

Теплофизика высоких температур, 2019, T. 57, № 2, стр. 304-307
Определение радиусов малых кластеров меди на основе моделирования процесса газофазной конденсации
А. Е. Коренченко 1, 2, *, А. Г. Воронцов 1, Б. Р. Гельчинский 2, А. А. Жукова 3
1 Южно-Уральский государственный университет (НИУ)
г. Челябинск, Россия
2 Институт металлургии УрО РАН
г. Екатеринбург, Россия
3 Первый Московский государственный медицинский университет
им. И.М. Сеченова
Москва, Россия
* E-mail: korenchenkoae@susu.ru
Поступила в редакцию 23.04.2018
После доработки 10.10.2018
Принята к публикации 09.05.2018
Аннотация
Проведено молекулярно-динамическое исследование зависимости радиусов малых металлических кластеров, образующихся в процессе газофазной конденсации меди, от количества атомов в них. Радиусы вычисляются по данным молекулярно-динамического моделирования о количестве столкновений между кластерами по формулам молекулярно-кинетической теории. Обнаружено, что зависимость радиусов от числа частиц имеет вид, близкий к классическому n1/3. Однако средние радиусы кластеров оказываются примерно на 40% больше, чем в случае, когда кластер считается каплей массивной жидкости.
ВВЕДЕНИЕ
Металлические дисперсные порошки широко используются на практике, поэтому изучению процессов конденсации металлических наночастиц посвящено большое количество исследований. Экспериментальные исследования функций распределения наночастиц по размерам показали, что процесс роста частиц отличается от классического присоединения атома к кластеру [1, 2] и на некоторых стадиях конденсации наблюдается коагуляция, происходящая вследствие столкновений малых частиц [3]. Для макроскопического моделирования коагуляции необходимо определить частоту столкновений кластеров между собой, для чего, помимо их скоростей и концентраций, следует знать величины сечений взаимодействия.
В литературе описано несколько способов определения сечения взаимодействия кластеров. Самый простой из них основан на представлении о кластере как о сгустке массивной жидкости – приближение “жидкой капли” [4]. В этом приближении радиус ${{R}_{n}}$ n-атомного кластера вычисляется по формуле
(1)
${{R}_{n}} = {{\left( {\frac{{3{{m}_{1}}}}{{4\pi \rho }}} \right)}^{{{1 \mathord{\left/ {\vphantom {1 3}} \right. \kern-0em} 3}}}}{{n}^{{{1 \mathord{\left/ {\vphantom {1 3}} \right. \kern-0em} 3}}}},$Оценка применимости соотношения (1) к кластерам малого размера $(n < {\text{5}}0)\quad$ не проводилась, хотя хорошо известно, что малые кластеры являются рыхлыми и неоднородными объектами [5], поэтому использование (1) должно давать большую погрешность.
В методе “твердых сфер” радиусы кластеров выражаются через радиусы вращения, рассчитанные по данным молекулярно-динамического (МД) моделирования как ${{R}_{{n,hs}}} = \sqrt {{5 \mathord{\left/ {\vphantom {5 2}} \right. \kern-0em} 2}} {{R}_{{n,{\text{gyr}}}}} + {{R}_{1}}.$ Здесь ${{R}_{{n{\text{,gyr}}}}}$ – усредненный по ансамблю радиус вращения; ${{R}_{1}}$ – эквивалентный радиус атома, равный 0.5d; d – межатомное расстояние, на котором притяжение между атомами сменяется отталкиванием [6].
В [5] предложено определять радиус кластера через точку в середине переходной области между плотным ядром и окружающим паром. Этот метод нельзя применять к малым кластерам, так как они не имеют плотного ядра.
До настоящего времени в литературе не был рассмотрен прямой метод определения сечения взаимодействия по данным МД-моделирования, рассчитанным по формуле молекулярно-кинетической теории (МКТ)
где ${{Z}_{{n,m}}}$ – частота столкновений между n- и m-атомными кластерами, ${{C}_{n}}{\text{ и }}{{C}_{m}}$ – концентрации, ${{\text{v}}_{{n,m}}}$ – относительная скорость теплового движения кластеров [7].Цель настоящей работы заключается в статистическом анализе результатов МД моделирования процесса конденсации пара меди для определения радиусов малых кластеров исходя из частоты столкновений.
