

Сенсорные системы, 2020, T. 34, № 3, стр. 188-200
Три тактики генной терапии двух врожденных заболеваний сетчатки. Обзор
Е. М. Максимова 1, *, П. В. Максимов 1
1 Федеральное государственное бюджетное учреждение науки
Институт проблем передачи информации им. А.А. Харкевича РАН
127051 Москва, Большой Каретный пер., 19, Россия
* E-mail: maximova@iitp.ru
Поступила в редакцию 02.03.2020
После доработки 06.04.2020
Принята к публикации 27.04.2020
Аннотация
Приведены литературные данные о генетических причинах двух врожденных заболеваний сетчатки – LCA2 и LCA10 (детская прогрессирующая слепота) и о современных способах их лечения. Причиной LCA2 является разрыв зрительного цикла из-за дефекта гена RPE65, экспрессирующегося в клетках пигментного эпителия сетчатки (RPE). Для лечения разработана и применяется усиливающая генная терапия (augmentation therapy): векторное субретинальное введение нормального гена RPE65. LCA10 – цилиопатия, вызванная мутацией гена CEP290, экспрессирующегося в основании связывающей реснички (CC) фоторецептора. Для лечения применяется “антисмысловая” терапия, устраняющая интронную (дефектную) мутацию в молекуле пре-мРНК в процессе синтеза белка CEP290. Рассказано о проекте “BRILLIANCE” – первом испытании прямого редактирования генома методом CRISPR/Cas9 прямо в теле пациента LCA10, анонсированном в номере Nature за 2020 г.
ВВЕДЕНИЕ
Врожденные патологии сетчатки связаны с мутацией генов, обеспечивающих разные этапы зрительного процесса. Большинство заболеваний рано или поздно оканчиваются слепотой в результате дегенерации зрительных рецепторов. До недавнего времени эти патологии были неизлечимы. С развитием генетических методов диагностики и генной инженерии появились принципиальная возможность и надежда на генную терапию, по крайней мере, некоторых из этих заболеваний (Максимова, 2010; Bainbridge et al., 2008a; Bennett, 2017; Hastie, Samulski, 2015; Hollander et al., 2010; Cideciyan, 2010; Sanagala et al., 2017).
Врожденный амавроз Лебера (Leber congenital amaurosis – LCA) – наиболее частая форма врожденной детской слепоты. Эта патология была впервые описана в 1869 г. доктором Т. Лебером и впоследствии получила его имя (Leber, 1869). Симптомы LCA – прогрессирующее ухудшение зрения в сумерках, нистагм, сужение поля зрения, дегенеративные изменения в сетчатке в области наибольшей плотности палочек – проявляются уже в первые годы жизни и к 20–30 годам заканчиваются слепотой (Jacobson et al., 2008; Lorenz et al., 2000). Частота встречаемости LCA в Европе и США – от двух до трех случаев на 100 000 новорожденных. По генетической диагностике LCA оказался не одним заболеванием, а большой группой гетерогенных аутосомальных рецессивных расстройств со сходными признаками зрительной патологии. В настоящее время более 400 мутаций, по крайней мере в 23 генах, связывают с разными формами LCA. Им стали присваивать номера. Ниже расскажем о генетических причинах и способах генной терапии LCA2 и LCA10. Выявленные генетические нарушения составляют 70% от общего количества заболеваний (Hollander et al., 2010; Takkar et al., 2018). По недавним исследованиям в странах восточной Азии (Корее, Пакистане, Саудовской Аравии, Южной Индии и популяции Хань Китая) спектры мутаций у людей, страдающих LCA, несколько отличны от выявленных на Западе, что, по-видимому, отражает этнические особенности (Liu, Bu, 2017; Seong et al., 2008; Li et al., 2009; McKibbin et al., 2010; Sundaresan et al., 2009; Li et al., 2011).
LCA2. УСИЛИВАЮЩАЯ ГЕННАЯ ТЕРАПИЯ
Одна из форм амавроза Лебера – LCA2 – обусловлена нарушением зрительного цикла и вызванной этим прогрессивной билатеральной дегенерацией сначала палочек, а затем и колбочек (Lorenz et al., 2000).
Зрительный процесс начинается с поглощения кванта света зрительным пигментом родопсином, встроенным в липидную мембрану дисков наружных сегментов фоторецепторов. Он составляет 90% строительных белков этой мембраны (Nathans, 1990). Молекула родопсина состоит из белка – опсина и связанного с ним хромофора (ретиналя) – остатка альдегида витамина А. При поглощении кванта света происходит цис-транс изомеризация этой молекулы и отщепление хромофора. Продукты распада родопсина переносятся из рецептора в клетки пигментного эпителия (RPE – retinal pigment epithelium), подстилающего сетчатку и тесно связанного с ней функционально. Там при участии специального фермента RPE65 происходит восстановление цис-ретиналя и его воссоединение с опсином – восстановление молекулы родопсина – и последующая транспортировка восстановленного пигмента в рецептор. Это так называемый зрительный цикл (Каламкаров, Островский, 2002; Redmond et al., 1998; Wolf, 2005).
Мутация гена RPE65 (картированного у человека в первой, а у мыши в третьей хромосоме) приводит к тому, что родопсин не восстанавливается. Тем самым нарушается зрительный цикл (Gu et al., 1997; Redmond et al., 2005). В результате происходит постепенное разрушение дисков наружных сегментов, сначала палочек, а потом и колбочек, оканчивающееся гибелью фоторецепторов. Наступает слепота.
Усиливающая генная терапия (augmentation gene therapy). Первый успех генной терапии LCA2 был достигнут в 2001 г. Взрослой собаке Swedish Briard dog, слепой по причине часто встречающегося у этой породы заболевания сетчатки, аналогичного LCA2 человека, было возвращено зрение векторным введением под сетчатку одного глаза нормального гена в надежде, что результат его деятельности превысит результат деятельности дефектного (Narfström et al., 1989; Aguirre et al., 1998; Veske et al.,1999; Acland et al., 2001; Petersen-Jones, Komáromy, 2015). С тех пор положительные стабильные результаты были получены еще на 50 собаках (в том числе и на более крупных) и на модели мыши с нокаутом Rpe65 (Redmond, Hamel, 2000; Bemelmans et al., 2006; Pang et al., 2006; Bennicelli et al., 2008; Cideciyan, 2010; Takkar et al., 2018).
