих активацию лимфоцитов, вовлеченных в развитие аутоиммунной провокации [14], а также возрастает содержание веществ, способствующих повышению проницаемости эпителиального барьера кишечника [17, 18].
В связи с предположением, что интенсификация свободнорадикального окисления способствует усугублению дисбиоза кишечника и является одним из главных патогенетических механизмов развития синдрома “протекающей кишки” и аутоиммунных реакций, было изучено влияние на микробиом кишечника N-ацетилцистеина (антиоксиданта, который является предшественником глутатиона) и детоксикации РФК [19]. Обработка N-ацетилцистеином приводила к отчетливым сдвигам в микробном сообществе кишечника, проявляющимся в виде изменений β-разнообразия и бактериальной представленности на уровне семейств. Установлено относительное увеличение бактериальных семейств Akkermansiaceae, Erysipelotrichaceae и Muribaculaceae и уменьшения Rikenellaceae. Добавление N-ацетилцистеина также приводило к обогащению на уровне рода Akkermansia и Turicibacter, в то время как произошло сокращение Alistypes [8].
Было показано, что за счет дисбиоза кишечника нарушается равновесие между Th17 и Treg [20], прослеживается проникновение микробных антигенов в подслизистую оболочку кишечника и системный кровоток, что и способствует развитию аутореактивности [21].
В табл. 1 мы суммировали некоторые изменения бактериальной представленности кишечной микробиоты при самых распространенных аутоиммунных заболеваниях. Так, установлено, что при многих системных аутоиммунных патологиях происходит изменение соотношения Firmicutes к Bacteroidetes (отношение F/B) и β-разнообразия кишечного микробиома [8]. Однако при СД1Т установлено увеличение отношения F/B, тогда как при аутоиммунных заболеваниях щитовидной железы и системной красной волчанке отмечается снижение данного соотношения (табл. 1 ) [8].
Таблица 1.
Специфические девиации бактериальной представленности кишечной микробиоты при некоторых аутоиммунных заболеваниях
| Аутоиммунные заболевания | Бактериальная представленность в кишечнике | Ссылка | |
|---|---|---|---|
| Повышение | Понижение | ||
| СД1Т | ● Bacteroides ● Streptococcus ● Blautia ● Bilophila ● Parabacteroides ● Ruminococcus ● Veillonella ● Clostridium ● Escherichia Примечание: некоторые исследования показали увеличение соотношения F/B, снижение Proteobacteria и Bacteroidetes либо не показали значимых различий. Наличие обратной зависимости количества аутоантител от Streptococcus и Ruminococcaceae |
● Bifidobacterium ● Roseburia ● Faecalibacterium ● Lachnospira ● Haemophilus ● Dialister ● Acidaminococcus |
|
| Воспалительные заболевания кишечника (неспецифический язвенный колит) | ● Адгезивно-инвазивная E. coli (AIEC) ● Proteobacteria ● Fusobacteria ● Bacteroides sp. увеличиваются у пациентов с ВЗК за счет видов Prevotella |
● Akkermansia muciniphila ● F. prausnitzii, ● Bacteroides uniforms |
[22] |
| Ревматоидный артрит | ● Увеличение соотношения F/B ● Prevotella copri ● Clostridium ● Lactobacillus salivarius |
[23] | |
| Аутоиммунные заболевания щитовидной железы | ● Bacteroidetes ● Bacteroides ● Lachnospiraceae ● Bacteroides fragilis |
● Firmrcutes ● Bifidobacterium, Lactobacillus ● Снижение соотношение F/B |
[24] |
| Системная красная волчанка | ● Enterobacteriaceae и Enterococcaceae ● Снижение соотношения F/B |
● Ruminococcaceae | [25, 26] |
Важно отметить, что при системной красной волчанке наблюдается транслокация Enterococcus gallinarum и Lactobacillus reuteri в некишечные ткани, и именно это может иметь решающее значение для развития системных аутоиммунных реакций [27].
Очевидно, что микробиота кишечника участвует в патогенезе различных кишечных и системных аутоиммунных патологиий [28]. Одним из главных защитных механизмов от аутоагрессии является формирование кишечного барьера, целостность которого обеспечивает защиту от адаптивных иммунных реакций против микробиоты здорового человека [29]. Когда интактные комменсалы или патогены проникают через кишечный барьер, ряд защитных механизмов препятствует доступу бактерий в системный кровоток [7]. Одним из главных механизмов является функционирование брыжеечных лимфатических узлов [29]. Такие механизмы имеют значения только при кишечной или сосудистой патологии, при химиотерапии или при отсутствии функциональной врожденной иммунной системы [30], хотя недавние исследования показывают, что кишечные комменсалы могут находиться в лимфоидных тканях ЖКТ и у здоровых лиц. Поэтому остаются вопросы, насколько транслокация патобионтов критична для стимуляции системного аутоиммунитета [31]. Однако вне сомнения, метаболическая активность патобионтов будет играть не меньшую роль в аутоиммунной провокации, чем их непосредственная транслокация.
2. МЕТАБОЛИЧЕСКИЙ ПРОФИЛЬ КИШЕЧНОЙ МИКРОБИОТЫ ПРИ АУТОИММУННЫХ ЗАБОЛЕВАНИЯХ
Особенности кинуренинового метаболизма Trp
В последние два десятилетия сформировалась теория, согласно которой метаболизм Trp через кинурениновый путь вовлечен в иммунную функцию организма [32]. Известно, что Trp является эссенциальной аминокислотой для организма человека и утилизируется по доминантному кинурениновому пути (~95% катаболизма Trp) и трем минорным путям: 1) серотониновому (гидроксилирование Trp), 2) триптаминовому (декарбоксилирование Trp) и 3) индольному (трансаминирование Trp) [32].
Все метаболиты обмена Trp являются сигнальными молекулами и вовлечены как в регуляцию quorum sensing (QS) в микробиотической экосистеме кишечника, так и в регуляцию метаболизма и работы иммунной системы организма человека [33]. Метаболиты кинуренинового пути обмена Trp являются универсальными сигнальными молекулами [34], и большая часть Trp (около 90–95%) утилизируется печенью именно по кинурениновому пути [35]. Остальной Trp попадает в кровоток и доступен для использования клетками периферических тканей, такими как эндотелиальные клетки сосудов, фибробласты и иммунные клетки, обеспечивающие врожденный иммунитет [36]. При иммунной стимуляции срабатывает так называемый “кинурениновый переключатель” [37], и большая часть Trp поступает в иммунокомпетентные клетки и ими метаболизируется по кинурениновому пути. Так, показано, что после иммунной стимуляции клетки иммунной системы генерируют высокие уровни внутриклеточной хинолиновой кислоты и, следовательно, НАД+. В свою очередь, данное событие вовлечено в индукцию синтеза белков цитоскелета и регуляцию подвижности иммунных клеток [38, 39].