СХЕМА МД-ЭКСПЕРИМЕНТА
Исследование формирования наночастиц проводилось методом молекулярной динамики, реализованным в пакете для многопроцессорных вычислений LAMMPS [8]. Система состояла из атомов буферного газа (Аr) и атомов металла (Сu). Взаимодействие атомов меди описывалось потенциалом погруженного атома [9]. Применение потенциала [9] к исследованию молекулы Cu2 дало значения 2.44 эВ для энергии диссоциации и 2.14 Å для длины связи молекулы. Экспериментальные данные составляют 2.01 ± 0.08 эВ и 2.219 Å соответственно [10]. Таким образом, погрешности равны ~15% для энергии и ~5% для межатомного расстояния. Проведенное исследование позволяет предположить, что потенциал [9] приемлемо описывает малые кластеры меди. Взаимодействия Аr–Аr и Cu–Ar описывались потенциалом типа Леннард-Джонса с параметрами е = = 0.0123 эВ, а = 3.76 Å. Радиус отсечки потенциалов составлял 10 Å. Временнóй шаг в системе был равен 3 фс.
Атомы помещались в кубическую ячейку, на краях ячейки задавались периодические граничные условия. Термостат применялся только для атомов инертного газа. Расчеты были проведены для температур аргона ${{T}_{{{\text{Ar}}}}} = 30{\text{0 и 1500 К }}$ и плотностей пара меди ${{\rho }_{{{\text{Cu}}}}}$ = 0.1–0.3 кг/м3. Соотношение между количеством атомов аргона и атомов металла составляло 3 : 1 при количестве атомов металла, равном 5000. Указанные системы частиц рассматривались в течение времен порядка 500 нс. Вычисления проводились для различных начальных конфигураций атомов с тем, чтобы набрать достаточный объем выборки для статистического анализа (не менее 100 столкновений на каждое значение ${{\sigma }_{{n,m}}}$). Специальные начальные конфигурации, содержащие n-атомные кластеры и атомы меди и аргона, были созданы, чтобы получить достаточные для анализа значения частот ${{Z}_{{n,1}}},$ ${{Z}_{{n,2}}}$ и ${{Z}_{{n,3}}},$ n > 10. С этой целью сто n-атомных кластеров (n > 10) случайным образом размещались в ячейке, скорости их центров масс были распределены по закону Максвелла, а координаты и скорости атомов в системе центра масс кластера были взяты из результатов предыдущих МД-расчетов. Дополнительные атомы меди и аргона размещались в ячейке случайным образом, их скорости были распределены по Максвеллу.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Выбор расстояния Δ для совместного анализа траекторий. Выделить столкновение в процессе движения атомов/кластеров, которые взаимодействуют друг с другом посредством протяженных потенциалов, является непростой задачей. Столкновением между кластерами будем называть событие, заключающееся в сближении атомов, принадлежащих этим кластерам, на расстояние, равное или меньшее некоторого заданного Δ [11]. Значение Δ определяется видом потенциала, однако строгого количественного критерия для расчета Δ нет. Между тем правильный выбор Δ очень важен, так как зафиксированное количество столкновений между кластерами и, следовательно, вычисленное сечение взаимодействия напрямую зависят от значения Δ. Здесь нужно учесть то, что большое значение Δ ведет к учету случаев пролета атомов на значительном расстоянии друг от друга без истинного столкновения. Однако расстояние Δ нельзя выбирать и слишком малым, так как при этом кратковременное отдаление атома от кластера будет считаться распадом кластера, а последующее приближение – новым взаимодействием, что также приведет к необоснованному увеличению частоты столкновений.
Критерий выбора Δ удобно сформулировать на примере взаимодействия двух атомов. Типичный димер, являющийся результатом столкновения атомов, образуется следующим образом: атомы начинают сильно притягиваться друг к другу и сближаются. При сближении притяжение сменяется отталкиванием, атомы тормозятся и затем могут разлететься или сформировать долгоживущий димер, в котором атомы вращаются вокруг центра масс [12]. Таким образом, необходимо подобрать Δ, чтобы столкновением считалось такое сближение атомов, при котором наблюдалось бы взаимодействие отталкивательной частью потенциала.
Для формулировки критерия правильного определения Δ рассмотрим функции распределения димеров меди по времени жизни (рис. 1), полученные для $\Delta = 5$ Å (сплошная линия) и $\Delta = 4.95$ Å (пунктир). Под временем жизни димера подразумевается интервал времени, в течение которого расстояние между центрами масс его атомов равно величине, меньшей или равной $\Delta .$ Функция распределения димеров по времени жизни $f\left( \tau \right)$ найдена на интервале $0 < \tau < 1.5{\text{ п с }}$ с шагом дискретизации $\delta \tau = 15{\text{ ф с }}{\text{.}}$ Полученная функция нормировалась так, чтобы площадь под кривой была равна единице. Промежуток времени, в течение которого набиралась статистика, составил 100 нс.