В феврале 2007 г. начались клинические испытания генной терапии на людях в Великобритании, а в конце того же года в США. Уже в 2008 г. были опубликованы первые результаты этих испытаний (Bainbridge et al., 2008b; Hauswirth et al., 2008; Maguire et al., 2008). В них приняли участие двенадцать LCA2 пациентов разных возрастов. Клетки пигментного эпителия были “инфицированы” нормальным геном RPE65 при помощи специально разработанного вектора – вируса rAAV2 (recombinant adeno-associated virus 2) (Warrington, Herzog, 2006; Bennicelli et al., 2008). Каждому пациенту под сетчатку одного глаза, который видел хуже, был введен нормальный ген. Были испробованы разные дозы: малая – 1.5 × 1010 векторных частиц, средняя – 4.8 × 1010 векторных частиц, высокая – 1.5 × 1011 векторных частиц на 1 мл. Через 2–4 нед улучшение зрения в инъецированном глазу отмечено у половины пациентов: появилась зрачковая реакция (восстановилась светочувствительность), прекратился нистагм, появилась способность ориентироваться в пространстве при низком (скотопическом) уровне освещения. Это свидетельствовало о том, что нормальный ген RPE65 встроился в клетки пигментного эпителия и заработал. Результат был зависим от дозы и возраста пациента. Все пациенты отмечали субъективные улучшения, а у детей (6–12 лет) восстановилось практически нормальное зрение (Maguire et al., 2008). Впоследствии клинические испытания начались в Иерусалиме и во Франции. Положительный эффект был стабилен в течение 1–3 лет наблюдения (Simonelli et al., 2010; Jacobson et al., 2012; Bainbridge et al, 2015; Le Meur et al., 2018). Никаких значительных иммунных ответов не было зарегистрировано ни в одном исследовании. Не обнаружено и вредных последствий хирургического вмешательства. Успех обусловлен точным знанием генетической причины заболевания и разработкой технологии соответствующей генной терапии, впоследствии названной усиливающей (augmentation gene therapy) (Maguire et al., 2008; 2009; Warrington, Herzog, 2006; Bainbridge et al., 2008b; Hauswirth et al., 2008; Cideciyan et al., 2009; Jacobson et al., 2012; Pennesi et al., 2018).
Однако у прооперированных людей болезнь продолжала развиваться: продолжал действовать и дефектный ген (Cideciyan et al., 2013). Через некоторое время многие пациенты обратились с просьбой провести повторную операцию на другом глазу (ранее лучшем), который со временем стал видеть хуже, чем инъецированный. Поскольку векторное введение нормального гена – это введение чужеродного белка, вначале было опасение, что его повторное введение может вызвать аллергическую реакцию (анафилактический шок). Контрольные опыты на больших обезьянах с повторным введением вируса во второй глаз показали, что это безопасно (Maguire et al., 2008). Глаз, сетчатка – предпочтительный орган для генной терапии, поскольку, как и мозг, имеет гематоретинальный барьер, препятствующий выработке антител на введение чужеродного белка.
В 2016 г. была проведена первая фаза клинических испытаний на людях по повторному векторному введению нормального гена во второй глаз (Bennett et al., 2016). Операция прошла без вредных последствий и привела к длительному улучшению зрения, как по субъективным ощущениям, так и по объективной оценке состояния сетчатки, ЭРГ и кортикальных реакций (ЭЭГ). В 2020 г. был опубликован обзор работ по усиливающей генной терапии LCA2 (включающий сравнение зрительных функций инъецированного и контрольного глаза, всего 164 глаза 82 пациентов). На основании статистического анализа терапевтической значимости полученных результатов авторы констатируют улучшение световой чувствительности и остроты зрения на срок до двух лет, отсутствие вредных последствий оперативного вмешательства в течение пяти лет. В то же время отмечено, что на этом фоне развитие заболевания продолжается (Weleber et al., 2016; Wang et al., 2020).
После публикаций об успешных клинических испытаниях генной усиливающей терапии (субретинальном векторном введении нормального гена RPE65 ) “Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США” (Food and Drug Administration (FDA)) в декабре 2017 г. разрешила и утвердила коммерческое использование этого фармакологического агента (voretigene neparvovec AAV2-hRPE65v2). Его коммерческое название Luxturna (Hussain et al., 2019). Девять лечебных центров в США проводят лечение LCA2 методом генной усиливающей терапии – билатерального субретинального введения Luxturna.
КРИТЕРИИ ПРИ ВЫБОРЕ ОЧЕРЕДНОГО ЗАБОЛЕВАНИЯ ДЛЯ ПРОВЕДЕНИЯ УСИЛИВАЮЩЕЙ ГЕННОЙ ТЕРАПИИ
Операциям на людях предшествует длительная проверка метода на культуре клеток (Burnight et al., 2014), на лабораторных животных-моделях (Acland et al., 2001), иногда выведение трансгенных животных, за неимением соответствующих диких форм (Redmond, Hamel, 2002); разработка вируса-вектора, эффективно “поражающего” нужные клетки-мишени и обладающего соответствующей “грузоподъемностью” (Cargo capacity), не токсичного, не приводящего к побочным мутациям (Bennicelli et al., 2008). Все это требует серьезных капитальных вложений, поиска инвестиций. Поэтому при выборе очередного заболевания для проведения генной терапии руководствуются следующими критериями.
– Генетическая патология должна приводить к действительно серьезным нарушениям зрения.
– Кассета, содержащая нормальный ген и адрес его доставки, должна удовлетворять грузоподъемности доступных векторов.
– Выбранные векторы должны эффективно и стабильно “поражать” первичную популяцию клеток.
– Модельное животное с соответствующим фенотипом должно быть доступно для демонстрации доказательства правоты концепции.
– Для проведения клинических испытаний количество пациентов должно быть не менее 25 человек.
– У пациентов должно быть сохранно необходимое количество клеток – первичных мишеней, которые можно обновить доставкой здоровых генов.
LCA10 – ДРУГАЯ ФОРМА АМАВРОЗА ЛЕБЕРА. ВЫБОР МЕТОДА ГЕННОЙ ТЕРАПИИ
LCA10 выявляется у 20–30% пациентов с LCA. Если причина LCA2 – это нарушение одной из функций клеток пигментного эпителия сетчатки в результате мутации гена RPE65, то причина LCA10 кроется в нарушении метаболизма в самом фоторецепторе из-за мутации гена CEP290. Это наиболее часто мутирующий ген, с его мутациями связаны многие цилиопатии разных органов, включая сетчатку. Тридцать шесть известных мутаций CEP290 повинны, по крайней мере, в трети всех форм LCA. Первичный дефект выявляется в цилии (connecting cilium (CC)), расположенной между внутренним и наружным сегментами фоторецептора (Drivas, Bennett, 2014; Coppieters et al., 2010). При LCA10 выявленная мутация CEP290 (p.Cys998X, известная также как c.2991+1655 A>G) приводит к нарушению интрафлагеллярного транспорта (IFT) – транспорта белковых и липидных молекул (в том числе родопсина) между внутренним и наружным сегментами палочки. (Dulla et al., 2018). В Европе и США по меньшей мере 2000 пациентов имеют эту специфическую мутацию.