Ключевой фермент кинуренинового пути – индоламин-2,3-диоксигеназа 1 (IDO1) – один из основных регуляторов функционирования иммунных клеток, который контролирует баланс между стимуляцией и подавлением активности иммунного ответа в очагах локального воспаления и имеет, таким образом, отношение к широкому спектру аутоиммунных и воспалительных заболеваний [39, 40].
Первый этап кинуренинового пути включает окисление индольного кольца и катализируется двумя дифференцированно распределенными в организме человека ферментами: триптофан-2,3-диоксигеназой (TDO) (преимущественно экспрессируется в печени и головном мозге) и IDO (экспрессируется в большинстве периферических тканей) [41] (рис. 2).
Рис. 2.
Особенности кинуренинового метаболизма Trp. Под действием N-формилкинуренинформамидазы N-формилкинуренин превращается в кинуренин. Далее метаболизм кинуренина может пойти по одному из двух путей: 1) превращение в кинуреновую и антраниловую кислоты с помощью кинуренинаминотрансферазы (KAT) и кинурениназы (KYNU) соответственно, или 2) образование нейротоксического метаболита 3-гидроксикинуренина, который далее трансаминируется в ксантуреновую кислоту KAT. Затем 3-гидроксикинуренин превращается KYNU в иммуномодулирующий метаболит – 3-гидроксиантраниловую кислоту, которая может самопроизвольно превращаться в никотиновую кислоту через 3-гидроксиантранилат-3,4-диоксигеназу (3-HAAO). Далее 3-гидроксиантранилат превращается в нестабильный промежуточный 2-амино-3-карбоксимуконовый полуальдегид, который спонтанно превращается в агонист рецепторов N-метил-D-аспартата (NMDA) – хинолиновую кислоту или превращается в нейропротекторную пиколиновую кислоту после ферментативного декарбоксилирования с помощью 2-амино-3-карбоксимуконат-семиальдегиддекарбоксилазы (ACMSD). Хинолиновая кислота подвергается декарбоксилированию с образованием никотиновой кислоты, которая является предшественником никотиамидаадениндинуклеотида (НАД+).
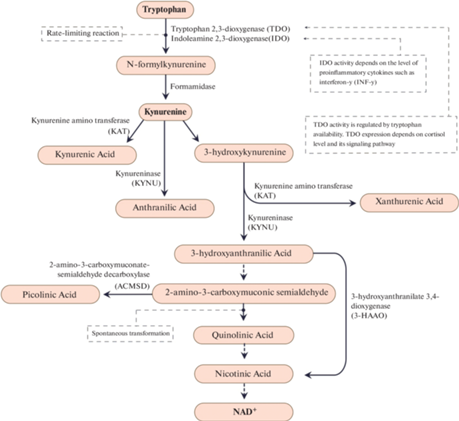
Рис. 3.
Влияние КЦЖК на целостность кишечного барьера и синтез провоспалительных цитокинов энтероцитами. При взаимодействии КЦЖК с GPCR43 происходит ассоциация β-аррестина с ингибитором (I-κB) и ингибируется фосфорилирование последнего, диссоциация I-κB и NF-κB не происходит. Результатом ингибирования пути NF-κB является снижение синтеза провоспалительных цитокинов. Градиент концентрации кислорода, поддерживаемый в том числе за счет окисления бутирата, способствует поддержанию целостности кишечного барьера. Эффекты бутирата связаны со снижением уровней фактора некроза опухоли-α (TNF-α), IL-6 и подавлением активации инфламмосомы Nod-подобного рецептора семейства NALP 3 (NLRP3) через G-белок ассоциированный рецептор 109А (GPR109A).
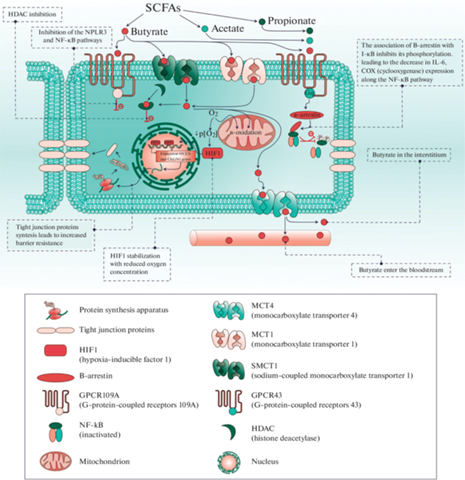
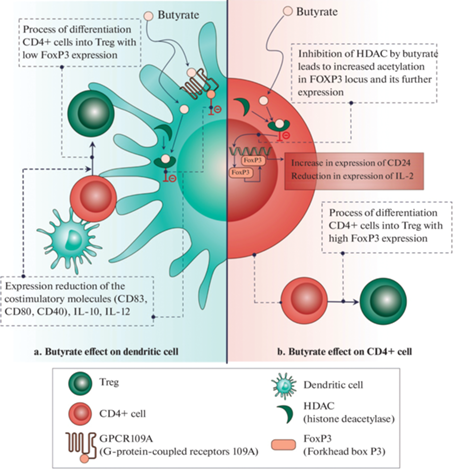
Рис. 4.
(a) – влияние бутирата на DC, ведущее к дифференцировке CD4+ клеток в Treg с низкой экспрессией FoxP3. (b) – влияние бутирата на CD4+ клетки с их дальнейшей дифференцировкой в Treg с высокой экспрессией FoxP3.
TDO и IDO – ферменты-гемопротеины, которые превращают Trp в N-формилкинуренин в регуляторных скорость-лимитирующих реакциях. Активность IDO зависит от уровня провоспалительных цитокинов. Так, одним из самых мощных индукторов экспрессии IDO является интерферон-γ (INF-γ) [42]. Активность TDO регулируется доступностью Trp, при этом промотор данного регуляторного фермента содержит GRE (элемент глюкокортикоидной регуляции в ДНК), следовательно, экспрессия TDO зависит от уровня кортизола и активности его сигнального пути. Так, известно, что при стрессе происходит индукция экспрессии TDO [32].