Рис. 1.
Функция распределения димеров меди по временам жизни при ${{T}_{{{\text{Ar}}}}} = 1500{\text{ K,}}$ ${{\rho }_{{{\text{Cu}}}}} = 0.25{\text{ к г }}{{{\text{м }}}^{{ - 3}}}{\text{:}}$ сплошная линия – $\Delta = 5$ Å, пунктир – $4.95.$
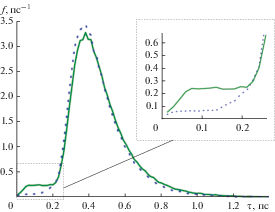
На графике функции, построенной для $\Delta = 5$ Å, видно, что при малых значениях τ имеется препик или плато, тогда как на графике для $\Delta = 4.95$ Å такая особенность отсутствует. Анализ показывает, что плато формируется димерами, атомы которых не сближались, а пролетали мимо практически без отклонения. Если постепенно уменьшать значение Δ, то можно добиться, чтобы такие димеры не рассматривались при анализе. Исходя из этого сформулировано следующее правило для выбора значения Δ: это наибольшее расстояние, при котором атомы, пролетающие мимо друг друга без искривления траекторий, не дают вклада в статистику столкновений (отсутствует плато при малых τ в функции распределения). Расчеты показали, что для потенциала погруженного атома, используемого для описания взаимодействия атомов меди, это расстояние можно выбрать равным $\Delta = 4.95$ Å.
Определение радиусов кластеров. Анализ зависимости радиуса кластера от числа атомов в нем осуществлялся следующим образом. По данным о количестве столкновений между n-атомными кластерами и атомами, о значениях скоростей теплового поступательного движения и значениях концентраций вычислялось сечение взаимодействия кластер–атом ${{\sigma }_{{n,m}}}$ по формуле (2) с m = 1. При этом относительная скорость кластеров находилась как ${{\text{v}}_{{n,m}}} = \sqrt {\bar {v}_{n}^{2} + \bar {v}_{m}^{2}} $ [7], где ${{\bar {v}}_{n}}$ и ${{\bar {v}}_{m}}$ – средние скорости теплового поступательного движения центра масс кластеров, найденные прямым усреднением по ансамблю. Вычислялся “эффективный” радиус атома меди как ${{R}_{{\text{1}}}} = 0.5\sqrt {{{{{\sigma }_{{{\text{1}}{\text{.1}}}}}} \mathord{\left/ {\vphantom {{{{\sigma }_{{{\text{1}}{\text{.1}}}}}} \pi }} \right. \kern-0em} \pi }} .$ Далее рассчитывались радиусы кластеров с $n > {\text{1}}$ как ${{R}_{n}} = \sqrt {{{{{\sigma }_{{n,1}}}} \mathord{\left/ {\vphantom {{{{\sigma }_{{n,1}}}} \pi }} \right. \kern-0em} \pi }} - {{R}_{1}}.$
Такой же алгоритм применялся для вычисления радиусов кластеров по данным о сечениях столкновений кластер–димер ${{\sigma }_{{n,2}}}$ и кластер–тример ${{\sigma }_{{n,3}}}.$
На рис. 2 представлены зависимости радиусов кластеров от количества атомов в них. Символами на рис. 2 показаны радиусы, полученные из анализа частот столкновений кластеров с атомами, димерами и тримерами. МД-моделирование проводилось для 29 начальных конфигураций с начальными температурами 300 и 1500 K и плотностями пара меди 0.1–0.3 кг/м3. Интервал времени для подсчета количества столкновений выбран равным 10 нс. Это значение удовлетворяет двум условиям: интервал должен быть малым настолько, чтобы на его протяжении концентрации и средние скорости не менялись значительно, и должен обеспечить количество столкновений, достаточно большое для статистического анализа. Время рассмотрения эволюции каждой конфигурации составляло ~500 нс, так что оказалось возможным получить несколько (верхний предел равен 50) значений сечения. Погрешности, показанные на графике, получены в результате статистической обработки этих данных.
Рис. 2.
Зависимости радиусов кластеров от числа атомов в них при $\Delta = 4.95$ Å: 1 – аппроксимация $R = 2.46{{(n - 0.052)}^{{0.31}}},$ 2 – (1), 3 – ${{T}_{{{\text{Ar}}}}}$ = 1500 K, 4 – 300.