Строение фоторецептора позвоночных. Наружные сегменты фоторецепторов позвоночных – высокоспециализированные производные первичной реснички (primary cilium), сформировавшиеся в эволюции для эффективного захвата квантов света. В наружном сегменте палочки стопкой упакованы двухслойные диски, в мембрану которых встроен родопсин (рис. 1). Во внутреннем сегменте фоторецептора сосредоточены многочисленные митохондрии, аппарат Гольджи, ретикулюм и прочие внутриклеточные органеллы биохимической кухни, производящей белки и липиды, необходимые наружному сегменту клетки. Наружный и внутренний сегменты связаны ресничкой (connecting cilium (СС)). По связывающей ресничке осуществляется транспорт в обе стороны белковых, липидных и прочих молекул, так называемый интрафлагеллярный транспорт (IFT) (Prevo et al., 2017). Диски наружного сегмента постоянно обновляются: с апикального конца палочки, каждый день отслаивается порядка 10% старых дисков, которые захватываются пальцеобразными выростами клеток пигментного эпителия, а новые диски образуются в основании наружного сегмента. Процесс пиноцитоза старых дисков называется шеддинг (shedding). Он подчинен циркадному ритму: утром происходит захват отработанных дисков палочек, а вечером – колбочек (Young, 1967; Pazour et al., 2002; Wheway et al., 2014). Обновление наружных сегментов требует постоянного притока липидов и протеинов, синтезируемых во внутреннем сегменте фоторецепторов, поэтому сохранность цилии (CC), по которой осуществляется транспорт в обе стороны белковых, липидных молекул (IFT), очень важен для нормальной функции рецептора (Prevo et al., 2017).
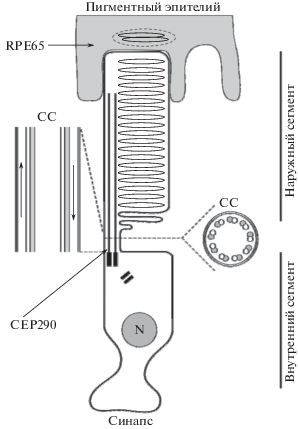
Рис. 1.
Схематическое изображение палочки, ее связывающей реснички (CC) в поперечном и продольном разрезе (увеличено) и клетка пигментного эпителия (RPE) с включением старых дисков (стриптосома). На продольном разрезе CC стрелками показаны направления интрафлагеллярного транспорта (intraflagellar transport (IFT)). Положение внутри палочки белков RPE65 и CEP290 указано стрелками. N – nucleus – ядро палочки; CC – connecting cilium – связывающая ресничка.
Длина наружного сегмента в норме остается постоянной благодаря равновесию процессов отслоения и фагоцитоза старых дисков и образования новых. Новые диски производит цилия из эктосом. Образовывать множество эктосом – пузырьков, заключенных в мембрану, – свойство всех цилий разных органов (Long, Huang, 2020). Эктосомы, возможно, используются для молекулярного обмена внутри клеток. В фоторецепторной цилии эктосомы не отшнуровываются от цилии, а постепенно удлиняясь и уплощаясь, превращаются в новые диски наружного сегмента фоторецептора. Механизм этого явления открыт недавно. Показано, что отделению эктосом от цилии фоторецептора препятствует белок периферин (Salinas et al., 2017). Раньше считалось, что новые диски образуются из складок наружной мембраны палочки (Ding et al., 2015).
Цилии (primary cilia) – эволюционно сохранные клеточные структуры, характерные для клеток многих органов (Винников, 1971; Заварзин, 1976; Satir, 2010; Rosenbaum, Witman, 2002; Long, Huang, 2020). Цилия фоторецептора состоит из аксонемы, образуемой девятью дуплетами микротрубочек, окруженной наружной мембраной (9 × 2 + 0) (рис. 1). Вдоль реснички различают три части. Ее основание – базальный комплекс и корешок – находятся в апикальном конце внутреннего сегмента фоторецептора, средняя часть – связующая цилия (СС) – образует мостик между наружным и внутренним сегментами, а дистальная часть продолжается в наружный сегмент (Insinna, Besharse, 2008). IFT (интрафлагеллярный транспорт) осуществляется по микротрубочкам CC в обе стороны (рис. 1). По одной микротрубочке в каждом из девяти дуплетов происходит движение в антероградном направлении – от внутреннего сегмента к наружному – перенос молекул при помощи моторных белков кинезинов (kinesin 2 motor proteins). Так транспортируются и молекулы опсина, где они затем включаются во вновь собранные мембраны дисков (Insinna, Besharse, 2008, Insinna et al., 2009; Marszalek et al., 2000; Pazour et al., 2002). По другой соседней микротрубочке в ретроградном направлении – от наружного сегмента к внутреннему – осуществляется движение молекул при помощи белка – цитоплазматического динеина (cytoplasmic dynein 2) (Rosenbaum, Witman, 2002). Транспорт между двумя сегментами имеет критическое значение для формирования и функции фоторецептора (Pazour et al., 2002). Для организации нормального процесса транспортировки молекул необходим белок CEP290, локализованный в основании связывающей цилии фоторецептора (Duijkers et al., 2018; Betleja, Cole, 2010). При LCA10 выявленная мутация CEP290 (мутация p.Cys998X, также известная как мутация c.2991+1655 A>G) приводит к нарушению синтеза белка CEP290 и прекращению деятельности моторных кинезинов в антероградном направлении IFT – к наружному сегменту. Нарушаются транспорт опсина и его встраивание во вновь образованные диски. Происходит рассогласование процессов шеддинга и обновления наружных сегментов палочек, что приводит к их укорочению, деградации и гибели (рис. 2) (Wheway et al., 2014). Так, снижение уровня CEP290 может привести к дистрофии сетчатки (Dulla et al., 2018). По данным томографии сетчатки у пациентов с данной мутацией гена CEP290 нарушена нормальная архитектура периферии сетчатки (преимущественно палочковой) – утоньшен наружный ядерный слой (где в норме располагаются ядра палочек). Фовеальная и парафовеальная области (преимущественно колбочковые) при этом дольше остаются сохранными (как и в случае LCA2), что оказывается существенным при выборе места для субретинальных инъекций при генной терапии (Hussain et al., 2019; Peng et al., 2017; Sheck et al., 2018). Отрабатывалась методика усиливающей терапии LCA10 на культуре индуцированных плюрипотентных стволовых клеток (iPSC-Induced pluripotent stem cell) человека. Продемонстрирована способность лентивирусных векторов, несущих полноразмерный CEP290, восстанавливать дефект цилиогенеза, наблюдаемый в фибробластах, полученных от пациента с LCA10 (Burnight et al., 2014). Несмотря на обнадеживающие результаты, внимание исследователей переключилось на другую идею генной терапии.
Рис. 2.
Схема “антисмысловой” терапии (“antisense” therapy) LCA10 при помощи синтетического препарата – короткой цепочки нуклеотидов – (QR-110), или сепофарсена (По Dulla et al., 2018).