Множество исследований посвящены изучению роли метаболитов кинуренинового пути обмена Trp в иммунной регуляции [28, 33, 36], а также анализу влияния кинуренина, кинуреновой кислоты, антраниловой кислоты, хинолиновой кислоты [25] и НАД+ на работу иммунокомпетентных клеток. Эти работы показывают, что именно нарушение метаболического звена кинуренинового обмена является одним из ключевых механизмов аутоиммунных провокаций [37].
Trp сигнальные молекулы оказывают свои эффекты AhR-зависимым (то есть через AhR-арилгидрокарбоновые рецепторы) [38] и AhR-независимым образом [34]. AhR в большом количестве представлены в энтероцитах и в иммунных клетках [35]. Было установлено, что эффекты, опосредованные активацией AhR, вносят значительный вклад в поддержание целостности кишечного барьера, тем самым способствуя развитию иммунотолерантности [38].
В качестве лигандов AhR выступают как эндогенные (кинуренин, кинуреновая и ксантуреновая кислоты и др.), так и микробные метаболиты Trp (индол и его производные, а также триптамин). После активации AhR с лигандом транслоцируется в ядро и активирует экспрессию генов, ответственных за синтез IL – IL-6 и IL-22 и Cyp1A1 (цитохрома P450 1A1), а также важнейших ферментов кинуренинового пути – IDO и TDO, косвенно регулируя отношения между комменсальной микробиотой и хозяином [44].
Роль метаболизма короткоцепочечных жирных кислот (КЦЖК) в развитии аутоиммунных заболеваний
Одними из наиболее изученных микробных метаболитов, обладающих иммуносупрессивным действием, являются КЦЖК, такие как бутират, ацетат и пропионат. Данные метаболиты оказывают как прямое действие, проникая внутрь клетки через специальные транспортеры, так и опосредованное – через рецепторы, ассоциированные с G-белками [45].
КЦЖК попадают внутрь энтероцитов через натрий-зависимый монокарбоксилатный транспортер (sodium-coupled monocarboxylate transporter 1, SMCT1) и монокарбоксилатный транспортер 1-го типа (monocarboxylate transporter 1, MCT1). Внутри клетки данные метаболиты могут выступать в качестве энергетических субстратов, а в кровотоке (посредством MCT4) оказывают системные эффекты. Кроме того, бутират и пропионат могут ингибировать гистондеацетилазы (HDAC) 1-го и 3-го типов, за счет чего снижается деацетилирования как гистоновых, так и негистоновых белков [46]. В результате данных модификаций изменяется экспрессия генов, продукты которых ответственны за развитие воспаления [47]. Опосредованное действие осуществляется через рецепторы, связанные с G-белками (GPCR41, GPCR43, GPCR109A) [48], однако с рецептором к ниацину – GPR109A может связаться только бутират [49]. Через активацию данных рецепторов по пути NF-kB КЦЖК снижают синтез провоспалительных цитокинов, белков ответа острой фазы воспаления и циклооксигеназы-2 [50].
КЦЖК, являясь одними из главных энергетических субстратов для колоноцитов, способствуют снижению концентрации кислорода в эпителии толстой кишки, поскольку большое количество кислорода идет на их метаболизм. В результате происходит стабилизация фактора, индуцированного гипоксией 1 (hypoxia-inducible factor 1, HIF1). Транслокация в ядро HIF1, являющегося транскрипционным фактором, индуцирует экспрессию генов OCLN (Occludin) и CLDN1 (Claudin 1), кодирующих белки плотных контактов – окклюдин и клаудин-1, что ведет к поддержанию целостности эпителиального барьера [51]. По нашему мнению, поддержание целостности барьера является одним из ведущих механизмов противодействия аутоагрессии, поскольку повышение проницаемости кишечника однозначно приведет к повышению транслокации бактериальных ДНК и липополисахаридов (ЛПС) в кровь. Также важно подчеркнуть значимость уменьшения локального воспаления, опосредованного снижением концентрации провоспалительных цитокинов. Мы считаем, что снижение биомассы бактерий, продуцирующих КЦЖК, должно привести к гиперпродукции провоспалительных цитокинов, опосредованной интенсификацией сигналинга по пути NF-kB, вследствие чего происходит гиперстимуляция кинуренинового пути в иммунных клетках. В свою очередь, срабатывание “кинуренинового переключателя” приводит к увеличению образования целого ряда различных сигнальных молекул Trp обмена в организме человека [26].
3. ПОТЕНЦИАЛЬНЫЕ МЕХАНИЗМЫ ИНДУКЦИИ АУТОИММУННОЙ АГРЕССИИ, ОПОСРЕДОВАННЫЕ МИКРОБИОТОЙ КИШЕЧНИКА
Молекулярная мимикрия
До сих пор остается не до конца ясным, является ли дисбиоз причиной или следствием аутоиммунных патологий. Многообразие перекрестных связей между микробиотой и хозяином обуславливает ее влияние на множество процессов, протекающих в организме: поддержание энергетического гомеостаза, регуляцию иммунных реакций, метаболизм ксенобиотиков и т.д. Несмотря на широкую распространенность аутоиммунных заболеваний, репертуар аутоантигенов является крайне ограниченным [53]. Интересным является то, что некоторые антигены микроорганизмов гомологичны человеческим аутоантигенам и могут стать индукторами развития различных аутоиммунных заболеваний. Данный механизм получил название “молекулярной мимикрии”. Так, гомология между мембранными липоолигосахаридами Campilobacter jejuni и человеческими ганглиозидами (G) GM1 и GD1a индуцирует развитие синдрома Гийена–Барре, что было показано в исследовании на животных моделях [54]. Подобный механизм также характерен для патогенеза аутоиммунных заболеваний щитовидной железы, рассеянного склероза, системной красной волчанки и др. [2, 55, 56].
Активация иммунокомпетентных клеток
Другим механизмом, способствующим развитию аутоиммунных заболеваний, является опосредованная микробиотой активация иммунокомпетентных клеток (Th17) в кишечнике и стимуляция выработки провоспалительных цитокинов, что имеет двойственное значение. С одной стороны, это способствует поддержанию кишечного барьера в борьбе с патогенными микроорганизмами, а с другой стороны – провоцирует аутоиммунную агрессию [57]. Стоит также подчеркнуть, что при развитии аутоиммунных заболеваний наблюдается повышенная активность Th17 на фоне сниженной активности Treg. Данное соотношение может регулироваться как самой микробиотой, так и ее метаболитами. Например, КЦЖК способствуют увеличению числа Treg в слизистой оболочке кишечника [58] за счет влияния на дендритные клетки [59]. Таким образом, дисбиоз кишечника может увеличивать соотношение Th17/Treg, тем самым индуцируя провоспалительные реакции, способствующие развитию аутоиммунных заболеваний.