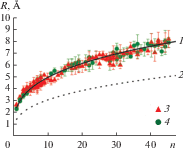
Анализ графика на рис. 2 позволяет сделать следующие выводы: в рассмотренных условиях радиусы кластеров не проявляют зависимости от температуры или плотности пара; данные хорошо аппроксимируются функцией
где показатель степени при n близок к классическому кубическому корню. График, полученный из анализа частоты столкновений, лежит намного выше графика, построенного для “жидкой капли”. Так, оценка, проведенная по (3), дает, что при n = 50 расчетная средняя плотность кластера меди меньше плотности жидкой меди в 3.5 раза. Так как используемый потенциал правильно описывает плотность твердой фазы [9] и равновесных молекул, можно заключить, что такая низкая плотность является свойством малых кластеров в процессе их роста. Это связано с тем, что состояние вещества в малых кластерах (n < 50) похоже на газообразное, при котором атомы могут двигаться во всем объеме кластера. Визуализации движения атомов в малых кластерах, полученных в процессе конденсации, подтверждают это.ЗАКЛЮЧЕНИЕ
Таким образом, “эффективный” радиус кластера, содержащего n атомов, удовлетворительно описывается формулой ${{R}_{n}} = 0.5\Delta {{n}^{{{1 \mathord{\left/ {\vphantom {1 3}} \right. \kern-0em} 3}}}}.$
Разность между радиусами кластеров, полученными в настоящей работе, и результатами применения формулы (1) уменьшается (в процентном отношении) с увеличением n. Так, для $n = {\text{2}}$ разность составляет ~40.5% от значения радиуса кластера, и уменьшается до 34% для $n = 45.$ Оценка для кластеров бóльшего размера показывает уменьшение относительной разности, например, для $n = {\text{1}}0{\kern 1pt} 000$ значение разности составляет ~25%. Иначе говоря, в пределе при увеличении n плотность кластера, возможно, будет приближаться к плотности объемной жидкости.
Работа выполнена при поддержке Правительства РФ (Постановление № 211 от 16.03.2013 г.), соглашение № 02.A03.21.0011, а также частично в рамках выполнения госзадания ИМЕТ УрО РАН (тема № 0396-2017-0008).
Список литературы
Woehl T.J., Park C., Evans J.E., Ilke Arslan, Ristenpart W.D., Browning N.D. Direct Observation of Aggregative Nanoparticle Growth: Kinetic Modeling of the Size Distribution and Growth Rate // Nano Lett. 2014. V. 14. P. 373.
Романов Н.А., Номоев А.В., Калашников С.В. Исследование функций распределения наночастиц по размерам. Механизм образования наночастиц, полученных методом испарения электронным пучком // Вестн. БГУ. 2013. Т. 3. С. 93.
Вараксин А.Ю. Кластеризация частиц в турбулентных и вихревых двухфазных потоках // ТВТ. 2014. Т. 52. № 5. С. 777.
Смирнов Б.М. Процессы с участием кластеров и малых частиц в буферном газе // УФН. 2011. Т. 181. С. 713.
Angélil R., Diemand J., Tanaka K.K., Tanaka H. Properties of Liquid Clusters in Large-scale Molecular Dynamics Nucleation Simulations // J. Chem. Phys. 2014. V. 140. Iss. 7. 074303.
Venkatesh R., Lucchese R.R., Marlow W.H., Schulte J. Thermal Collision Rate Constants for Small Nickel Clusters of Size 2–14 Atoms // J. Chem. Phys. 1995. V. 102. P. 7683.
IUPAC, Compendium of Chemical Terminology – the Gold Book. 2nd ed. 1997.
Plimpton S. Fast Parallel Algorithms for Short–Range Molecular Dynamics // J. Comput. Phys. 1995. V. 117. Iss. 1. P. 1.
Foiles S.M., Baskes M.I., Daw M.S. Embedded-atom-method Functions for the fcc Metals Cu, Ag, Au, Ni, Pd, Pt, and their Alloys // Phys. Rev. B. 1986. V. 33. P. 7983.
Morse M.D. Clusters of Transition-metal Atoms // Chem. Revs. 1986. V. 86. P. 1049.
Stillinger F.H. Rigorous Basis of the Frenkel-Band Theory of Association Equilibrium // J. Chem. Phys. 1963. V. 38. P. 1486.
Korenchenko A.E., Vorontsov A.G., Gel’chinskii B.R., Sannikov G.P. Statistical Analysis of Dimer Formation in Supersaturated Metal Vapor Based on Molecular Dynamics Simulation // Physica A. 2018. V. 496. P. 147.
Дополнительные материалы отсутствуют.
Инструменты
Теплофизика высоких температур