Сверху – схема “починки” интронной мутации в молекуле пре-мРНК при превращении ее в зрелую мРНК в процессе синтеза силиарного белка CEP290. Внизу слева – дегенерирующая палочка в результате нарушенного синтеза белка CER290. Внизу справа – палочка, восстановленная “антисмысловой” терапией.
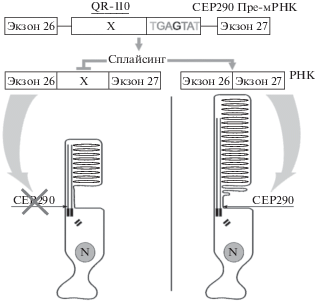
“Антисмысловая” терапия (“antisense” therapy). Для лечения LCA10 разработана новая стратегия генной терапии сообразно выявленной мутации СЕР290 (c.2991+1655A>G) – антисмысловая терапия (antisense therapy). Этот метод лечения основан на выключении/остановке синтеза белка, участвующего в развитии заболевания. В процессе синтеза белка матричная РНК (mRNA) выступает в качестве посредника между генетической информацией в ДНК и аминокислотной последовательностью белка (Abramowicz, Gos, 2019). При мутации (c.2991 + 1655A> G) в гене CEP290 в процессе транскрипции в молекуле незрелой матричной РНК (мРНК) при превращении ее в зрелую мРНК создаются сильные сайты для сплайсинга11 с дефектным интроном, узнаваемые сплайсинговым механизмом. Дефектный интрон препятствует нормальному сплайсингу и построению нормального белка CEP290. “Починка” интронной мутации – интересная задача для генной терапии (Duijkers et al., 2018) (рис. 2). Наиболее перспективным подходом оказалось использование “антисмысловых” олигонуклеотидов (antisense oligonucleotides – AONs) (Siva et al., 2014; Havens, Hastings, 2016). Синтезирован химический аналог короткой цепочки РНК, которая нацелена на связывание сайта сплайсинга на пре-мРНК и модификацию содержания экзона в мРНК22. Именно так и работает препарат сепофарсен (QR-110) – короткая цепочка олигонуклеотидов. Он направлен на восстановление генетического дефекта в пре-матричной РНК, при ее превращении в зрелую мРНК, что позволяет ему функционировать как нормальная мРНК “дикого типа” (рис. 2). В ходе испытаний QR-110 был протестирован на культуре фибробластов, на трехмерных сетчаточных органоидах, произведенных из индуцированных плюрипотентных стволовых клеток (iPSC), на животных (мышах, кроликах и обезьянах) и, наконец, на людях (Dulla et al., 2018). В 2017 г. интернациональный центр клинических испытаний начал оценивать последствия интравитреальных инъекций QR-110. Результаты значительного улучшения зрения у одного пациента обусловили проведение промежуточных анализов у всех пациентов, принимавших участие в исследовании. Десять пациентов показали значительное улучшение остроты зрения и способности обнаруживать вспышки света (Dulla et al., 2018). Через три месяца после субретинального введения препарата QR-110 (сепофарсена) около 60% пациентов продемонстрировали клинически значимый эффект улучшения зрения и способности ориентироваться в условиях слабой освещенности. Препарат сепофарсен (QR-110) производит голландская биотехнологическая компания ProQR, основанная в 2012 г. и специализирующаяся на разработке орфанных препаратов (orphan drug) для лечения редких тяжелых врожденных заболеваний. FDA и European Medicines Agency включили сепофарсен в категорию orphan drugs (Aronson, 2006). Согласно закону, компании, занимающиеся разработкой препаратов для лечения заболевания, которым страдают менее 200 000 человек в Соединенных Штатах, могут продавать их без конкуренции в течение семи лет и могут получать стимулирующие налоговые льготы (Pollack, 1990). Законы об орфанных препаратах существуют также в Австралии, Сингапуре и Японии.
МЕТОД РЕДАКТИРОВАНИЯ ГЕНОМА
“Антисмысловая” терапия LCA10 не является формально генной терапией, поскольку редактированию подвергается молекула пре-мРНК, а не ДНК. Существуют технологии редактирования собственно ДНК (Sanagala et al., 2017). Ключевой среди технологий редактирования генов, благодаря простоте, высокой эффективности и универсальности, стала молекулярная технология, известная как CRISPR/Cas9, разработанная в 2012 г. американскими и французскими учеными Дженнифер Дудна и Эммануэль Шарпантье и усовершенствованная Фэн Чжаном и соавт. (Doudna, Charpentier, 2014).
Cas9 – это белок, ассоциированный с одноцепочечной РНК c регулярно расположенными фрагментами – группами коротких палиндромных повторов (CRISPR33). В природе система CRISPR/Cas9 встречается у некоторых бактерий как часть их иммунной системы. Фрагменты ДНК (части растерзанного вируса) появляются от вирусных атак и сохраняются в бактериальном геноме как часть массива CRISPR. Белок Cas9 использует эти фрагменты для распознавания будущих “врагов” и их уничтожения, разрезая их генетический материал. Поскольку комплекс CRISPR/CAS9 становится частью бактериального генома, он передается потомству.
Сведения о CRISPR в геноме бактерий появились в 1980-х годах, к середине 2000-х стала понятна их функция, и началось изучение молекулярных механизмов их действия (Jinek et al., 2012). Возникла мысль о применении функциональных возможностей CRISPR/CAS9 в эукариотических клетках. В настоящее время технология на основе упрощенной природной системы CRISPR/Cas9 с изменяемой “направляющей РНК” (guide RNA) для вырезания определенных мест в геноме эффективно работает практически на всех видах клеток и применяется очень широко (Du et al., 2016). Генное редактирование при помощи CRISPR/Cas9 использует одноцепочечную РНК для поиска интересующего фрагмента ДНК (мутации). CRISPR идентифицирует ген-мишень через спейсерные последовательности, а Cas9 способствуют расщеплению генов-мишеней и позволяют производить “редактирование гена” ДНК при помощи новой цепи ДНК потенциально функционального белка.
В журнале Nature в марте 2020 г. появилось сообщение о первой попытке устранения мутации, ответственной за развитие LCA10, непосредственно в ДНК, – редактирования генома – прямо в теле человека при помощи технологии CRISPR/ CAS9 (Ledford, 2020). В исследовании, получившем название “BRILLIANCE”, будут оцениваться безопасность, переносимость и эффективность экспериментального препарата AGN-151587 (EDIT-101), произведенного биотехнологической компанией ProQR, специально для лечения врожденного амавроза Лебера 10 (LCA10). Исследование проводится компаниями Allergan и Editas Medicine. Взрослые и дети в возрасте от 3 до 17 лет (около 18 человек, разделенные на три группы) получат разовую дозу AGN-151587 в один глаз субретинально (для доставки механизма редактирования генов непосредственно к фоторецепторным клеткам). В предыдущих клинических испытаниях технология CRISPR/Cas9 использовалась для редактирования геномов клеток, удаленных из организма. Первый пациент в проекте BRILLIANCE открывает новую эру – использование лекарств CRISPR для длительного лечения тяжелых заболеваний, не только таких редких, как LCA10, но и пигментных ретинитов, возрастной макулярной дистрофии и прочих, от которых страдают миллионы людей.