Увеличение проницаемости кишечного барьера
В недавних исследованиях было продемонстрировано, что аутоиммунные заболевания ассоциированы с развитием синдрома “дырявой кишки” и повышенной бактериальной транслокацией, вследствие которых развивается хроническое вялотекущее системное воспаление [60]. Проникновение в кровь и соответственно в ткани такого патобионта, как Enterococcus gallinarum, способствует развитию системной красной волчанки и аутоиммунного гепатита. При этом имеет значение не только увеличение концентрации циркулирующих бактериальных ЛПС в крови, но и индуцируемая E. gallinarum избыточная экспрессия на гепатоцитах эндогенного ретровирусного гликопротеина 70 и β2-гликопротеина I, которая в дальнейшем у генетически предрасположенных животных способствует развитию аутоиммунных реакций [61].
4. МЕХАНИЗМЫ РЕГУЛЯЦИИ АУТОИММУННОЙ АГРЕССИИ
Влияние бутирата на иммунокомпетентные клетки
Одним из важнейших факторов, имеющих значение в поддержании аутотолерантности, является функционирование Treg клеток. Их способность к регуляции иммунных реакций возрастает за счет усиления экспрессии FoxP3 (forkhead box P3), индукции дифференцировки наивных CD4+ T-клеток в Treg под действием толерогенных дендритных клеток (DС). Важно отметить, что КЦЖК могут влиять на оба этих процесса [62].
В экспериментах in vitro исследовалось влияние ацетата, пропионата и бутирата на индуцированную ЛПС активацию DC, полученных из моноцитов человека. Было показано снижение экспрессии костимулирующих молекул (CD83, CD80, CD40), IL-10 и IL-12, а также снижение метаболической активности DC. При этом наиболее значимый эффект оказывал бутират, и в меньшей мере пропионат и ацетат. Культивирование DC, предварительно обработанных КЦЖК, с наивными CD4+ T-лимфоцитами усиливало дифференцировку последних в Treg, однако их фенотип в данном случае характеризовался низкой продукцией FoxP3 [63]. Подобные эффекты бутират реализует путем ингибирования HDAC и через активацию GPR109A, которые в большом количестве представлены на DC, но отсутствуют на лимфоидных клетках [49].
Также было установлено, что в наивных CD4+ T-клетках бутират индуцирует экспрессию FoxP3 – транскрипционного фактора, способствующего активации и экспансии Treg [64]. FoxP3 подавляет выработку IL-2, но при этом увеличивает экспрессию CD25 – высокоаффинного рецептора данного цитокина [65]. Хотя функция Treg регулируется далеко не одним компонентом, а более сложной системой, включающей в себя другие транскрипционные факторы, генетические и эпигенетические элементы, а также сигналы тканевого микроокружения, FoxP3 является все же ключевым игроком [66]. Предполагается, что подобное влияние бутирата осуществляется благодаря ингибированию HDAС, а как следствие – усиленному ацетилированию в локусе FOXP3 [66].
Влияние метаболитов обмена Trp на иммунокомпетентные клетки
Значительные изменения концентрации метаболитов кинуренинового пути, а также микробных производных Trp выявляются при различных патологических состояниях: ожирении [26], аутоиммунных [67] и онкологических заболеваниях [68]. Так, при ожирении было показано повышение концентрации хинолиновой и кинуреновой кислот, кинуренина, 5-гидроксииндол-3-ацетата, индол-3-лактата, индол-3-ацетата и индол-3-бутирата в сыворотке крови, что говорит об интенсификации всех путей метаболизма Trp в организме [26]. Для рассеянного склероза характерно снижение концентрации Trp в сыворотке крови при одновременном повышении уровня кинуренина [67]. Подобные изменения связаны не столько с истощением пула Trp, сколько с повышенной его утилизацией для восполнения потребности иммунной системы в продукции НАД+ [29].
Все больше данных указывают на то, что НАД+, известный как молекула, участвующая в энергетическом метаболизме, является одним из основных модуляторов иммунно-метаболических взаимосвязей. Концентрация НАД+ отражает его потребление и синтез de novo по кинурениновому пути, а также уровень экспрессии генов, кодирующих НАД-зависимые ферменты [69]. НАД+ участвует в модификации белков и сигнальных молекул, выступая в качестве кофермента сиртуинов (SIRTs), поли(АДФ-рибозо)-полимеразы (PARPs), АДФ-рибозотрансферазы (ARTs) и CD38/CD157 [70].
CD38 локализуется как на поверхности активированных Т-клеток, так и в их цитоплазме, а также обнаруживается в макрофагах, нейтрофилах и DC. Было показано, что при системной красной волчанке статистически значимо повышается экспрессия данной молекулы в CD8+ T-лимфоцитах, NK-клетках и DC [71]. CD38 проявляет свойства АДФ-рибозилциклазы и циклической АДФ-рибозилгидролазы в реакциях образования циклического АДФ-рибозила (цАДФР) и АДФР соответственно, однако преимущественно выступает в качестве НАД-гликогидролазы в реакциях гидролиза НАД+ до АДФР и никотинамида. Продукты данных реакций участвуют в регуляции концентрации Ca2+ в цитоплазме, что необходимо для активации Т-клеток. Такие же свойства проявляет и CD157, однако обладает меньшим сродством к НАД+. Работа данных ферментов сопровождается истощением пула НАД+, что снижает его доступность для SIRTs, PARPs и ARTs [72].
Повышение доступности НАД+ для SIRT1 возможно характеризует его роль в патогенезе аутоиммунных заболеваний в качестве провоспалительного агента. Так, установлено, что SIRT1 деацетилирует транскрипционный фактор FoxP3, определяющий состояние Treg, что приводит к его деградации [73]. Также было показано, что SIRT1 связывает и деацетилирует транскрипционный фактор RORγt – главный регулятор дифференцировки Th17. Таким образом, происходит ингибирование функции Treg на фоне усиления дифференцировки Th17, что ведет к изменению соотношения данных клеточных популяций [74].
Как говорилось выше, синтез кинуренинов контролируется IDO. При некоторых онкологических заболеваниях наблюдается повышенная экспрессия IDO, что позволяет опухолевым клеткам уклоняться от иммунного надзора и объясняет резистентность таких опухолей к иммунотерапии [23]. Ингибиторы IDO были предложены в качестве агентов для преодоления лекарственной устойчивости опухоли. Возможно, противоположный подход, способствующий повышению экспрессии фермента, был бы эффективен в борьбе с аутоиммунными заболеваниями. Действительно, было показано, что при стимуляции экспрессии IDO, опосредованной TGF-β и IFN-γ, в DC происходит формирование толерогенного фенотипа и дифференцировка наивных CD4+ T-клеток в Treg, а также ингибируется переход Treg в провоспалительные Th17 [75].
В ответ на TGF-β происходит фосфорилирование двух иммунорецепторных тирозиновых ингибирующих мотивов (ITIMs) киназами Src. Благодаря этому образуются сайты стыковки для связывания и активации тирозинфосфатаз SHP-1 и SHP-2, после чего они дефосфорилируют киназу IRAK, сдвигая баланс передачи сигналов NF-κB по противовоспалительному пути [23]. При высокой концентрации IL-6 в начале воспалительной реакции IDO связывается с фактором SOCS3, что способствует его протеасомной деградации и, как следствие, снижению продукции кинуренина, обеспечивающей истощение Trp во внеклеточной среде, а также долгосрочных влияний, опосредованных AhR и сигналингом через фосфорилирование ITIM1 и ITIM2 [76].
ЗАКЛЮЧЕНИЕ
Несмотря на то, что механизмы влияния кишечной микробиоты и ее дисбиоза на развитие аутоиммунных заболеваний точно не определены, очевидной является зависимость системных аутоиммунных реакций от разнообразия и состава кишечного микробиома. Аберрантный кишечный микробиом способствует развитию оксидативного стресса в кишечнике, барьерной дисфункции, воспалению и аутореактивности. Кроме того, изменение таксономического состава микробиоты влияет на ее метаболический профиль и способствует увеличению или, напротив, снижению продукции определенных сигнальных и иммуннорегуляторных молекул, влияющих на дифференцировку, созревание и миграцию иммунных клеток. С другой стороны, повышение проницаемости кишечника, связанное с дисбиозом и изменением продукции КЦЖК и метаболитов триптофана, является одним из основных механизмов развития воспалительной реакции и аутоагрессии.
Список литературы
Nasonov EL, Aleksandrova EN, Novikov AA (2015) Autoimmune Rheumatic Diseases – Problems of Immunopathology and Personalized Treatment. Proceed Vestn Ross Akad Medi Nauk 70: 169–182. https://doi.org/10.15690/vramn.v70i2.1310
Wang L, Wang FS, Gershwin ME (2015) Human autoimmune diseases: a comprehensive update. J Intern Med. 278: 369–395. https://doi.org/10.1111/joim.12395
Bach JF (2018) The hygiene hypothesis in autoimmunity: the role of pathogens and commensals. Nat Rev Immunol 18: 105–120. https://doi.org/10.1038/nri.2017.111
Schroeder BO, Bäckhed F (2016) Signals from the gut microbiota to distant organs in physiology and disease. Nat Med 10: 1079–1089. https://doi.org/10.1038/nm.4185
Sharon G, Sampson TR, Geschwind DH, Mazmanian SK (2016) The Central Nervous System and the Gut Microbiome. Cell 167(4): 915–932. https://doi.org/10.1016/j.cell.2016.10.027
Levy M, Kolodziejczyk AA, Thaiss CA, Elinav E (2017) Dysbiosis and the immune system. Nat Rev Immunol 17(4): 219–232. https://doi.org/10.1038/nri.2017.7
Wang H, Wang G, Banerjee N, Liang Y, Du X, Boor PJ, Hoffman KL., Khan MF (2021) Aberrant Gut Microbiome Contributes to Intestinal Oxidative Stress, Barrier Dysfunction, Inflammation and Systemic Autoimmune Responses in MRL/Lpr Mice. Front Immunol 12: 651191. https://doi.org/10.3389/fimmu.2021
Greiling TM, Dehner C, Chen X, Hughes K, Iñiguez AJ, Boccitto M, Ruiz DZ, Renfroe SC, Vieira SM, Ruff WE (2018) Commensal Orthologs of the Human Autoantigen Ro60 as Triggers of Autoimmunity in Lupus. Sci Transl Med 10: eaan2306. https://doi.org/10.1126/scitranslmed.aan2306
Ruff WE, Dehner C, Kim WJ, Pagovich O, Aguiar CL, Yu AT, Roth AS, Vieira SM, Kriegel C, Adeniyi O (2019) Pathogenic Autoreactive T and B Cells Cross-React with Mimotopes Expressed by a Common Human Gut Commensal to Trigger Autoimmunity. Cell Host Microbe 26: 100–113.e8. https://doi.org/10.1016/j.chom.2019.05.003
Horai R, Zárate-Bladés CR, Dillenburg-Pilla P, Chen J, Kielczewski JL, Silver PB, Jittayasothorn Y, Chan CC, Yamane H, Honda K (2015) Microbiota-Dependent Activation of an Autoreactive T Cell Receptor Provokes Autoimmunity in an Immunologically Privileged Site. Immunity 43: 343–353. https://doi.org/10.1016/j.immuni.2015.07.014
Ruff WE, Kriegel MA (2015) Autoimmune Host-Microbiota Interactions at Barrier Sites and Beyond. Trends Mol Med 21: 233–244. https://doi.org/10.1016/j.molmed.2015.02.006
Cayres LC, de Salis, Rodrigues GSP, Lengert A, Biondi APC, Sargentini LD, Brisotti JL, Gomes E, de Oliveira GL (2021) Detection of Alterations in the Gut Microbiota and Intestinal Permeability in Patients With Hashimoto Thyroiditis. Front Immunol 12: 579140. https://doi.org/10.3389/fimmu.2021.579140