В начале эры электронной микроскопии (60-е годы 20-го века) был колоссальный бескорыстный интерес (любопытство) к исследованию того, что впервые открылось глазу на фотографиях электронных срезов. Бросались в глаза удивительные структуры, одинаковые в разных объектах: клетках разных тканей, разных органов разных животных. Так, впервые увидели поперечнополосатые митохондрии, аппарат Гольджи, хлоропласты и другие органеллы клеток. В таких разных клетках, как фоторецепторы позвоночных, клетки внутреннего уха, клетки почечного ресничного эпителия, были обнаружены геометрически правильные структуры, названные впоследствии ресничками. По тонкому строению они были похожи на жгутик хламидомонады и сперматозоида. Эта ресничка или цилия (cilium), круглая в поперечном сечении, имела внутри девять пар микротрубочек (9×2 +0), расположенных по кругу вблизи охватывающей ресничку мембраны, в некоторых органах в ресничке была еще и центральная пара (9×2 +1). Именно эта “картинка” и бросалась в глаза (рис. 1). К настоящему времени на базе этих наивных наблюдений выросла фундаментальная наука о биохимии, генетике, развитии, эволюции и функциях органеллы. В ХХI веке результаты ее оказались востребованы в медицине.
С чистого любопытства по поводу странного паттерна, который был обнаружен в ДНК некоторых бактерий, началось фундаментальное исследование, приведшее к одному из самых мощных прорывов в современной генетике, созданию CRISPR технологии, которая дала возможность редактировать ДНК практически любого организма с беспрецедентной точностью и легкостью. Неисповедимы пути развития науки.
ИСТОЧНИК ФИНАНСИРОВАНИЯ
Работа выполнена при финансовой поддержке в рамках темы госзадания ИППИ РАН “Сенсомоторные механизмы организации поведения животных и человека (нейрофизиология, поведение, механизмы эволюции поведения, математическое моделирование и применение изученных закономерностей в диагностике заболеваний)”.
Список литературы
Винников Я.А. Цитологические и молекулярные основы рецепции. Л.: Наука, 1971. 298 с.
Заварзин A.A. Основы частной цитологии и сравнительной гистологии многоклеточных животных. Л.: Наука, 1976. 411 с.
Каламкаров Г.Р., Островский М.А. Молекулярные механизмы зрительной рецепции. М.: Наука, 2002. 279 с.
Максимова Е.М. Последние достижения в области восстановления зрения при сетчаточной недостаточности у млекопитающих. Сенсорные системы. 2010. Т. 24. № 3. С. 188–197.
Abramowicz A., Gos M. Splicing mutations in human genetic disorders: examples, detection, and confirmation. J Appl Genetics. 2019. V. 60 (2). P. 231. https://doi.org/10.1007/s13353-019-00493-z
Acland G.M., Aguirre G.D., Ray J., Zhang Q., Aleman T.S., Cideciyan A.V., Pearce-Kelling S.E., Anand V., Zeng Y., Maguire A.M., Jacobson S.G., Hauswirth W.W., Bennett J. Gene therapy restores vision in a canine model of childhood blindness. Nat Genet. 2001. V. 28 (1). P. 92–95. https://doi.org/10.1038/ng0501-92
Aguirre G., Baldwin V., Pearce-Kelling S., Narfstrom K., Ray K., Acland G. Congenital stationary night blindness in the dog: common mutation in the RPE65 gene indicates founder effect. Mol Vis. 1998. V. 4 (23). P. 1–7.
Aronson J.K. Rare diseases and orphan drugs. Br J Clin Pharmacol. 2006. V. 61 (3). P. 243–245. https://doi.org/10.1111/j.1365-2125.2006.02617.x
Bainbridge J., Ali R. Gene therapy for inherited childhood blindness shows promise. Expert Rev. Ophthalmol. 2008a. 3 (4). P. 357–359. https://doi.org/10.1586/17469899.3.4.357
Bainbridge J.W., Mehat M.S., Sundaram V. Long-term effect of gene therapy on Leber’s congenital amaurosis. N. Engl. J. Med. 2015. V. 372 (20). P. 1887–1897. https://doi.org/10.1056/NEJMoa1414221
Bainbridge J.W., Smith A.J., Barker S.S., Robbie S., Henderson R., Balaggan K., Viswanathan A., Holder G.E., Stockman A., Tyler N., Peterson-Jones S., Battacharya S.S., Thrasher A.J., Fitzke F.W., Carter B.J., Rubin G.S., Moore A.T., Ali R.R. Effect of gene therapy on visual function in Leber’s congenital amaurosis. N. Engl. J. Med. 2008b. V. 358 (21). P. 2231–2239. https://doi.org/10.1056/NEJMoa0802268
Bemelmans A.-P., Kostic C., Crippa S.V., Hauswirth W.W., Lem J., Munier F.L., Seeliger M.W, Wenzel A., Arsenijevic Y. Lentiviral Gene Transfer of Rpe65 Rescues Survival and Function of Cones in a Mouse Model of Leber Congenital Amaurosis. PLoS Med. 2006. V. 3 (10). P. 1892–1903. https://doi.org/10.1371/journal.pmed.0030347
Bennett J. Taking Stock of Retinal Gene Therapy: Looking Back and Moving Forward. Mol. Ther. 2017. V. 25 (5). P. 1076–1094. https://doi.org/10.1016/j.ymthe.2017.03.008
Bennett J., Wellman J., Marshall K.A., McCague S., Ashtari M., DiStefano-Pappas J., Elci O.U., Chung D.C., Sun J., Wright J.F., Cross D.R., Aravand P., Cyckowski L.L., Bennicelli J.L., Mingozzi F., Auricchio A., Pierce E.A., Ruggiero J., Leroy B.P., Simonelli F., High K.A., Maguire A.M. Safety and durability of effect of contralateral-eye administration of AAV2 gene therapy in patients with childhood-onset blindness caused by RPE65 mutatons: a follow-on phase 1 trial. Lancet. 2016. V. 388 (10045). P. 661–672. https://doi.org/(16)30371-3https://doi.org/10.1016/S0140-6736
Bennicelli J., Wright J.F., Komaromy A., Jacobs J.B., Hauck B., Zelenaia O., Mingozzi F., Hui D., Chung D., Rex T.S., Wei Z., Qu G., Zhou S., Zeiss C., Arruda V.R., Acland G.M., Dell’Osso L.F., High K.A., Maguire A.M., Bennett J. Reversal of blindness in animal models of leber congenital amaurosis using optimized AAV2-mediated gene transfer. Mol Ther. 2008. V. 16 (3). P. 458–465. https://doi.org/10.1038/sj.mt.6300389
Betleja E., Cole D.G. Ciliary Trafficking: CEP290 Guards a Gated Community. Curr. Biol. 2010. V. 20 (21). P. R928–R931. https://doi.org/10.1016/j.cub.2010.09.058
Burnight E.R., Wiley L.A., Drack A.V., Braun T.A., Anfinson K.R., Kaalberg E.E., Halder J.A., Affatigato L.M., Mullins R.F., Stone E.M., Tucker B.A. CEP290 gene transfer rescues Leber congenital amaurosis cellular phenotype. Gene. Ther. 2014. V. 21 (7). P. 662–672. https://doi.org/10.1038/gt.2014.39
Cideciyan A.V. Leber Congenital Amaurosis due to RPE65 Mutations and its Treatment with Gene Therapy. Prog. Retin. Eye. Res. 2010. V. 29 (5). P. 398–427. https://doi.org/10.1016/j.preteyeres.2010.04.002
Cideciyan A.V., Hauswirth W.W., Aleman T.S., et al. Vision 1 year after gene therapy for Leber’s congenital amaurosis. N. Engl. J. Med. 2009. V. 361 (7). P.725–727. https://doi.org/10.1056/NEJMc0903652
Cideciyan A.V., Jacobson S.G., Beltran W.A., Sumaroka A., Swider M., Iwabe S., Roman A.J., Olivares M.B., Schwartz S.B., Komáromy A.M., Hauswirth W.W., Aguirre G.D. Human retinal gene therapy for Leber congenital amaurosis shows advancing retinal degeneration despite enduring visual improvement. Proc. Natl. Acad. Sci. U S A. 2013. V. 110 (6). P. E517–E525. https://doi.org/10.1073/pnas.1218933110
Coppieters F., Lefever S., Leroy B.P., de Baere E.B. CEP290, a gene with many faces: mutation overview and presentation of CEP290base. Hum. Mutat. 2010. V. 31 (10). P. 1097–1108. https://doi.org/10.1002/humu.21337
Ding J.‑D., Salinas R.Y., Arshavsky V.Y. Discs of mammalian rod photoreceptors form through the membrane evagination mechanism. J. Cell. Biol. 2015. V. 211 (3). P. 495–502. https://doi.org/10.1083/jcb.201508093
Drivas T.G., Bennett J. CEP290 and the Primary Cilium. Retinal Degenerative Diseases. Advances in Experimental Medicine and Biology, vol 801. New York. Springer, 2014. P. 519–525. https://doi.org/10.1007/978-1-4614-3209-8_66
Du Q.-S., Cui J., C.-jie Zhang, He K.Visualization analysis of CRISPR/Cas9 gene editing technology studies. J. Zhejiang. Univ. Sci. B. 2016. V.17 (10). P. 798–806. https://doi.org/10.1631/jzus.B1601985
Duijkers L., van den Born I., Neidhardt J., Bax N.M., Pierrache L.H.M., Klevering B.J., Collin R.W.J., Garanto A. Antisense Oligonucleotide-Based Splicing Correction in Individuals with Leber Congenital Amaurosis due to Compound Heterozygosity for the c.2991+1655A>G Mutation in CEP290. Int. J. Mol. Sci. 2018. V. 19 (3). P. 753–760. https://doi.org/10.3390/ijms19030753
Dulla K., Aguila M., Lane A., Jovanovic K., Parfitt D.A., Schulkens I., Chan H.L., Schmidt I., Beumer W., Vorthoren L., Collin R.W.J., Garanto A., Duijkers L., Brugulat-Panes A., Semo M., Vugler A.A., Biasutto P., Adamson P., Cheetham M.E. Splice-Modulating Oligonucleotide QR-110 (sepofarsen) Restores CEP290mRNA and Functionin Human c.2991+1655A>G LCA10 Models. Mol. Ther. Nucleic. Acids. 2018. V. 12. P. 730–740. https://doi.org/10.1016/j.omtn.2018.07.010
Doudna J.A., Charpentier E. The new frontier of genome engineering with CRISPR-Cas9. Review. Science. 2014. V. 346, Issue 6213, 1258096.https://doi.org/10.1126/science.1258096
Gu S.M., Thompson D.A., Srikumari C.R., Lorenz B., Finckh U., Nicoletti A., Murthy K.R., Rathmann M., Kumaramanickavel G., Denton M.J., Gal A. Mutations in RPE65 cause autosomal recessive childhood-onset severe retinal dystrophy. Nat. Genet. 1997. V. 17 (2). P. 194–197. https://doi.org/10.1038/ng1097-194
Hastie E., Samulski R.J. Adeno-associated virus at 50: a golden anniversary of discovery, research, and gene therapy success–a personal perspective. Hum. Gene. Ther. 2015. V. 26 (5). P. 257–265. https://doi.org/10.1089/hum.2015.025
Hauswirth W.W., Aleman T.S., Kaushal S., Cideciyan A.V., Schwartz S.B., Wang L, Conlon T.J., Boye S.L., Flotte T.R., Byrne B.J., Jacobson S.G. Treatment of leber congenital amaurosis due to RPE65 mutations by ocular subretinal injection of adeno-associated virus gene vector: short-term results of a phase I trial. Hum. Gene. Ther. 2008. V. 19 (10). P. 979–990. https://doi.org/10.1089/hum.2008.107
Havens M.A., Hastings M.L. Splice-switching antisense oligonucleotides as therapeutic drugs. Nucleic. Acids. Res. 2016. V. 44 (14). P. 6549–6563. https://doi.org/10.1093/nar/gkw533
Hollander A.I., Black A., Bennett J., Cremers F.P. Lighting a candle in the dark: advances in genetics and gene therapy of recessive retinal dystrophies. J. Clin. Invest. 2010. V. 120 (9). P. 3042–3053. https://doi.org/10.1172/JCI42258
Hussain R.M., Tran K.D., Maguire A.M., Berrocal A.M. Subretinal Injection of Voretigene Neparvovec-rzyl in a Patient With RPE65-Associated Leber’s Congenital Amaurosis. Ophthalmic Surg Lasers Imaging Retina. 2019. V. 50 (10). P. 661–663. https://doi.org/10.3928/23258160-20191009-01
Insinna C., Besharse J.C. Intraflagellar Transport and the Sensory Outer Segment of Vertebrate Photoreceptors. Dev Dyn. 2008. V. 237 (8). P. 1982–1992. https://doi.org/10.1002/dvdy.21554
Insinna C., Humby M., Sedmak T., Wolfrum U., Besharse J.C. Different Roles For KIF17 and Kinesin II In Photoreceptor Development and Maintenance. Dev Dyn. 2009. V. 238 (9). P. 2211–2222. https://doi.org/10.1002/dvdy.21956