Yuan X, Wang R, Han B, Sun CJ, Chen R, Wei H, Chen L, Du H, Li G, Yang Y (2022) Functional and Metabolic Alterations of Gut Microbiota in Children with New-Onset Type 1 Diabetes. Nat Commun 13: 6356. https://doi.org/10.1038/s41467-022-33656-4
Maffeis C, Martina A, Corradi M, Quarella S, Nori N, Torriani S, Plebani M, Contreas G, Felis GE (2016) Association between Intestinal Permeability and Faecal Microbiota Composition in Italian Children with Beta Cell Autoimmunity at Risk for Type 1 Diabetes. Diabetes Metab Res Rev 32: 700–709. https://doi.org/10.1002/dmrr.2790
Siljander H, Honkanen J, Knip M (2019) Microbiome and Type 1 Diabetes. EBioMedicine 46: 512–521. https://doi.org/10.1016/j.ebiom.2019.06.031
Walters WA, Xu Z, Knight R (2014) Meta-Analyses of Human Gut Microbes Associated with Obesity and IBD. FEBS Lett 588: 4223–4233. https://doi.org/10.1016/j.febslet.2014.09.039
Viladomiu M, Kivolowitz C, Abdulhamid A, Dogan B, Victorio D, Castellanos JG, Woo V, Teng F, Tran NL, Sczesnak A (2017) IgA-Coated E. Coli Enriched in Crohn’s Disease Spondyloarthritis Promote TH17-Dependent Inflammation. Sci Transl Med 9: eaaf9655. https://doi.org/10.1126/scitranslmed.aaf9655
Zhang X, Zhang D, Jia H, Feng Q, Wang D, Liang D, Wu X, Li J, Tang L, Li Y (2015) The Oral and Gut Microbiomes Are Perturbed in Rheumatoid Arthritis and Partly Normalized after Treatment. Nat Med 21: 895–905. https://doi.org/10.1038/nm.3914
Taneja V (2014) Arthritis Susceptibility and the Gut Microbiome. FEBS Lett 588: 4244–4249. https://doi.org/10.1016/j.febslet.2014.05.034
Gong B, Wang C, Meng F, Wang H, Song B, Yang Y, Shan Z (2021) Association Between Gut Microbiota and Autoimmune Thyroid Disease: A Systematic Review and Meta-Analysis. Front Endocrinol (Lausanne) 12: 774362. https://doi.org/10.3389/fendo.2021.774362
Mu Q, Zhang H, Liao X, Lin K, Liu H, Edwards MR, Ahmed SA, Yuan R, Li L, Cecere TE (2017) Control of Lupus Nephritis by Changes of Gut Microbiota. Microbiome 5: 73. https://doi.org/10.1186/s40168-017-0300-8
Dei-Cas I, Giliberto F, Luce L, Dopazo H, Penas-Steinhardt A (2020) Metagenomic Analysis of Gut Microbiota in Non-Treated Plaque Psoriasis Patients Stratified by Disease Severity: Development of a New Psoriasis-Microbiome Index. Sci Rep 10: 12754. https://doi.org/10.1038/s41598-020-69537-3
Mondanelli G, Iacono A, Carvalho A, Orabona C, Volpi C, Pallotta MT, Matino D, Esposito S, Grohmann U (2019) Amino Acid Metabolism as Drug Target in Autoimmune Diseases. Autoimmun Rev 18: 334–348. https://doi.org/10.1016/j.autrev.2019.02.004
Dadvar S, Ferreira DMS, Cervenka I, Ruas JL (2018) The Weight of Nutrients: Kynurenine Metabolites in Obesity and Exercise. J Int Med 284: 519–533. https://doi.org/10.1111/joim.12830
Moffett JR, Arun P, Puthillathu N, Vengilote R, Ives JA, Badawy AAB, Namboodiri AM (2020) Quinolinate as a Marker for Kynurenine Metabolite Formation and the Unresolved Question of NAD+ Synthesis During Inflammation and Infection. Front Immunol 11: 31. https://doi.org/10.3389/fimmu.2020.00031
Shestopalov AV, Shatova OP, Karbyshev MS, Gaponov AM, Moskaleva NE, Appolonova SA, Tutelyan AV, Makarov VV, Yudin SM, Roumiantsev SA (2021) “Kynurenine Switch” and Obesity. Bull Siber Med 20: 103–111. https://doi.org/10.20538/1682-0363-2021-4-103-111
Hevia A, Milani C, López P, Cuervo A, Arboleya S, Duranti S, Turroni F, González S, Suárez A, Gueimonde M (2014) Intestinal Dysbiosis Associated with Systemic Lupus Erythematosus. mBio 5: e01548-14. https://doi.org/10.1128/mBio.01548-14
Badawy AAB (2017) Kynurenine Pathway of Tryptophan Metabolism: Regulatory and Functional Aspects. Int J Tryptophan Res 10: 1178646917691938. https://doi.org/10.1177/1178646917691938
Badawy AAB (2019) Tryptophan Metabolism: A Versatile Area Providing Multiple Targets for Pharmacological Intervention. Egypt J Basic Clin Pharmacol 9: https://doi.org/10.32527/2019/101415. 10.32527/2019/101415
Yuasa HJ, Ball HJ (2015) Efficient Tryptophan-Catabolizing Activity Is Consistently Conserved through Evolution of TDO Enzymes, but Not IDO Enzymes. J Exp Zool B Mol Dev Evol 324: 128–140. https://doi.org/10.1002/jez.b.22608
Alvarado DM, Chen B, Iticovici M, Thaker AI, Dai N, VanDussen KL, Shaikh N, Lim CK, Guillemin GJ, Tarr PI, Ciorba MA (2019) Epithelial Indoleamine 2,3-Dioxygenase 1 Modulates Aryl Hydrocarbon Receptor and Notch Signaling to Increase Differentiation of Secretory Cells and Alter Mucus-Associated Microbiota. Gastroenterology 157: 1093–1108.e11. https://doi.org/10.1053/j.gastro.2019.07.013
Cecchi M, Paccosi S, Silvano A, Eid AH, Parenti A (2021) Dexamethasone Induces the Expression and Function of Tryptophan-2-3-Dioxygenase in SK-MEL-28 Melanoma Cells. Pharmaceuticals (Basel) 14: 211. https://doi.org/10.3390/ph14030211
Savitz J (2020) The Kynurenine Pathway: A Finger in Every Pie. Mol Psychiatry 25: 131–147. https://doi.org/10.1038/s41380-019-0414-4
Shatova OP, Shestopalov AV (2023) Tryptophan Metabolism: A New Look at the Role of Tryptophan Derivatives in the Human Body. Biol Bull Rev 13: 81–91. https://doi.org/10.1134/S2079086423020068