Jacobson S.G., Cideciyan A.V., Aleman T.S., Sumaroka A., Windsor E.A.M., Schwartz S.B., Heon E., Stone E.M. Photoreceptor Layer Topography in Children with Leber Congenital Amaurosis Caused by RPE65 Mutations. Invest. Ophthalmol. Vis. Sci. 2008. V. 49 (10). P. 4573–4577. https://doi.org/10.1167/iovs.08-2121
Jacobson S.G., Cideciyan A.V., Ratnakaram R., Heon E., Schwartz S.B., Roman A.J., Peden M.C., Aleman T.S., Boye S.L., Sumaroka A., Conlon T.J., Calcedo R., Pang J.-J., Erger K.E., Olivares M.B., Mullins C.L., Swider M., Kaushal S., Feuer W.J., Iannaccone A., Fishman G.A., Stone E.M., Byrne B.J., Hauswirth W.W. Gene Therapy for Leber Congenital Amaurosis Caused by RPE65 Mutations Safety and Efficacy in 15 Children and Adults Followed Up to 3 Years. Arch Ophthalmol. 2012. V. 130 (1). P. 9–24. https://doi.org/10.1001/archophthalmol.2011.298
Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012. V. 17. 337 (6096). P. 816–821. https://doi.org/10.1126/science.1225829
Le Meur G., Lebranchu P., Billaud F., Adjali O., Schmitt S., Bézieau S., Péréon Y., Valabregue R., Ivan C., Darmon C., Moullier P., Rolling F., Weber M. Safety and Long-Term Efficacy of AAV4 Gene Therapy in Patients with RPE65 Leber Congenital Amaurosis. Mol Ther. 2018. V. 26 (1). P. 256–268. https://doi.org/10.1016/j.ymthe.2017.09.014
Leber T. Uber retinitis pigmentosa und angeborene amaurose. von Graefe’s archives. Ophthalmology. 1869. V. 15. P. 1–25.
Ledford H. CRISPR treatment inserted directly into the body for first time. Nature. 2020. V. 579 (7798). P. 185–190. https://doi.org/10.1038/d41586-020-00655-8
Li L., Xiao X., Li S., Jia X., Wang P., Guo X., Jiao X., Zhang Q., Hejtmancik J. F. Detection of Variants in 15 Genes in 87 Unrelated Chinese Patients with Leber Congenital Amaurosis. PLoS ONE. 2011. V. 6 (5). https://doi.org/10.1371/journal.pone.0019458
Li Y., Wang H., Peng J., Gibbs R.A., Lewis R.A., et al. Mutation survey of known LCA genes and loci in the Saudi Arabian population. Invest. Ophthalmol. Vis. Sci. 2009. V. 50 (3). P. 1336–1343. https://doi.org/10.1167/iovs.08-2589
Liu J., Bu J. A Gene Scan Study of RPE65 in Chinese Patients with Leber Congenital Amaurosis. Chin. Med. J. (Engl). 2017. V. 130 (22). P. 2709–2712. https://doi.org/10.4103/0366-6999.218007
Long H., Huang K. Transport of Ciliary Membrane Proteins. Front. Cell. Dev. Biol. 2020. V. 7. P. 381–390. https://doi.org/10.3389/fcell.2019.00381
Lorenz B., Gyurus P., Preising M., Bremser D., Gu S., Andrassi M., Gerth C., Gal A. Early-onset severe rod-cone dystrophy in young children with RPE65 mutations. Invest. Ophthalmol. Vis. Sci. 2000. V. 41 (9). P. 2735–2742.
Maguire A.M., High K.A., Auricchio A., Wright J.F., Pierce E.A., Testa F. Age-dependent effects of RPE65 gene therapy for Leber’s congenital amaurosis:a phase 1 dose-escalation trial. Lancet. 2009. V. 374. P. 1597–1605. https://doi.org/10.1016/S0140-6736
Maguire A.M., Simonelli F., Pierce E.A., et al. Safety and efficacy of gene transfer for Leber’s congenital amaurosis. N. Engl. J. Med. 2008. V. 358 (21). P. 2240–2248. https://doi.org/10.1056/NEJMoa0802315
Marszalek J.R., Liu X., Roberts E.A., Marth J.D., Williams D.S., Goldstein L.S.B. Genetic Evidence for Selective Transport of Opsin and Arrestin by Kinesin-II in Mammalian Photoreceptors. Cell. 2000. V. 102 (2). P. 175–187.https://doi.org/10.1016/S0092-8674
McKibbin M., Ali M., Mohamed M.D., Booth A.P., Bishop F. Genotype-phenotype correlation for leber congenital amaurosis in Northern Pakistan. Arch. Ophthalmol. 2010. V. 128 (1). P. 107–113. https://doi.org/10.1001/archophthalmol.2010.309
Narfström K., Wrigstad A., Nilsson S.E. The Briard dog: a new animal model of congenital stationary night blindness. Br. J. Ophthalmol. 1989. V.73 (9). P.750–756. https://doi.org/10.1136/bjo.73.9.750
Nathans J. Determinants of visual pigment absorbance: identification of the retinylidene Schiff’s base counterion in bovine rhodopsin. Biochemistry. 1990. V. 29 (41). P. 9746–9752. https://doi.org/10.1021/bi00493a034
Pang J.J., Chang B., Kumar A., Nusinowitz S., Noorwez S.M., Li J., Rani A., Foster T.C., Chiodo V.A., Doyle T., Li H., Malhotra R., Teusner J.T., McDowell J.H., Min S.H., Li Q., Kaushal S., Hauswirth W.W. Gene Therapy Restores Vision-Dependent Behavior as Well as Retinal Structure and Function in a Mouse Model of RPE65 Leber Congenital Amaurosis. Mol. Ther. 2006. V. 13 (3). P. 565–572. https://doi.org/10.1016/j.ymthe.2005.09.001
Pazour G.J., Baker S.A., Deane J.A., Cole D.G., Dickert B.L., Rosenbaum J.L., Witman G.B., Besharse J.C. The intraflagellar transport protein, IFT88, is essential for vertebrate photoreceptor assembly and maintenance. J. Cell. Biol. 2002. V. 157 (1). P. 103–113. https://doi.org/10.1083/jcb.200107108
Peng Y., Tang L., Zhou Y. Subretinal Injection: A Review on the Novel Route of Therapeutic Delivery for Vitreoretinal Diseases. Ophthalmic. Res. 2017. V. 58 (4). P. 217–226. https://doi.org/10.1159/000479157
Pennesi M.E., Weleber R.G., Yang P., Whitebirch C., Thean B., Flotte T.R., Humphries M., Chegarnov E., Beasley K.N., Stout J.T., Chulay J.D. Results at 5 years after gene therapy for RPE65-deficient retinal dystrophy. Hum. Gene. Ther. 2018. V. 29 (12). P. 1428–1437. https://doi.org/10.1089/hum.2018.014
Petersen-Jones S.M., Komáromy A.M. Dog Models for Blinding Inherited Retinal Dystrophies. Hum. Gene. Ther. Clin Dev. 2015. V. 26 (1). P. 15–26. https://doi.org/10.1089/humc.2014.155
Pollack A. Orphan Drug Law Spurs Debate. The New York Times. 1990.
Prevo B., Scholey J.M., Peterman E.J.G. Intraflagellar Transport: Mechanisms of Motor Action, Cooperation and Cargo Delivery. FEBS J. 2017. V. 284 (18). P. 2905–2931. https://doi.org/10.1111/febs.14068
Redmond T.M., Poliakov E., Yu S., Tsai J.Y., Lu Z., Gentleman S. Mutation of key residues of RPE65 abolishes its enzymatic role as isomerohydrolase in the visual cycle. Proc. Natl. Acad. Sci. U S A. 2005. V. 102 (38). P. 13658–13663. https://doi.org/10.1073/pnas.0504167102
Redmond T.M., Yu S., Lee E., Bok D., Hamasaki D., Chen N., Goletz. P., Ma J.X., Crouch R.K., Pfeifer K. Rpe65 is necessary for production of 11-cis-vitamin A in the retinal visual cycle. Nat. Genet. 1998. V. 20 (4). P. 344–351. https://doi.org/10.1038/3813
Redmond T.M. and Hamel C.P. Genetic analysis of RPE65: from human disease to mouse model. Methods. Enzymol. 2000. V. 316. P. 705–724. https://doi.org/10.1016/s0076-6879
Rosenbaum J.L., Witman G.B. Intraflagellar transport. Nat. Rev. Mol. Cell. Biol. 2002. V. 3 (11). P. 813–825.
Salinas R.Y., Pearring J.N., Ding J.-D., Spencer W.J., Hao Y., Arshavsky V.Y. Photoreceptor discs form through peripherin-dependent suppression of ciliary ectosome release. JCB. 2017. V. 216 (5). P. 1489–1499. https://doi.org/10.1083/jcb.201608081
Sanagala R., Moola A.K., Bollipo Diana R.K. A review on advanced methods in plant gene targeting. J. Genet. Eng. Biotechnol. 2017. V. 15 (2). P. 317–321. https://doi.org/10.1016/j.jgeb.2017.07.004
Satir P., Pedersen L.B., Christensen S.T. The primary cilium at a glance. J. Cell. Sci. 2010. V. 123 (Pt 4). P. 499–503. https://doi.org/10.1242/jcs.050377
Seong M.W., Kim S.Y., Yu Y.S., Hwang J.M., Kim J.Y. Molecular characterization of Leber congenital amaurosis in Koreans. Mol. Vis. 2008. V. 14. P. 1429–1436.
Sheck L., Davies W.I.L., Moradi P., Robson A.G., Kumaran N., Liasis A.C., Webster A.R., Moore A.T., Michaelides M. Leber Congenital Amaurosis Associated with Mutations in CEP290, Clinical Phenotype, and Natural History in Preparation for Trials of Novel Therapies. Ophthalmology. 2018. V. 125 (6). P. 894–903. https://doi.org/10.1016/j.ophtha.2017.12.013
Simonelli F., Maguire A.M., Testa F., Pierce E.A., Mingozzi F., Bennicelli J.L., Rossi S., Marshall K., Banfi S., Surace E.M., Sun J., Redmond T.M., Zhu X., Shindler K.S., Ying G.S., Ziviello C., Acerra C., Wright J.F., McDonell J.W., High K.A., Bennett J., Auricchio A. Gene therapy for Leber’s congenital amaurosis is safe and effective through 1.5 years after vector administration. Mol. Ther. 2010. V. 18 (3). P. 643–650. https://doi.org/10.1038/mt.2009.277
Siva K., Covello G., Denti M.A. Exon-skipping antisense oligonucleotides to correct missplicing in neurogenetic diseases. Nucleic. Acid. Ther. 2014. V. 24 (1). P. 69–86. https://doi.org/10.1089/nat.2013.0461
Sundaresan P., Vijayalakshmi P., Thompson S., Ko A.C., Fingert J.H., et al. Mutations that are a common cause of Leber congenital amaurosis in northern America are rare in southern India. Mol. Vis. 2009. V.15. P. 1781–1787.
Takkar B., Bansal P., Venkatesh P. Leber’s Congenital Amaurosis and Gene Therapy. Indian. J. Pediatr. 2018. V. 85 (3). P. 237–242. https://doi.org/10.1007/s12098-017-2394-1
Veske A., Nilsson S., Narfström K., Gal A. Retinal dystrophy of Swedish Briard/Briard-Beagle dogs is due to a 4-bp deletion in RPE65. Genomics. 1999. V. 57 (1). P. 57–61. https://doi.org/10.1006/geno.1999.5754
Wang X., Yu C., Tzekov R.T., Zhu Y., Li1 W. The effect of human gene therapy for RPE65-associated Leber’s congenital amaurosis on visual function: a systematic review and meta-analysis. Orphanet. J. Rare. Dis. 2020. V. 15 (1). P. 49–55. https://doi.org/10.1186/s13023-020-1304-1
Warrington K.H. Jr, Herzog R.W. Treatment of human disease by adeno-associated viral gene transfer. Hum. Genet. 2006. V. 119 (6). P. 571–603. https://doi.org/10.1007/s00439-006-0165-6
Weleber R.G., Pennesi M.E., David J.W., Kaushal Sh., Erker L.R., Jensen L., McBride M.T., Flotte T.R., Humphries M., Calcedo R., Hauswirth, W.W., Chulay J.D., Stout J.T. Results at 2 Years after Gene Therapy for RPE65-Deficient Leber Congenital Amaurosis and Severe Early-Childhood-Onset Retinal Dystrophy. Ophthalmology. 2016. V. 123 (7). P. 1606–1620. https://doi.org/10.1016/j.ophtha.2016.03.003
Wheway G., Parry D.A., Johnson C.A. The role of primary cilia in the development and disease of the retina. Organogenesis. 2014. V. 10 (1). P. 69–85. https://doi.org/10.4161/org.26710
Wolf G. Function of the Protein RPE65 in the Visual Cycle. Nutr. Rev. 2005. V. 63 (3). P. 97–100. https://doi.org/10.1111/j.1753-4887.2005.tb00127.x
Young R.W. The renewal of photoreceptor cell outer segments. J. Cell. Biol. 1967. V. 33 (1). P. 61–72. https://doi.org/10.1083/jcb.33.1.61
Дополнительные материалы отсутствуют.
Инструменты
Сенсорные системы