Jamshed L, Debnath A, Jamshed S, Wish JV, Raine JC, Tomy GT, Thomas PJ, Holloway AC (2022) An Emerging Cross-Species Marker for Organismal Health: Tryptophan-Kynurenine Pathway. Int J Mol Sci 23: 6300. https://doi.org/10.3390/ijms23116300
Chen Y, Guillemin GJ (2009) Kynurenine Pathway Metabolites in Humans: Disease and Healthy States. Int J Tryptophan Res 2: 1–19. https://doi.org/10.4137/ijtr.s2097
Krupa A, Kowalska I (2021) The Kynurenine Pathwa – New Linkage between Innate and Adaptive Immunity in Autoimmune Endocrinopathies. Int J Mol Sci 22: 9879. https://doi.org/10.3390/ijms22189879
Zádori D, Klivényi P, Szalárdy L, Fülöp F, Toldi J, Vécsei L (2012) Mitochondrial Disturbances, Excitotoxicity, Neuroinflammation and Kynurenines: Novel Therapeutic Strategies for Neurodegenerative Disorders. J Neurol Sci 322: 187–191. https://doi.org/10.1016/j.jns.2012.06.004
Silva S, Shimizu JF, Oliveira DM, de Assis LR, de Bittar C, Mottin M, Sousa BK, de P Mesquita NC, Regasini LO, Rahal P (2019) A Diarylamine Derived from Anthranilic Acid Inhibits ZIKV Replication. Sci Rep 9: 17703. https://doi.org/10.1038/s41598-019-54169-z
Marszalek-Grabska M, Walczak K, Gawel K, Wicha-Komsta K, Wnorowska S, Wnorowski A, Turski WA (2021) Kynurenine Emerges from the Shadows – Current Knowledge on Its Fate and Function. Pharmacol Ther 225: 107845. https://doi.org/10.1016/j.pharmthera.2021.107845
Gao J, Xu K, Liu H, Liu G, Bai M, Peng C, Li T, Yin Y (2018) Impact of the Gut Microbiota on Intestinal Immunity Mediated by Tryptophan Metabolism. Front Cell Infect Microbiol 8: 13. https://doi.org/10.3389/fcimb.2018.00013
Schiering C, Wincent E, Metidji A, Iseppon A, Li Y, Potocnik AJ, Omenetti S, Henderson CJ, Wolf CR, Nebert DW (2017) Feedback Control of AHR Signalling Regulates Intestinal Immunity. Nature 542: 242–245. https://doi.org/10.1038/nature21080
Neavin DR, Liu D, Ray B, Weinshilboum RM (2018) The Role of the Aryl Hydrocarbon Receptor (AHR) in Immune and Inflammatory Diseases. Int J Mol Sci 19: 3851. https://doi.org/10.3390/ijms19123851
Liu M, Nieuwdorp M, de Vos WM, Rampanelli E (2022) Microbial Tryptophan Metabolism Tunes Host Immunity, Metabolism, and Extraintestinal Disorders. Metabolites 12: 834. https://doi.org/10.3390/metabo12090834
Horiuchi H, Kamikado K, Aoki R, Suganuma N, Nishijima T, Nakatani A, Kimura I (2020) Bifidobacterium Animalis Subsp. Lactis GCL2505 Modulates Host Energy Metabolism via the Short-Chain Fatty Acid Receptor GPR43. Sci Rep 10: 4158. https://doi.org/10.1038/s41598-020-60984-6
Schilderink R, Verseijden C, Seppen J, Muncan V, van den Brink GR, Lambers TT, van Tol EA, de Jonge WJ (2016) The SCFA Butyrate Stimulates the Epithelial Production of Retinoic Acid via Inhibition of Epithelial HDAC. Am J Physiol Gastrointest Liver Physiol 310: 1138–1146. https://doi.org/10.1152/ajpgi.00411.2015
Fattahi Y, Heidari HR, Khosroushahi AY (2020) Review of Short-Chain Fatty Acids Effects on the Immune System and Cancer. Food Biosci 38: 100793. https://doi.org/10.1016/j.fbio.2020.100793
Parada Venegas D, De la Fuente MK, Landskron G, González MJ, Quera R, Dijkstra G, Harmsen HJM, Faber KN, Hermoso MA (2019) Corrigendum: Short Chain Fatty Acids (SCFAs)-Mediated Gut Epithelial and Immune Regulation and Its Relevance for Inflammatory Bowel Diseases. Front Immunol 10: 1486. https://doi.org/10.3389/fimmu.2019.01486
Singh N, Gurav A, Sivaprakasam S, Brady E, Padia R, Shi, Thangaraju M, Prasad PD, Manicassamy S, Munn DH (2014) Activation of Gpr109a, Receptor for Niacin and the Commensal Metabolite Butyrate, Suppresses Colonic Inflammation and Carcinogenesis. Immunity 40: 1486. https://doi.org/10.1016/j.immuni.2013.12.007
Huang W, Man Y, Gao C, Zhou L, Gu J, Xu H, Wan Q, Long Y, Chai L, Xu Y (2020) Short-Chain Fatty Acids Ameliorate Diabetic Nephropathy via GPR43-Mediated Inhibition of Oxidative Stress and NF-κ B Signaling. Oxid Med Cell Longev 4074832. https://doi.org/10.1155/2020/4074832
Kelly CJ, Zheng L, Campbell, EL, Saeedi B, Scholz CC, Bayless AJ, Wilson KE, Glover LE, Kominsky DJ, Magnuson A et al. (2015) Crosstalk between Microbiota-Derived Short-Chain Fatty Acids and Intestinal Epithelial HIF Augments Tissue Barrier Function. Cell Host Microbe 17(5): 662–671. https://doi.org/10.1016/j.chom.2015.03.005
Li YJ, Chen X, Kwan TK, Loh YW, Singer J, Liu Y, Ma J, Tan J, Macia L, Mackay CR (2020) Dietary Fiber Protects against Diabetic Nephropathy through Short-Chain Fatty Acid-Mediated Activation of G Protein-Coupled Receptors GPR43 and GPR109A. J Am Soc Nephrol 31: 1267–1281. https://doi.org/10.1681/ASN.2019101029
Williams SG, Wolin SL (2021) The Autoantigen Repertoire and the Microbial RNP World. Trends Mol Med 27: 422–435. https://doi.org/10.1016/j.molmed.2021.02.003
Koga M, Gilbert M, Li Yuki N (2015) Complex of GM1-and GD1a-Likelipo-Oligosaccharide Mimics GM1b, Inducing Anti-GM1b Antibodies. PLoS One 10: e0124004. https://doi.org/10.1371/journal.pone.0124004
Kiseleva EP, Mikhailopulo KI, Sviridov OV, Novik GI, Knirel YA, Dey ES (2011) The Role of Components of Bifidbacterium and Lactobacillus in Pathogenesis and Serologic Diagnosis of Autoimmune Thyroid Diseases. Benef Microbes 2: 139–154. https://doi.org/10.3920/BM2010.0011
Elsayed NS, Aston P, Bayanagari VR, Shukla SK (2022) The Gut Microbiome Molecular Mimicry Piece in the Multiple Sclerosis Puzzle. Front Immunol 13: 972160. https://doi.org/10.3389/fimmu.2022.972160
Kinashi Y, Hase K (2021) Partners in Leaky Gut Syndrome: Intestinal Dysbiosis and Autoimmunity. Front Immunol 12: 673708. https://doi.org/10.3389/fimmu.2021.673708
Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly YM, Glickman JN, Garrett WS (2013) The Microbial Metabolites, Short-Chain Fatty Acids, Regulate Colonic T Reg Cell Homeostasis. Science 341: 569–573. https://doi.org/10.1126/science.1241165
Masetti G, Ludgate M. (2020) Microbiome and Graves’ Orbitopathy. Eur Thyroid J 9: 78–85. https://doi.org/10.1159/000512255
Paray BA, Albeshr MF, Jan AT, Rather IA (2020) Leaky Gut and Autoimmunity: An Intricate Balance in Individuals Health and the Diseased State. Int J Mol Sci 21: 9770. https://doi.org/10.3390/ijms21249770
Manfredo Vieira S, Hiltensperger M, Kumar V, Zegarra-Ruiz D, Dehner C, Khan N, Costa FRC, Tiniakou E, Greiling T, Ruff W (2018) Erratum: The Report “Translocation of a Gut Pathobiont Drives Autoimmunity in Mice and Humans”. Science 359(6380): 1156–1161. https://doi.org/10.1126/science.aar7201
Kibbie JJ, Dillon SM, Thompson TA, Purba CM, McCarter MD, Wilson CC (2021) Butyrate Directly Decreases Human Gut Lamina Propria CD4 T Cell Function through Histone Deacetylase (HDAC) Inhibition and GPR43 Signaling. Immunobiology 226: 152126. https://doi.org/10.1016/j.imbio.2021.152126
Kaisar MM, Pelgrom LR, van der Ham AJ, Yazdanbakhsh M, Everts B (2017) Butyrate Conditions Human Dendritic Cells to Prime Type 1 Regulatory T Cells via Both Histone Deacetylase Inhibition and G Protein-Coupled Receptor 109A Signaling. Front Immunol 8: 1429. https://doi.org/10.3389/fimmu.2017.01429
Arpaia N, Campbell C, Fan X, Dikiy S, Van Der Veeken J, Deroos P, Liu H, Cross JR, Pfeffer K, Coffer PJ (2013) Metabolites Produced by Commensal Bacteria Promote Peripheral Regulatory T-Cell Generation. Nature 504: 451–455. https://doi.org/10.1038/nature12726
Wu Y, Borde M, Heissmeyer V, Feuerer M, Lapan AD, Stroud JC, Bates DL, Guo L, Han A, Ziegler SF (2006) FOXP3 Controls Regulatory T Cell Function through Cooperation with NFAT. Cell 126: 375–387. https://doi.org/10.1016/j.cell.2006.05.042
Raugh A, Allard D, Bettini M (2022) Nature vs. Nurture: FOXP3, Genetics, and Tissue Environment Shape Treg Function. Front Immunol 13: 911151. https://doi.org/10.3389/fimmu.2022.911151
Fathi M, Vakili K, Yaghoobpoor S, Tavasol, Jazi K, Mohamadkhani A, Klegeris A, McElhinney A, Mafi Z, Hajiesmaeili M (2022) Dynamic Changes in Kynurenine Pathway Metabolites in Multiple Sclerosis: A Systematic Review. Front Immunol 13: 1013784. https://doi.org/10.3389/fimmu.2022.1013784
Li H, Ning S, Ghandi M, Kryukov GV, Gopal S, Deik A, Souza A, Pierce K, Keskula P, Hernandez D (2019) The Landscape of Cancer Cell Line Metabolism. Nat Med 25: 850–860. https://doi.org/10.1038/s41591-019-0404-8
Navarro MN, Gómez de las Heras MM, Mittelbrunn M. (2022) Nicotinamide Adenine Dinucleotide Metabolism in the Immune Response, Autoimmunity and Inflammageing. Br J Pharmacol 179: 1839–1856. https://doi.org/10.1111/bph.15477
Grahnert A, Grahnert A, Klein C, Schilling E, Wehrhahn J, Hauschildt S (2011) NAD+: A Modulator of Immune Functions. Innate Immun 17: 212–233. https://doi.org/10.1177/1753425910361989
Burns M, Ostendorf L, Biesen R, Grützkau A, Hiepe F, Mei HE, Alexander T (2021) Dysregulated Cd38 Expression on Peripheral Blood Immune Cell Subsets in Sle. Int J Mol Sci 22: 2424. https://doi.org/10.3390/ijms22052424
Kar A, Mehrotra S, Chatterjee S (2020) CD38: T Cell Immuno-Metabolic Modulator. Cells 9: 1716. https://doi.org/10.3390/cells9071716
Kwon HS, Lim HW, Wu J, Schnölzer M, Verdin E, Ott M (2012) Three Novel Acetylation Sites in the Foxp3 Transcription Factor Regulate the Suppressive Activity of Regulatory T Cells. J Immunol 188: 2712–2721. https://doi.org/10.4049/jimmunol.1100903
Elkhal A, Biefer HRC, Heinbokel T, Uehara H, Quante M, Seyda M, Schuitenmaker JM, Krenzien F, Camacho V, De La Fuente MA (2016) NAD+ Regulates Treg Cell Fate and Promotes Allograft Survival via a Systemic IL-10 Production That Is CD4+ CD25+ Foxp3+ T Cells Independent. Sci Rep 6: 22325. https://doi.org/10.1038/srep22325
Pallotta MT, Orabona C, Volpi C, Vacca C, Belladonna ML, Bianchi R, Servillo G, Brunacci C, Calvitti M, Bicciato S (2011) Indoleamine 2,3-Dioxygenase Is a Signaling Protein in Long-Term Tolerance by Dendritic Cells. Nat Immunol 12: 870–878. https://doi.org/10.1038/ni.2077
Buonaguro L, Mayer-Mokler A, Flohr C, Reinhardt C, Singh-Jasuja H, Accolla R, Tosi G, Ma YT, Adams D, Valmori D (2017) Corrigendum to “New Vaccination Strategies in Liver Cancer”. Cytokine Growth Factor Rev 36: 125–129. https://doi.org/10.1016/j.Cytogfr.2017.06.010
Дополнительные материалы отсутствуют.
Инструменты
Российский физиологический журнал им. И.М. Сеченова